Introduction
Pancreatic cancer is a highly aggressive and almost
lethal neoplasm among the most intractable of human malignancies
with a 5-year survival rate of <10% (1,2).
Pancreatic cancer is the fourth leading cause of cancer-related
mortality worldwide. The lethality of pancreatic cancer is
characterized by the rapid invasion of the surrounding tissue,
early metastatic disease, and poor responses to standard
chemotherapy and radiotherapy (3).
Surgical resection and chemotherapeutic regimens, which include
gemcitabine, currently provide optimal clinical benefits.
Gemcitabine, a cytidine analog that inhibits DNA synthesis and DNA
repair, has been the reference drug for the treatment of this often
fatal disease for >10 years (4). However, gemcitabine only produces a
modest survival benefit (6–8 months) for patients with pancreatic
cancer (4). The combination of
5-fluorouracil, leucovorin, irinotecan and oxaliplatin (FOLFIRINOX)
has recently achieved more effective responses than gemcitabine in
patients with advanced pancreatic cancer (5). However, the use of FOLFIRINOX results
in greater toxicity than with treatment with gemcitabine and still
only achieves a median overall survival of approximately 11 months
(5). A number of novel anticancer
drugs have been introduced over the past decade with the aim of
improving the survival of patient with pancreatic cancer. However,
despite continuous efforts to develop novel agents, none of the
currently available chemotherapeutic agents have an objective
response rate >10% (6–8). Therefore, the development of novel
and effective therapeutic agents or novel effective combination
regimens is essential, and more effective and safe chemotherapeutic
treatments are required.
Ras transformation renders cells sensitive to
reactive oxygen species (ROS)-induced cell death (9) and pancreatic cancers exhibit an
extremely high mutation rate of K-ras (>90%) (10). Cotylenin A (CN-A), which has a
diterpenoid tricarbocyclic skeleton, is produced by
plant-pathogenic fungi and has been shown to induce the
differentiation of human myeloid leukemia cells and apoptosis or
the growth arrest of human carcinoma cells in the presence of
interferon-α or rapamycin, respectively (11,12).
We recently reported that CN-A significantly potentiated the
arsenic trioxide-induced inhibition of cell growth in human breast
cancer cells (13). We found that
this synergistic growth inhibitory effect of CN-A plus arsenic
trioxide was significantly reduced by treatment with
N-acetylcysteine (NAC), a typical ROS scavenger (13). These findings thus suggest that
CN-A acts as a potent sensi-tizer of the antitumor activity of ROS
inducers against solid tumors, including breast cancer and
pancreatic cancer.
Ferroptosis has recently emerged as a new form of
programmed cell death that has been characterized as non-apoptotic
peroxidation-induced cell death contingent on the availability of
iron and ROS (14–17). We recently reported that CN-A and
phenethyl isothiocyanate (PEITC; a dietary anticarcinogenic
compound and inducer of ROS) (18,19)
synergistically inhibited the proliferation of pancreatic cancer
MIAPaCa-2 and PANC-1 cells (20).
Combined treatment with CN-A and PEITC synergistically induced the
generation of ROS. Antioxidants (NAC and trolox), ferroptosis
inhibitors [ferrostatin-1 (Ferr-1) and liproxstatin-1 (Liprox)],
and the lysosomal iron chelator deferoxamine (DFO) abolished
synergistic cell death. An apoptosis inhibitor [Z-VAD-FMK (Z-VAD)]
and the necrosis inhibitor necrostatin-1 (Nec-1) did not inhibit
synergistic cell death (20).
These findings indicated that synergistic cell death induced by
combined treatment with CN-A and PEITC was mainly due to the
induction of ferroptosis, whereas CN-A or PEITC alone did not
induce the ferroptosis of pancreatic cancer cells. Therefore, there
may be more effective combination treatment regimens using CN-A and
other ROS inducers against pancreatic cancer cells.
Piperlongumine (PL) is a biologically active
alkaloid that largely exists in the long pepper (Piper
longum L.). PL selectively induces the death of numerous cancer
cell lines in vitro and in vivo, including pancreatic
cancer, breast cancer, and leukemia, but does not exhibit
anti-proliferative behavior in non-transformed cells (21–24).
Therefore, PL is an attractive molecule for use in the development
of novel treatment regimens for these types of cancer (25). PL directly binds to and inhibits
the antioxidant enzyme, glutathione S-transferase Pi 1, resulting
in elevated intracellular ROS levels and subsequent apoptotic cell
death in cancers with no apparent toxicity to normal cells
(21,25). Apart from the induction of
apoptosis, PL has been shown to cause cell cycle arrest (26), induce autophagy (27), and to inhibit migration and
invasion (28,29). However, the potential of PL to
induce ferroptosis, particularly in pancreatic cancer cells, has
not yet been demonstrated, at least to the best of our knowledge.
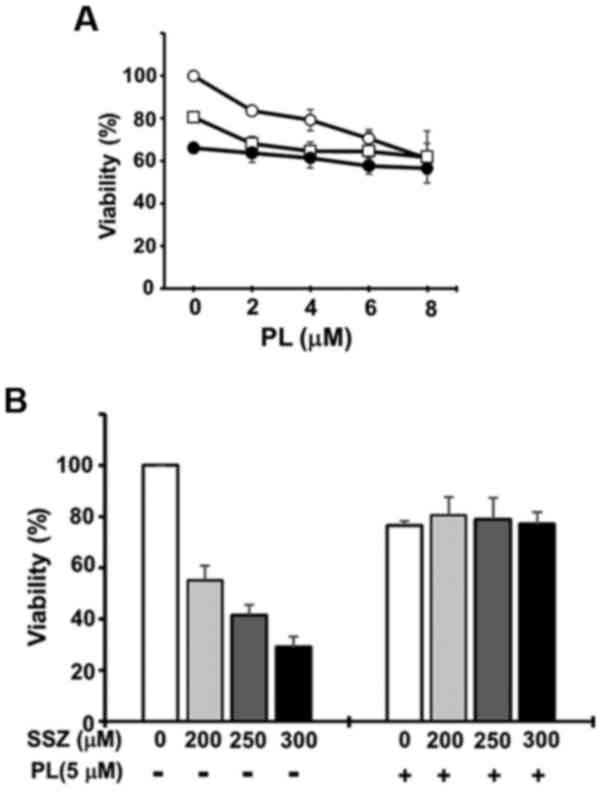
In this study, we report that PL alone induces the ferroptotic
death of pancreatic cancer cells and that a combined treatment with
PL plus CN-A and/or a lower dose of sulfasalazine (SSZ; approved
for clinical use for several diseases, such as rheumarthritis and
one of the known ferroptosis inducers) (30) may further induce the ferroptotic
death of pancreatic cancer cells.
Materials and methods
Cell culture
The human pancreatic cancer cell lines, MIAPaCa-2,
PANC-1, CFPAC-1, and BxPC-3, were purchased from ATCC (Manassas,
VA, USA). These pancreatic cancer cells were cultured in RPMI-1640
supplemented with 10% fetal bovine serum at 37°C in a humidified
atmosphere of 5% carbon dioxide in air. Mouse embryonic fibroblasts
(MEFs) were obtained from ATCC (ATCC CRL-2977) and cultured in
Dulbecco's modified Eagle's medium (DMEM) containing 0.1 mM
non-essential amino acids, 0.05 mM 2-mercaptoethanol and 10% fetal
bovine serum at 37°C in a humidified atmosphere of 5% carbon
dioxide in air.
Materials
CN-A was prepared as previously described (31). PL,
3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT),
NAC, Ferr-1, 3-methyladenine (3-MA), DFO, ciclopirox (CPX),
RPMI-1640 medium, DMEM and arsenic trioxide were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Z-VAD, Nec-1 and Liprox were
purchased from Selleckchem (Houston, TX, USA). PD146176 was
obtained from Cayman Chemical Co. (Ann Arbor, MI, USA). SSZ was
obtained from Tokyo Chemical Industry (Tokyo, Japan).
Anti-glutathione peroxidase 4 (GPX4) antibody (Cat. no. ab125066)
was obtained from Abcam (Tokyo, Japan). Anti-cleaved
poly(ADP-ribose) polymerase (PARP) (Cat. no. 5625), anti-cleaved
capase-3 (Cat. no. 9664), anti-LC3B (Cat. no. 2775) and anti-α/β
tubulin (Cat. no. 2148) antibodies were purchased from Cell
Signaling Technology (Danvers, MA, USA). Anti-p53 antibody (Cat.
no. sc-126) was obtained from Santa Cruz Biotechnology (Santa Cruz,
CA, USA).
Cell viability
Cell viability was assessed by MTT assay. The cells
were seeded at 3×104 cells/ml in a 24-well multi-dish.
After being cultured with or without the test compounds for 16 h,
500 μl of DMSO were added to each well to solubilize
formazan in viable cells. The plates were analyzed by measuring the
optical density at 540 nm, as previously described (12).
Measurement of glutathione (GSH)
levels
Cellular GSH contents were measured using a GSH-Glo™
Glutathine Assay kit (Promega, Madison, WI, USA) according to the
instructions provided by the manufacturer.
Western blot analysis
The cells were packed after washing with cold PBS
and then lysed at a concentration of 1×107 cells/ml in
lysis buffer CelLytic™-M (Sigma-Aldrich) supplemented with a
proteinase inhibitor cocktail and phosphatase inhibitor cocktail
1/2 (Sigma-Aldrich). Equal amounts of protein were separated on
10–20% SDS-polyacrylamide gels (Wako, Osaka, Japan). Proteins were
electrophoresed on gels and transferred onto Immobilon-P membranes
(Millipore, Bedford, MA, USA) using the primary antibodies. An
anti-rabbit or anti-mouse IgG HRP-linked antibody (Cell Signaling
Technology) was used as a secondary antibody (1:2,000 dilution).
The bands were identified by treatment with Immune-Star™ HRP
chemiluminescence (Bio-Rad Laboratories, Hercules, CA, USA) for 5
min at room temperature and detected using a FujiLumino Image
Analyser LAS-4000 system (Fuji Film Co., Ltd., Tokyo, Japan)
(13). All western blots shown are
representative of at least 3 independent experiments.
Preparation of positive control lysate
for cleaved PARP and cleaved caspase-3 in western blot
analysis
The MIAPaCa-2 cells were treated with 10
μg/ml CN-A and 3 μM arsenic trioxide for 72 h. Whole
cell lysate was obtained and used in western blot analysis as
previously described (13).
Measurement of ROS generation
The production of ROS was monitored using a Muse
cell analyzer (Millipore, Billerica, MA, USA), and the experimental
protocol followed the description provided with the kit (Muse
Oxidative Stress kit, Millipore). The MIAPaCa-2 cells were treated
with the indicated drugs for 4 h to induce ROS production, and ROS
levels were then measured using the Muse Oxidative Stress kit. Two
populations of cells are distinguished in this assay: ROS (−) (live
cells) and ROS (+) (cells exhibiting ROS).
Measurement of Annexin V-positive
cells
Annexin V staining and the detection of Annexin
V-positive cells were performed using the Muse cell analyzer, and
the experimental protocol followed the description provided with
the kit (Muse Annexin V & Dead Cell Assay kit) (Millipore). The
assay utilizes Annexin V to detect phosphatidylserine (PS) on the
external membranes of apoptotic cells.
Statistical analysis
Values were compared using a two-tailed Student's
t-test. Differences between the means were considered to be
significant when P-values were <0.05.
Results
PL induces the ferroptotic death of human
pancreatic cancer cells
The PANC-1 cells were pre-treated with the ROS
scavenger, NAC, the ferroptosis inhibitors (Ferr-1 and Liprox), or
the iron chelator, DFO, for 2 h, and were then further cultured in
the presence or absence of PL for 16 h. PL dose-dependently
decreased PANC-1 cell viability: 14 μM PL decreased cell
viability to approximately 10% of the control. Consistent with
findings from previous studies indicating that increased ROS levels
are critical for cell death induced by PL (21,22,25),
NAC (3 mM) almost completely abrogated the PL-induced decrease in
the viability of the PANC-1 cells (Fig. 1B). Ferroptosis is a recently
recognized ROS- and iron-dependent form of regulated cell death
that is accompanied by the accumulation of lipid peroxidation
products (21). Since this process
may be inhibited by lipid peroxidation inhibitors (Ferr-1 and
Liprox) and an iron chelator (DFO), we examined the effects of
these ferroptosis inhibitors. As shown in Fig. 1C and D, Ferr-1 (1 μM) and
Liprox (1 μM) significantly abolished PL (>8
μM)-induced cell death. DFO (0.2 mM) also potently inhibited
PL-induced cancer cell death. The viability of the PANC-1 cells
following treatment with PL (14 μM) in the presence of DFO
was still >60% (Fig. 1E). CPX
(another intracellular iron chelator) also significantly reduced
PL-induced cancer cell death (Fig.
1F). Similar results were also obtained when cell viability was
measured by trypan blue dye exclusion assay (data not shown).
Similar results were also obtained with the MIAPaCa-2 cells (data
not shown). Consistent with the established role for
lipoxygenase-catalyzed lipid hydroperoxidation for ferroptosis
(17,33), we observed that treatment with the
lipoxygenase inhibitor, PD146176, prevented the cancer cells from
undergoing PL-induced cell death (Fig.
1G). Intracellular GSH is a very important endogenous
antioxidant used in the defense against ferroptosis (16,33).
Thus, we examined whether PL can deplete GSH in the MIAPaCa-2
cells. The content of cellular GSH in the cells treated with 10
μM PL for 4 h was markedly depleted (Fig. 1H). Ferroptosis inducers
preferentially kill cancer cells that accumulate mutant p53 protein
(32,33). Regarding this point, we noted that
all of the pancreatic cancer cell lines used in this study
expressed mutant p53, although PL had no obvious effect on the
amount of p53 (Fig. 1I). PL did
not decrease GPX in pancreatic cancer cells (Fig. 1I). However, GSH is a co-factor for
GPX activity and GSH-depleting reagents inhibit GPX activity
(16). Since we actually found
that PL markedly depleted GSH (Fig.
1H), these results suggest that PL may inhibit GPX activity. PL
did not induce the expression of typical apoptotic markers, such as
cleaved PARP and cleaved caspase-3 (Fig. 1J). PL also did not affect the
LC3-II/LC3-I ratio, an indicator of autophagy induction (Fig. 1K). These results thus suggest that
PL alone induces cancer cell death through, at least in part, the
induction of ferroptosis in pancreatic cancer cells.
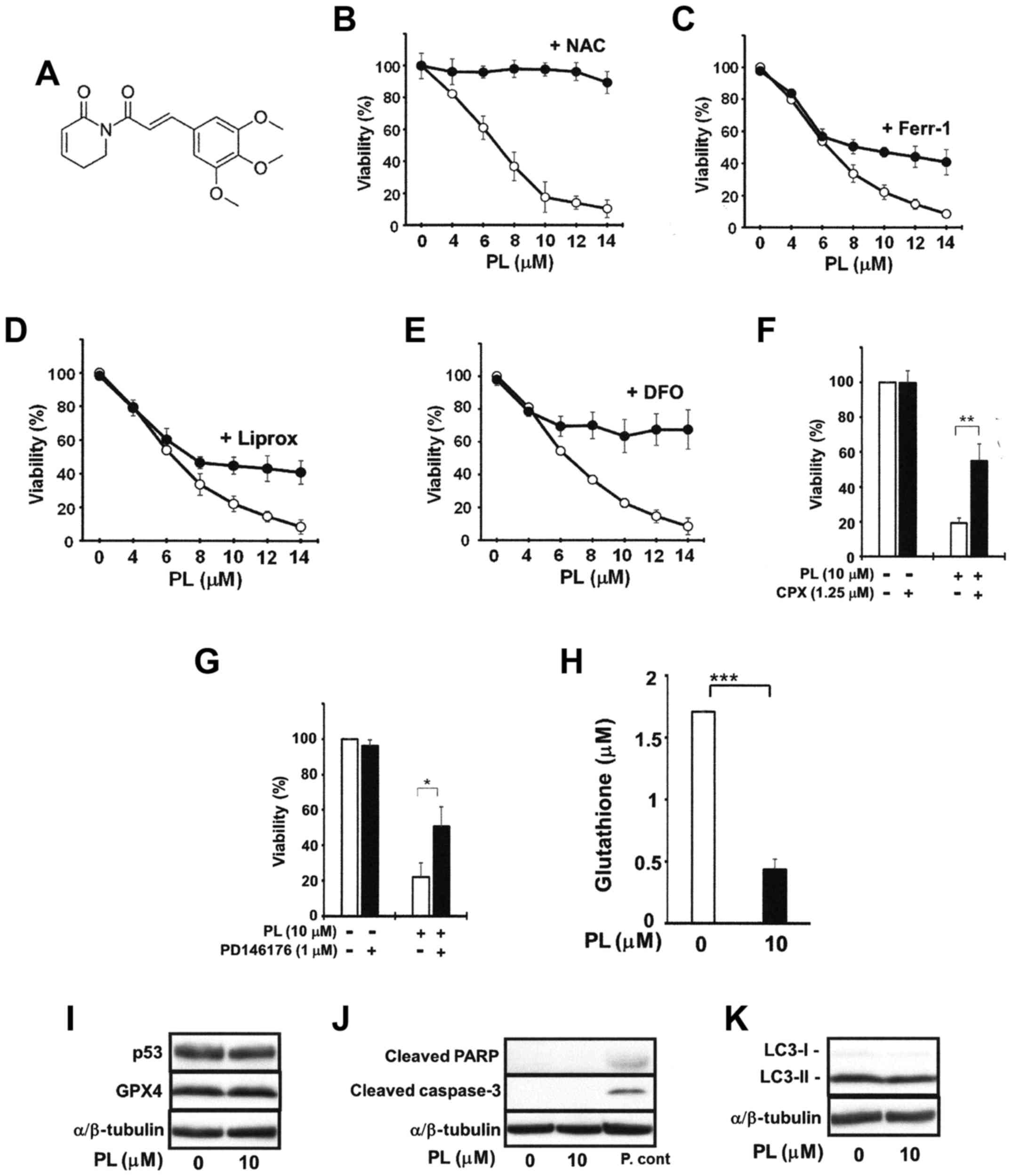 | Figure 1Effects of ferroptosis inhibitors on
the piperlongumine (PL)-induced death of PANC-1 cells. (A)
Structure of PL. PANC-1 cells (3×104 cells/ml) were
cultured with the indicated concentrations of PL in the absence
(open circles) or presence of (B) 3 mM N-acetylcysteine
(NAC), (C) 1 μM of ferrostatin-1 (Ferr-1), (D) 1 μM
of liproxstatin-1 (Liprox), (E) or 0.2 mM deferoxamine (DFO)
(closed circles) for 16 h. The MIAPaCa-2 cells were treated with 10
μM PL in the presence or absence of (F) 1.25 μM
ciclopirox (CPX) or (G) 1 μM PD146176 for 16 h. The cells
were treated with NAC, Ferr-1, Liprox, DFO, CPX, or PD146176 for 2
h prior to PL treatment. Cell viability was then assessed by MTT
assay. (H) Glutathione (GSH) levels in MIAPaCa-2 cells (8,000
cells/well) treated with PL for 4 h. Values are expressed as the
means ± standard deviation of 3 measurements. Western blot analyses
for (I) p53 and GPX4, (J) cleaved PARP and cleaved caspase-3, and
(K) LC3-I/II proteins. The MIAPaCa-2 cells were cultured with or
without 10 μM PL for 16 h. P.cont, positive control for
cleaved PARP and cleaved caspase-3 [MIAPaCa-2 cells were treated
with 10 μg/ml CN-A and 3 μM arsenic trioxide for 72 h
as previously described (13)].
Whole cell lysates were used for western blot analysis. The protein
expression of α/β-tubulin served as the loading control. Similar
results were obtained in two additional experiments. *P<0.05,
**P<0.01 and ***P<0.001. |
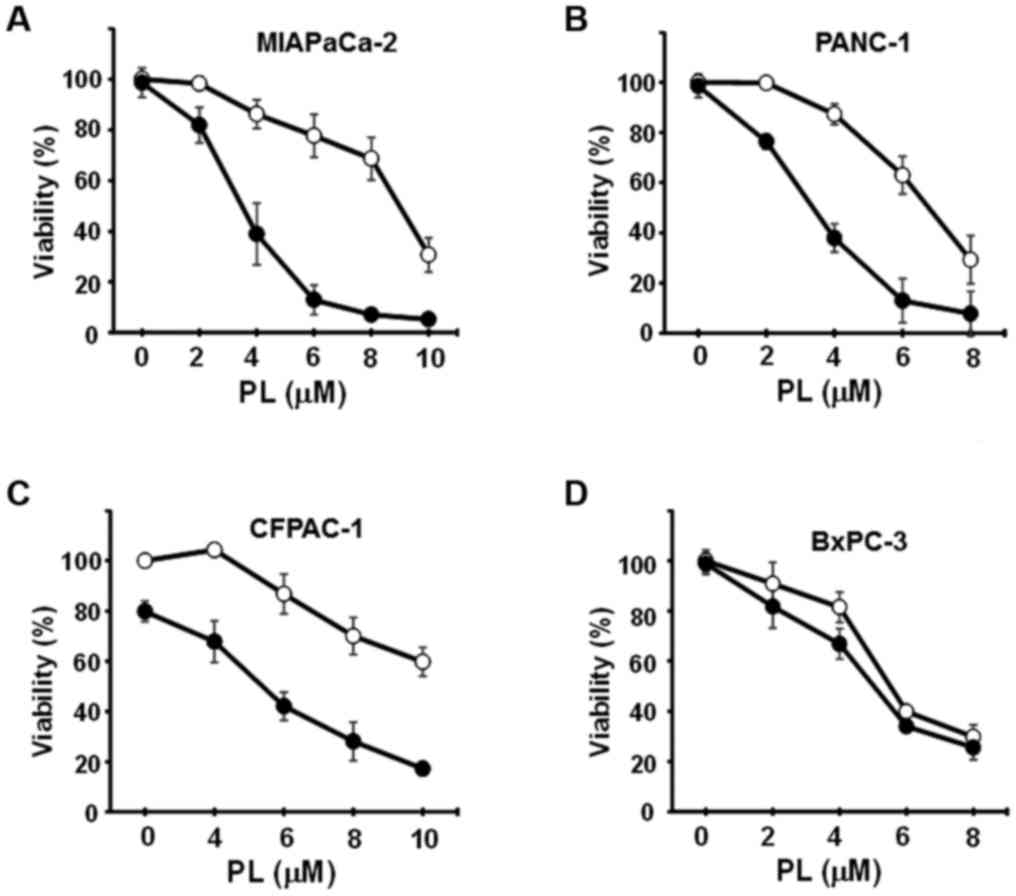
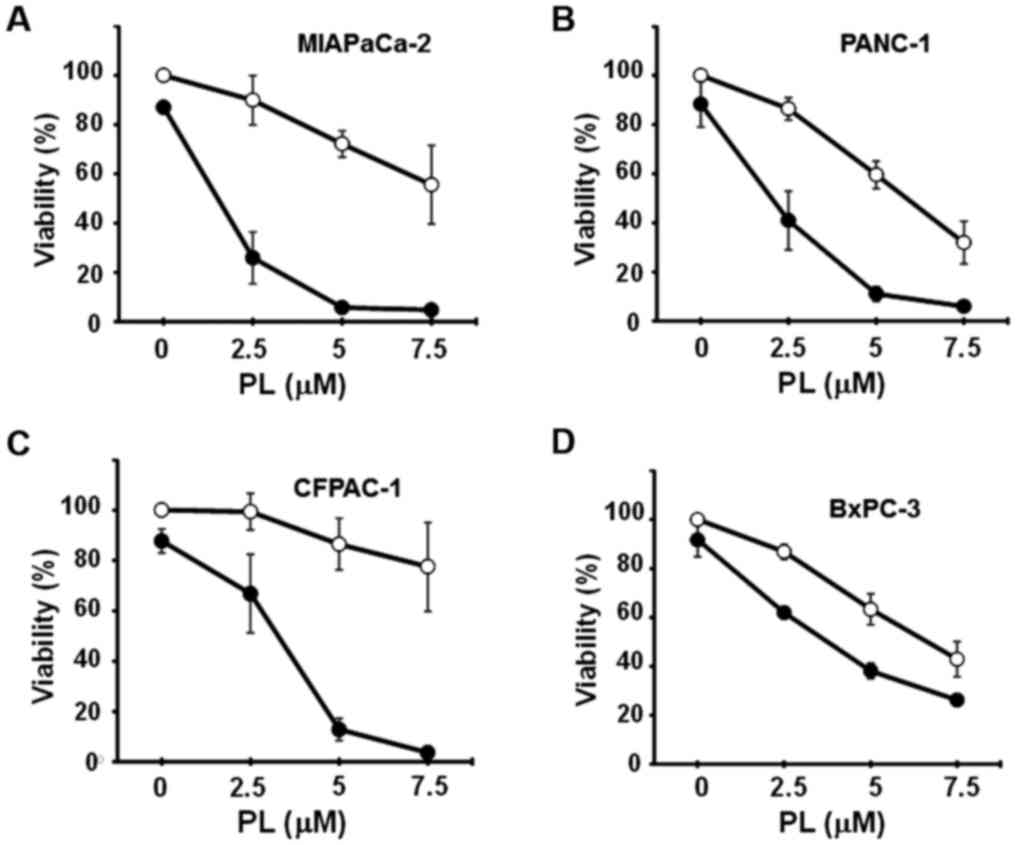
CN-A and PL synergistically induce the
death of pancreatic cancer MIAPaCa-2 and PANC-1 cells
We have recently reported that combined treatment
with CN-A and PEITC (another ROS inducer) induced the ferroptotic
death of MIAPaCa-2 and PANC-1 cells, whereas PEITC alone, even at
higher concentrations, did not (20). In this study, we examined whether
CN-A enhances the PL-induced death of pancreatic cancer cell lines.
The MIAPaCa-2, PANC-1, CFPAC-1 and BxPC-3 cells were cultured with
or without CN-A, PL, or CN-A plus PL for 16 h. Treatment with CN-A
alone was shown as a closed circle at the 0 μM PL position
(Fig. 2). CN-A and PL
synergistically reduced the viability of the MIAPaCa-2 and PANC-1
cells (Fig. 2A and B). Although PL
alone, even at a higher concentration (8 μM), reduced the
viability of the MIAPaCa-2 cells to ~70% and CN-A (15 μg/ml)
alone did not influence the viability of the MIAPaCa-2 cells, PL at
4, 6, or 8 μM reduced the viability of the MIAPaCa-2 cells
to ~40, 15, or 10%, respectively, in the presence of CN-A (15
μg/ml) (Fig. 2A). The
synergistic induction of cell death induced by CN-A plus PL was
also observed in the PANC-1 cells (Fig. 1B). CN-A and PL synergistically
induced the death of CFPAC-1 cells (Fig. 2C). On the other hand, CN-A only
slightly enhanced the PL-induced death of BxPC-3 cells, which did
not contain the K-ras mutation (Fig.
2D).
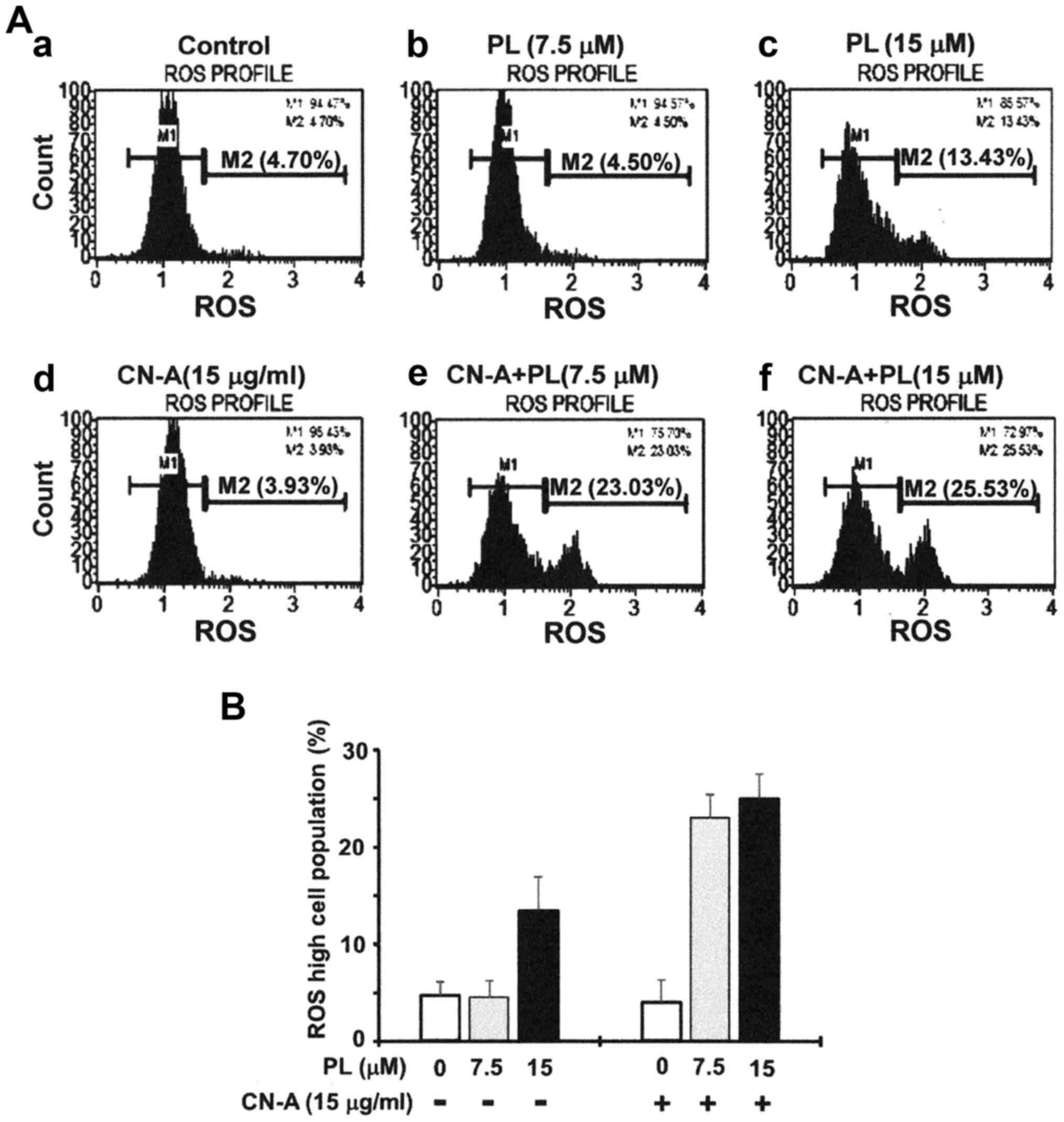
CN-A enhances the PL-induced ferroptotic
death of pancreatic cancer cells
We then investigated whether the synergistic
induction of cancer cell death induced by combined treatment with
CN-A and PL was due to the induction of ferroptosis. We measured
the generation of ROS using the Muse cell analyzer. The MIAPaCa-2
cells were treated with 15 μg/ml CN-A and/or 7.5 or 15
μM PL for 4 h in order to induce ROS generation, and ROS
levels were then examined using the Muse Oxidative Stress kit.
Although CN-A (15 μg/ml) or 7.5 μM PL alone did not
increase the population of ROS-positive cells (<5%), treatment
with CN-A (15 μg/ml) plus 7.5 μ PL synergistically
increased the population of these cells (>20%) (Fig. 3A and B). At a higher concentration
(15 μM), PL alone significantly increased the population of
ROS-positive cells (~13%), and treatment with PL (15 μM)
plus CN-A resulted in a further increase in the population of
ROS-positive cells (~25%) (Fig. 3A and
B).
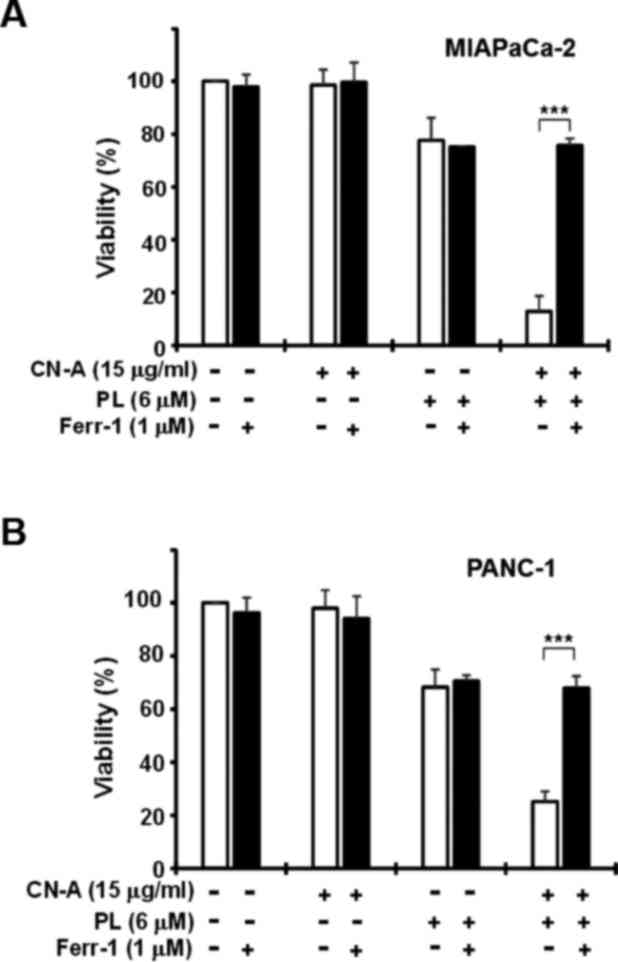
Treatment with CN-A (15 μg/ml) plus PL (6
μM) for 16 h markedly decreased PANC-1 cell viability to
~20–30%, while treatment with CN-A alone did not significantly
reduce cell viability and PL (6 μM) alone moderately reduced
viability to 55–70% (Fig. 4).
Treatment with the ROS scavenger, NAC (3 mM), almost completely
abrogated the reduction in cell viability induced by treatment with
PL alone (Figs. 1A and 4A) or CN-A plus PL (Fig. 4A). Treatment with the ferrop-tosis
inhibitor, Ferr-1 (1 μM), clearly abolished the CN-A plus
PL-induced death of the MIAPaCa-2 and PANC-1 cells (Fig. 5). Treatment with another
ferroptosis inhibitor, Liprox (1 μM), also protected the
PANC-1 cells from CN-A plus PL-induced death (Fig. 4B). The iron chelator, DFO (0.2 mM),
also markedly abollished CN-A plus PL-induced cancer cell death
(Fig. 4C). Similar results were
obtained with the MIAPaCa-2 cells (data not shown). These results
suggest that CN-A effectively enhances PL-induced ferroptotic
cancer cell death.
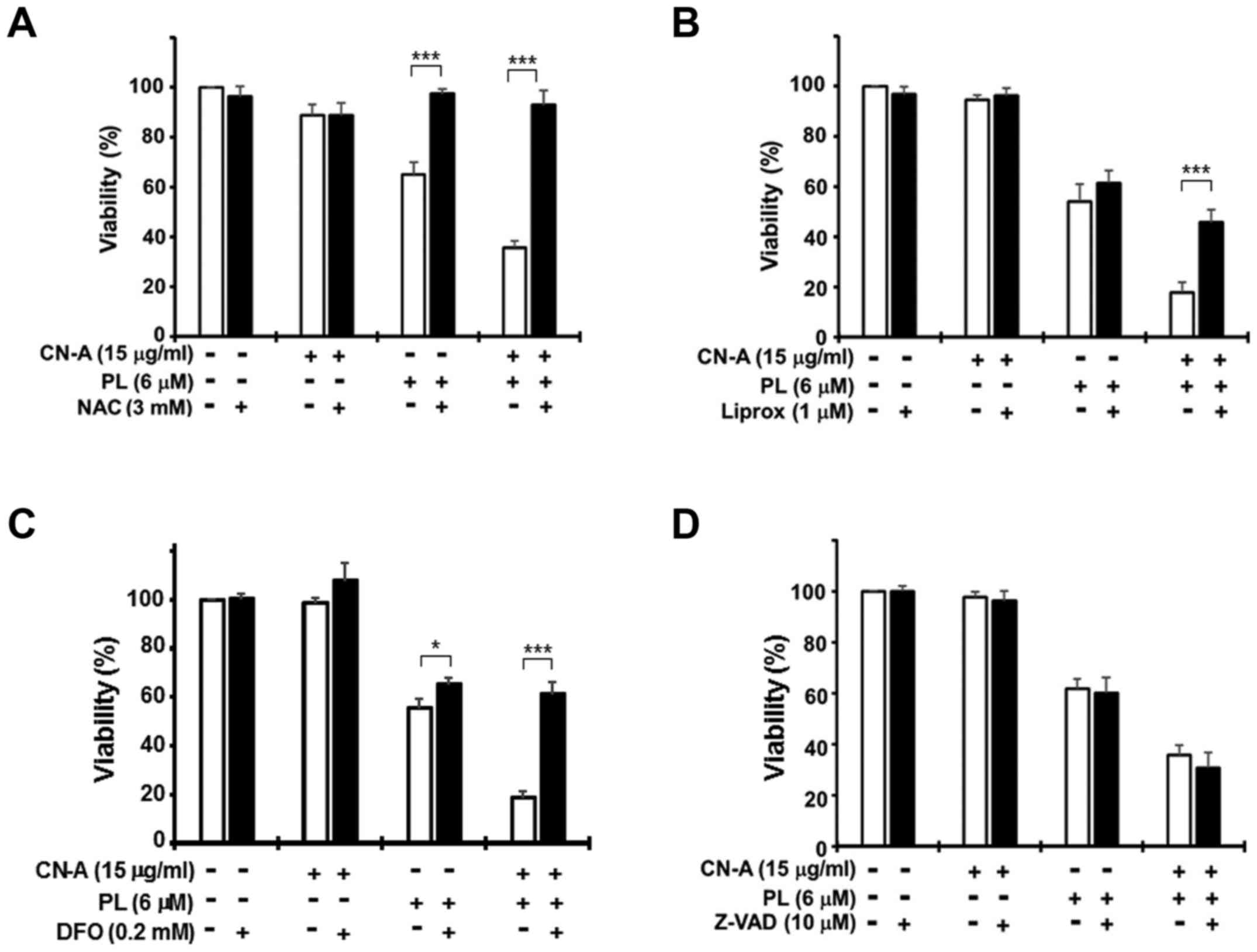 | Figure 4Effects of N-acetylcysteine
(NAC), liproxstatin-1 (Liprox), deferoxamine (DFO), or Z-VAD-FMK
(Z-VAD) on cotylenin A (CN-A) plus piperlongumine (PL)-induced cell
death. PANC-1 cells were treated with 15 μg/ml CN-A, 6
μM PL, or 15 μg/ml CN-A plus 6 μM PL in the
presence or absence of (A) 3 mM NAC, (B) 1 μM Liprox, (C)
0.2 mM DFO, or (D) 10 μM Z-VAD for 16 h. The cells were
treated with NAC, Liprox, DFO, or Z-VAD for 2 h prior to CN-A
and/or PL treatment. Cell viability was assessed by MTT assay.
Values are expressed as the means ± standard deviation of 3
measurements. *P<0.05 and
***P<0.001. |
Although PL (8 μM) or CN-A (15 μg/ml)
alone only induced the death of (Annexin V-positive) MIAPaCa-2
cells to a limited extent at 16 h, combined treatment with PL and
CN-A moderately induced (Annexin V-positive) MIAPaCa-2 cell death
(14.2%) (Fig. 3C and D). However,
DFO (0.2 mM) completely abolished the induction of (Annexin
V-positive) cell death indcued by CN-A plus PL (Fig. 3C and D), indicating that the
induction of (Annexin V-positive) cell death was an iron-dependent
form. Ferr-1 (1 μM) also abolished the induction of (Annexin
V-positive) cell death induced by CN-A plus PL (data not shown).
Furthermore, Z-VAD (a typical apoptosis inhibitor,) failed to
protect the cells against CN-A plus PL-induced death (Fig. 4D). Another apoptosis inhibitor,
Q-VD-OPH, also failed to protect the cells against CN-A plus
PL-induced cell death (data not shown). Combined treatment with
CN-A plus PL did not induce the expression of cleaved caspase-3
when the cells were examined using an apoptosis assay lit (R&D
Systems) (data not shown). These results thus suggest that CN-A
plus PL induced ferroptotic cancer cell death before the clear
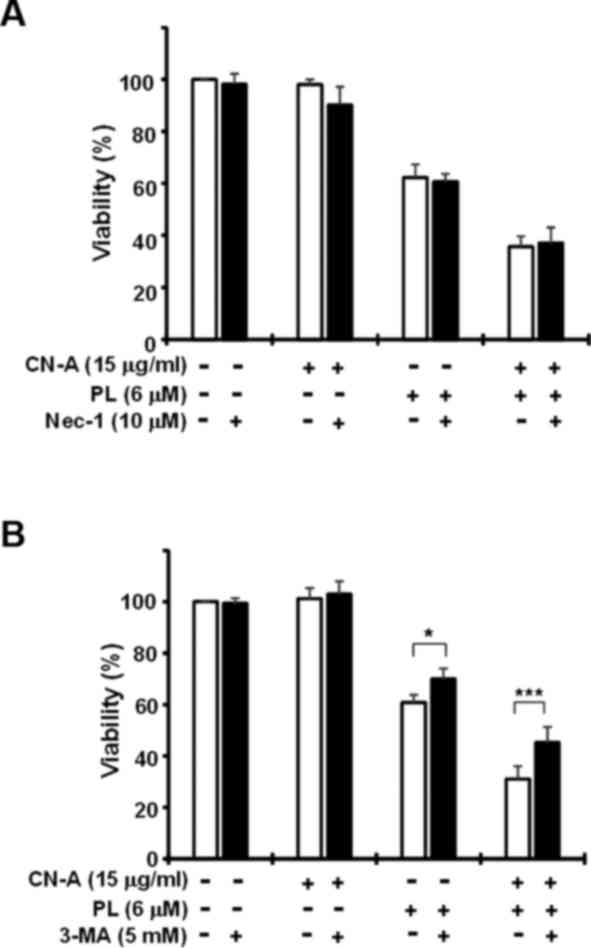
induction of apoptotic cell death. The necroptosis inhibitors,
Nec-1 (Fig. 6A) and Nec-1s (data
not shown), failed to suppress CN-A plus PL-induced cell death.
Since a recent study reported that ferroptosis was involved in an
autophagic cell death process (34), we examined the effects of the
autophagy inhibitor, 3-MA (Fig.
6B). 3-MA partially, but reproducibly prevented PL-induced cell
death and CN-A plus PL-induced cell death. Collectively, these
results suggest that synergistic cell death induced by combined
treatment with CN-A and PL was mainly due to the induction of
ferroptosis.
SSZ and PL synergistically induce the
death of pancreatic cancer cells
Our preliminary results revealed that at relatively
lower concentrations, erastin, one of the most representative
ferroptosis inducers, co-operated with PL and/or CN-A to induce
pancreatic cancer cell death (data not shown). We examined the
effects of combined treatment with PL and clinical drugs (SSZ,
sorafenib and auranofin) that have been reported to induce
ferroptosis (30), and found that
SSZ was the most effective enhancer of PL-induced ferroptotic
cancer cell death (Fig. 7 and data
not shown). We then investigated whether SSZ also potentiates
PL-induced cancer cell death using several pancreatic cancer cells.
The MIAPaCa-2, PANC-1, CFPAC-1 and BxPC-3 cells were cultured with
or without SSZ, PL, or SSZ plus PL for 16 h. Treatment with SSZ
alone was shown as a closed circle at 0 μM PL position
(Fig. 7). SSZ and PL
synergistically reduced the viability of the MIAPaCa-2, PANC-1 and
CFPAC-1 cells (Fig. 7A–C). SSZ
plus PL also more than additively induced the death of BxPC-3 cells
(Fig. 7D), whereas the effects of
this combination on viability were weaker than those in the
MIAPaCa-2, PANC-1 and CFPAC-1 cells. On the other hand, SSZ did not
enhance PEITC (a ROS inducer)-induced cell death and conversely
inhibited PEITC-induced cell death (data not shown). Furthermore,
PEITC inhibited the SSZ-induced death of pancreatic cancer cells
(data not shown).
SSZ enhances the PL-induced ferroptotic
death of pancreatic cancer cells
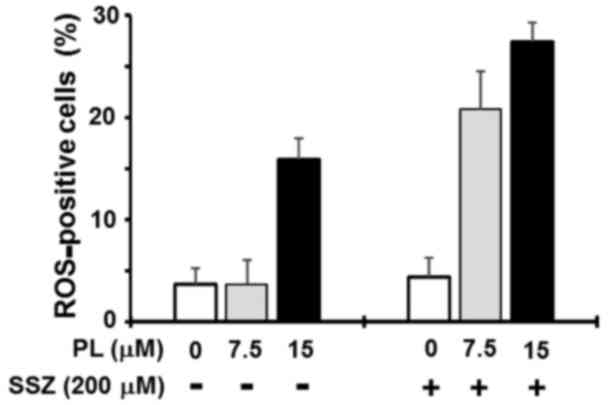
We then examined whether the synergistic induction
of cancer cell death induced by combined treatment with SSZ and PL
was due to the induction of ferroptosis. The MIAPaCa-2 cells were
treated with 200 μSSZ and/or 7.5 or 15 μ PL for 4 h
to induce ROS generation, and ROS production was then examined
using the Muse Oxidative Stress kit. Although SSZ at higher
concentrations (>300 μM) significantly increased the
population of ROS-positive cells in our assay system (data not
shown), SSZ (200 μM) alone did not increase the population
of ROS-positive cells (<5%) (Fig.
8). PL (7.5 μM) alone also did not increase the
population of ROS-positive cells (Fig.
8). By contrast, treatment with SSZ (200 μM) plus 7.5
μM PL markedly increased the population of ROS-positive
cells (>20%) (Fig. 8). At a
higher concentration (15 μM), PL alone significantly
increased the population of ROS-positive cells (~15%), and
treatment with PL (15 μM) plus SSZ (200 μM) resulted
in a further increase in the population of ROS-positive cells
(>25%) (Fig. 8). Treatment with
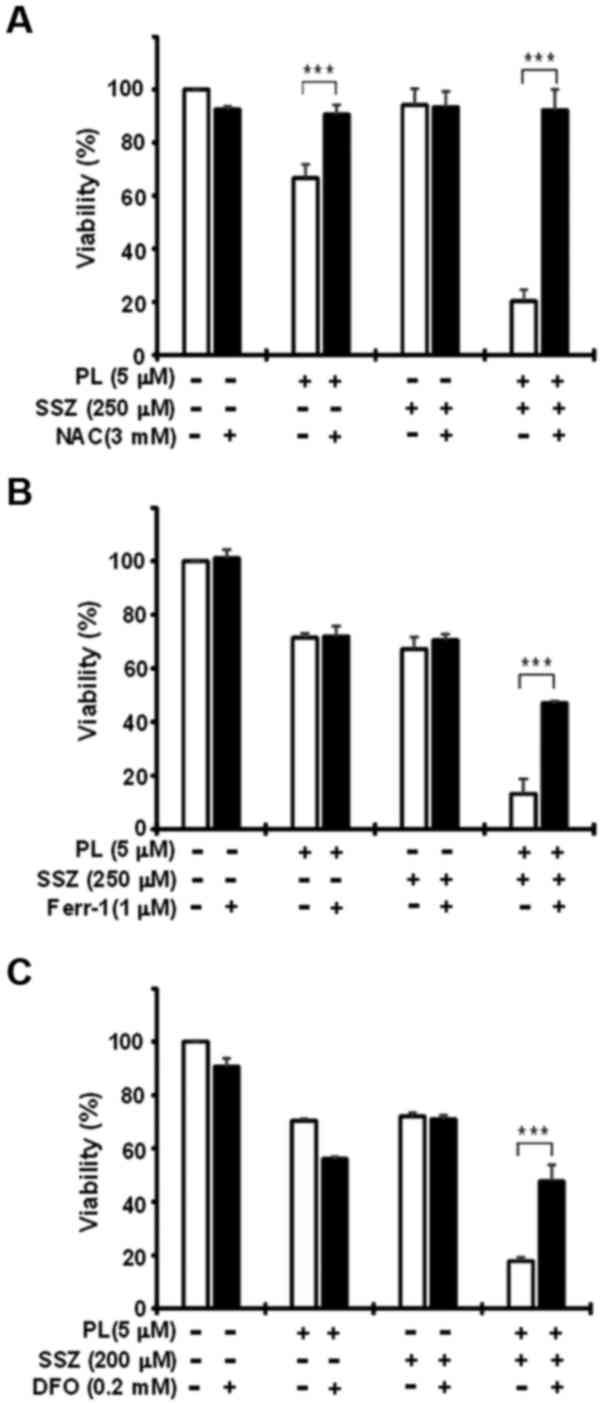
the ROS scavenger, NAC (3 mM), almost completely abolished the
reduction in cell viability induced by treatment with PL alone or
SSZ plus PL (Fig. 9A). The
ferroptosis inhibitor, Ferr-1 (1 μM), clearly abolished the
SSZ plus PL-induced death of PANC-1 cells (Fig. 9B). The co-addition of the iron
chelator, DFO (0.2 mM), significantly blocked cell death induced by
SSZ plus PL, demonstrating the iron dependency of cell death in
PANC-1 cells (Fig. 9C). Similar
results were obtained using the MIAPaCa-2 cells (data not shown).
These results suggest that SSZ also effectively enhances PL-induced
the ferroptotic death of pancreatic cancer cells.
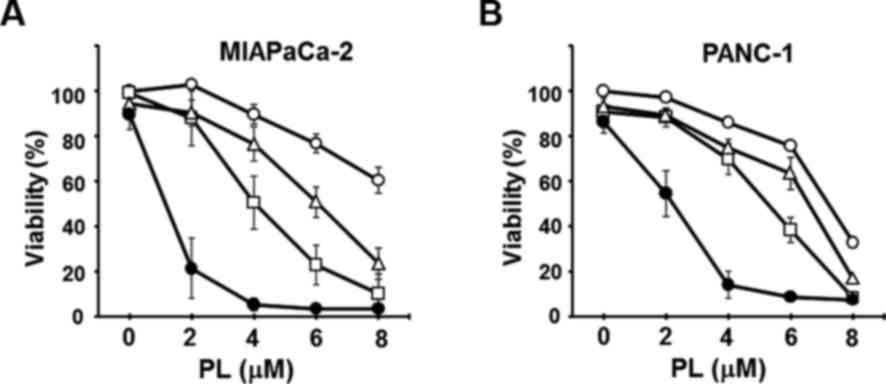
Triple combined treatment with PL, CN-A
and SSZ markedly inhibits the viability of pancreatic cancer
cells
We examined the effects of triple combined treatment
with PL, CN-A and SSZ on the viability of MIAPaCa-2 and PANC-1
cells (Fig. 10). We cultured
these cancer cells in the presence or absence of SSZ (100
μM), CN-A (15 μg/ml), or SSZ plus CN-A with or
without various concentrations of PL for 16 h. Although PL alone at
2 μM, CN-A alone at 15 μg/ml, or SSZ alone at 100
μM did not influence the viability of the MIAPaCa-2 cells,
and PL (2 μM) plus CN-A (15 μg/ml) or PL (2
μM) plus SSZ (100 μM) only slightly reduced viability
(~90%), triple combined treatment with PL (2 μM), CN-A (15
μg/ml) and SSZ (100 μM) synergistically reduced the
viability of the MIAPaCa-2 cells to ~20% (Fig. 10A). The synergistic induction of
cell death induced by triple combined treatment with PL, CN-A and
SSZ was also observed in the PANC-1 cells (Fig. 10B). The cancer cell death induced
by the triple combined treatment was also largely abolished by
pre-treatment with the ferroptosis inhibitor, Ferr-1 (data not
shown).
Effects of PL, CN-A and SSZ on the
viability of MEFs
We examined the effects of combined treatment with
CN-A and PL on non-transformed MEFs. The MEFs (without the addition
of 2-mecaptoethanol) were cultured with or without CN-A, PL, or
CN-A plus PL for 16 h. The viability of the MEFs was mildly and
dose-dependently decreased by PL, although the viability of the
MEFs was >60% in the presence of PL, even at a higher
concentration (8 μM) (Fig.
11A). Treatment with CN-A alone was shown as open square at 0
μM PL position. CN-A (15 μg/ml) or CN-A (15
μg/ml) plus SSZ (150 μM) did not co-operate with PL
to reduce the viability of the MEFs (Fig. 11A). Furthermore, we examined the
effects of PL (5 μM) on the SSZ-induced death of the MEFs.
The MEFs were cultured with or without PL (5 μM) in the
absence or presence of various concentrations of SSZ for 16 h.
Although SSZ and PL synergistically induced the death of the
pancreatic cancer cells (Fig. 7),
as described above, PL almost completely abolished the SSZ-induced
death of the MEFs (Fig. 11B).
Discussion
PL selectively induces the death of numerous cancer
cell lines, as well as cancerous tumors in animal models, including
pancreatic cancer, breast cancer and leukemia, but does not exhibit
anti-proliferative behavior in non-transformed cells, thus
rendering it a good candidate for cancer treatment (21–24).
PL directly binds to and inhibits the antioxidant enzyme,
glutathione S-transferase Pi 1, resulting in elevated intracellular
ROS generation and subsequent apoptotic cell death in cancers with
no apparent toxicity to normal cells (21,25).
Zou et al recently reported that PL interacted with
thioredoxin reductase 1 to induce the ROS-mediated apoptosis of
human gastric cancer cells (35).
In addition, PL has been shown to promote cell death by activating
several mechanisms, including apoptosis, autophagy and necrosis,
affecting the PI3K/AKT/mTOR, p38/JNK, MEK/ERK and NF-κB pathways
(22,27–29,36).
In the present study, we found that PL alone induced the death of
pancreatic cancer cells, at least in part, by the induction of
ferroptosis, the execution of which requires the accumulation of
ROS in an iron-dependent manner (16). Furthermore, we demonstrated that
PL-induced ferroptotic cell death was markedly enhanced by CN-A
and/or SSZ in pancreatic cancer cells. These results support the
potential of PL as an attractive therapeutic option that functions
through different mechanisms from those already reported in
pancreatic cancer cells.
We recently found that PEITC (a dietary
anticarcinogenic compound and ROS inducer) in the presence of CN-A
induced ferroptotic cancer cell death in pancreatic cancer cells,
whereas PEITC alone did not (20).
These findings indicate the potential of finding other effective
therapeutic compounds in combination with CN-A to induce the
ferroptotic death of pancreatic cancer cells. In the present study,
we demonstrated that co-treatment with PL and CN-A induced
synergistic cancer cell death in pancreatic cancer MIAPaCa-2 and
PANC-1 cells and that these effects on the viability of the cancer
cells were largely canceled by ferroptotic inhibitors and iron
chelator. We also unexpectedly found that the induction of
pancreatic cancer cell death by PL alone at relatively higher
concentrations was significantly reduced by ferroptotic inhibitors
and the iron chelator. These results suggest that PL induces
ferroptotic pancreatic cancer cell death and its effects are
markedly enhanced by CN-A. Furthermore, we demonstrated that PL and
SSZ (a clinical drug and ferroptosis inducer) synergistically
induced the death of pancreatic cancer MIAPaCa-2, PANC-1 and
CFPAC-1 cells (Fig. 7). Although
combined treatment with CN-A plus PEITC (20) or CN-A plus PL similarly induced
ferroptotic cancer cell death (Figs.
2 and 4Figure 5–6), the effects of combined treatment with
PEITC plus SSZ (data not shown) or PL plus SSZ (Figs. 7 and 9) differed markedly. PL and SSZ
synergistically induced cancer cell death and these effects were
largely abolished by the ferroptotic inhibitor, Ferr-1, or the iron
chelator, DFO (Figs. 7 and
9), whereas PEITC inhibited
SSZ-induced cancer cell death and SSZ did not enhance the
PEITC-induced death of pancreatic cancer cells (data not shown). We
found that the triple combined treatment with CN-A, PL and SSZ
markedly reduced the viability of the MIAPaCa-2 and PANC-1 cells
(Fig. 10). On the other hand, the
viability of non-transformed MEFs was mildly decreased by PL,
although the viability of the MEFs was >60% in the presence of
PL, even at a higher concentration (8 μM). CN-A or CN-A plus
SSZ did not co-operate with PL to reduce the viability of the MEFs.
Furthermore, PL almost completely abolished the SSZ-induced death
of the MEFs (Fig. 11). These
results suggest that PL is superior to PEITC as a partner in
combined treatment with CN-A plus SSZ as a possible treatment
regimen for pancreatic cancer.
The mechanisms underlying the interaction between
CN-A and PL have not yet been elucidated in detail. Ferroptosis is
regulated by several important events, such as ROS production, the
reduction of glutathione, the inhibition of lipid hydroperoxidase
GPX4 and the oxygenation of polyunsaturated
phosphatidylethanolamines by 15-lipoxygenase (16,17,32,33).
In this study, we reported that CN-A or SSZ (at lower
concentrations) enhanced ROS production induced by PL (Figs. 3A and 8). The effect of CN-A or SSZ on other
important ferroptosis-associated events in the presence of PL
remains to be determined. Kwon et al reported that heme
oxygenase-1 accelerated erastin-induced ferroptotic cell death
(37). Heme oxygenase-1 is a major
intracellular source of iron. Zinc protoporphyrin IV (a heme
oxygenase-1 inhibitor) prevented erastin-triggered ferroptotic
cancer cell death (37). Erastin
increases the mRNA levels of heme oxygenase-1 in HT-1080
fibrosarcoma cells (37). Lee
et al reported that heme oxygenase-1 influenced the
differential responses of breast cancer and normal cells to PL
(38). We found that CN-A (15
μg/ml) or PL (6 or 8 μM) markedly increased the mRNA
levels of heme oxygenase-1 for 16 h in the PANC-1 cells (data not
shown). These results suggest that CN-A- and/or PL-induced heme
oxygenase-1 plays a role in the induction of ferroptotic cancer
cell death induced by CN-A plus PL.
As previously demonstrated, combined treatment with
CN-A and PEITC strongly induced cell death within 1 day, whereas
treatment with synthetic CN-A derivatives (ISIR-005 and ISIR-042)
and PEITC did not exert synergistic effects on cell death (20). On the other hand, we previously
reported that the synthetic CN-A-derivatives, ISIR-005 and
ISIR-042, as well as CN-A in combination with certain bioactive
agents, such as rapamycin and arsenic trioxide synergistically
exerted antitumor effects in several cancer cells (12,39–41).
We observed that combined treatment with synthetic CN-A derivatives
(ISIR-005 and ISIR-042) and PL did not exert synergistic effects on
cell death (data not shown). These results suggest that CN-A, but
not the other CN-A derivatives, specifically interacts with PEITC
or PL, and induces the ferroptotic death of pancreatic cancer
cells. Since only CN-A among CN-A and its derivatives contains an
epoxide ring, we synthesized a CN-A derivative without the epoxide
ring and investigated whether the CN-A derivative without the
epoxide ring enhanced the PL-induced ferroptotic death of PANC-1
cells. The CN-A derivative without the epoxide ring did not enhance
the PL-induced ferroptotic death of PANC-1 cells (Yamaguchi et
al, unpublished data). These findings suggest that the epoxide
ring of CN-A plays an important role in enhancing PL-induced
ferroptotic cell death.
Since pancreatic cancer is among the most aggressive
human malignancies with a 5-year survival rate of <10% despite
the optimal treatments currently available, the development of
novel and effective therapeutic agents or new effective combination
regimens is essential, and more effective and safe chemotherapeutic
treatments are required. In this study, we demonstrated that PL
alone induced the ferroptotic death of pancreatic cancer cells and
that combined treatment with PL plus CN-A and/or a lower dose of
SSZ (approved for clinical use for several diseases, such as
rheumarthritis and one of the known ferroptosis inducers) may
further effectively induce the ferroptotic death of pancreatic
cancer cells. On the other hand, the synergistic induction of cell
death by PL and CN-A was not observed in MEFs, and SSZ did not
enhance cell death induced by PL plus CN-A in MEFs. These results
suggest that the triple combined treatment with PL, CN-A and SSZ is
very effective against pancreatic cancer. Viswanathan et al
(42) recently reported that
therapy-resistant cancer cells are characterized by a dysregulated
apoptotic cascade and exhibit an enhanced ability to undergo
ferroptosis. Therefore, the triple combined treatment with PL,
CN-A, and SSZ may also be effective against therapy-resistant
pancreatic cancer cells.
Acknowledgments
Not applicable.
Notes
[1]
Funding
This study was partly supported by a grant
(16K10459) from the Ministry of Education, Culture, Sports,
Science, and Technology of Japan and by a grant from the Shimane
University 'SUIGAN' project.
[2] Availability
of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
All authors contributed to the manuscript as
follows: Conceptualization, YY, TK and SK; methodology, YY and TK;
investigation, YY and TK; writing of original draft, YY and TK;
writing, revisions (reviewing) and editing, TK and SK; funding
acquisition and supervision, TK and SK. All authors have read and
approved the final manuscript.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Han X, Saiyin H, Zhao J, Fang Y, Rong Y,
Shi C, Lou W and Kuang T: Overexpression of miR-135b-5p promotes
unfavorable clinical characteristics and poor prognosis via the
repression of SFRP4 in pancreatic cancer. Oncotarget.
8:62195–62207. 2017.PubMed/NCBI
|
|
3
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al Groupe Tumeurs Digestives of Unicancer;
PRODIGE Intergroup: FOLFIRINOX versus gemcitabine for metastatic
pancreatic cancer. N Engl J Med. 364:1817–1825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kroep JR, Pinedo HM, van Groeningen CJ and
Peters GJ: Experimental drugs and drug combinations in pancreatic
cancer. Ann Oncol. 10(Suppl 4): 234–238. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jakstaite A, Maziukiene A, Silkuniene G,
Kmieliute K, Gulbinas A and Dambrauskas Z: HuR mediated
post-transcriptional regulation as a new potential adjuvant
therapeutic target in chemotherapy for pancreatic cancer. World J
Gastroenterol. 21:13004–13019. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhong S, Qie S, Yang L, Yan Q, Ge L and
Wang Z: S-1 mono-therapy versus S-1 combination therapy in
gemcitabine-refractory advanced pancreatic cancer. A meta-analysis
(PRISMA) of randomized control trial Medicine (Baltimore).
96:e76112017.
|
|
9
|
Shaw AT, Winslow MM, Magendantz M, Ouyang
C, Dowdle J, Subramanian A, Lewis TA, Maglathin RL, Tolliday N and
Jacks T: Selective killing of K-ras mutant cancer cells by small
molecule inducers of oxidative stress. Proc Natl Acad Sci USA.
108:8773–8778. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hezel AF, Kimmelman AC, Stanger BZ,
Bardeesy N and Depinho RA: Genetics and biology of pancreatic
ductal adenocarcinoma. Genes Dev. 20:1218–1249. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Honma Y, Ishii Y, Yamamoto-Yamaguchi Y,
Sassa T and Asahi K: Cotylenin A, a differentiation-inducing agent,
and IFN-alpha cooperatively induce apoptosis and have an antitumor
effect on human non-small cell lung carcinoma cells in nude mice.
Cancer Res. 63:3659–3666. 2003.PubMed/NCBI
|
|
12
|
Kasukabe T, Okabe-Kado J, Kato N, Sassa T
and Honma Y: Effects of combined treatment with rapamycin and
cotylenin A, a novel differentiation-inducing agent, on human
breast carcinoma MCF-7 cells and xenografts. Breast Cancer Res.
7:R1097–R1110. 2005. View Article : Google Scholar
|
|
13
|
Kasukabe T, Okabe-Kado J, Kato N, Honma Y
and Kumakura S: Cotylenin A and arsenic trioxide cooperatively
suppress cell proliferation and cell invasion activity in human
breast cancer cells. Int J Oncol. 46:841–848. 2015. View Article : Google Scholar
|
|
14
|
Yang WS and Stockwell BR: Synthetic lethal
screening identifies compounds activating iron-dependent,
nonapoptotic cell death in oncogenic-RAS-harboring cancer cells.
Chem Biol. 15:234–245. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar :
|
|
17
|
Latunde-Dada GO: Ferroptosis: Role of
lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta.
1861:1893–1900. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiao D, Powolny AA, Moura MB, Kelley EE,
Bommareddy A, Kim SH, Hahm ER, Normolle D, Van Houten B and Singh
SV: Phenethyl isothiocyanate inhibits oxidative phosphorylation to
trigger reactive oxygen species-mediated death of human prostate
cancer cells. J Biol Chem. 285:26558–26569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hong Y-H, Uddin MH, Jo U, Kim B, Song J,
Suh DH, Kim HS and Song YS: ROS accumulation by PEITC selectively
kills ovarian cancer cells via UPR-mediated apoptosis. Front Oncol.
5:1672015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kasukabe T, Honma Y, Okabe-Kado J, Higuchi
Y, Kato N and Kumakura S: Combined treatment with cotylenin A and
phenethyl isothiocyanate induces strong antitumor activity mainly
through the induction of ferroptotic cell death in human pancreatic
cancer cells. Oncol Rep. 36:968–976. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Raj L, Ide T, Gurkar AU, Foley M, Schenone
M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, et al:
Selective killing of cancer cells by a small molecule targeting the
stress response to ROS. Nature. 475:231–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dhillon H, Mamidi S, McClean P and Reindl
KM: Transcriptome analysis of piperlongumine-treated human
pancreatic cancer cells reveals involvement of oxidative stress and
endoplasmic reticulum stress pathways. J Med Food. 19:578–585.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jin HO, Park JA, Kim HA, Chang YH, Hong
YJ, Park IC and Lee JK: Piperlongumine downregulates the expression
of HER family in breast cancer cells. Biochem Biophys Res Commun.
486:1083–1089. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Alpay M, Yurdakok-Dikmen B, Kismali G and
Sel T: Antileukemic effects of piperlongumine and alpha lipoic acid
combination on Jurkat, MEC1 and NB4 cells in vitro. J Cancer Res
Ther. 12:556–560. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Harshbarger W, Gondi S, Ficarro SB, Hunter
J, Udayakumar D, Gurbani D, Singer WD, Liu Y, Li L, Marto JA, et
al: Structural and biochemical analyses reveal the mechanism of
glutathione S-transferase pi 1 inhibition by the anti-cancer
compound piperlongumine. J Biol Chem. 292:112–120. 2017. View Article : Google Scholar :
|
|
26
|
Chen SY, Liu GH, Chao WY, Shi CS, Lin CY,
Lim YP, Lu CH, Lai PY, Chen HR and Lee YR: Piperlongumine
suppresses proliferation of human oral squamous cell carcinoma
through cell cycle arrest, apoptosis and senescence. Int J Mol Sci.
17:6162016. View Article : Google Scholar :
|
|
27
|
Wang Y, Wang JW, Xiao X, Shan Y, Xue B,
Jiang G, He Q, Chen J, Xu HG, Zhao RX, et al: Piperlongumine
induces autophagy by targeting p38 signaling. Cell Death Dis.
4:e8242013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu QR, Liu JM, Chen Y, Xie XQ, Xiong XX,
Qiu XY, Pan F, Liu D, Yu SB and Chen XQ: Piperlongumine inhibits
migration of glioblastoma cells via activation of ROS-dependent p38
and JNK signaling pathways. Oxid Med Cell Longev. 2014:6537322014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen Y, Liu JM, Xiong XX, Qiu XY, Pan F,
Liu D, Lan SJ, Jin S, Yu SB and Chen XQ: Piperlongumine selectively
kills hepatocellular carcinoma cells and preferentially inhibits
their invasion via ROS-ER-MAPKs-CHOP. Oncotarget. 6:6406–6421.
2015.PubMed/NCBI
|
|
30
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sassa T, Tojyo T and Munakata K: Isolation
of a new plant growth substance with cytokinin-like activity.
Nature. 227:3791970. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiang L, Kon N, Li T, Wang S-J, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hangauer MJ, Viswanathan VS, Ryan MJ, Bole
D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL,
et al: Drug-tolerant persister cancer cells are vulnerable to GPX4
inhibition. Nature. 551:247–250. 2017.PubMed/NCBI
|
|
34
|
Ma S, Henson ES, Chen Y and Gibson SB:
Ferroptosis is induced following siramesine and lapatinib treatment
of breast cancer cells. Cell Death Dis. 7:e23072016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zou P, Xia Y, Ji J, Chen W, Zhang J, Chen
X, Rajamanickam V, Chen G, Wang Z, Chen L, et al: Piperlongumine as
a direct TrxR1 inhibitor with suppressive activity against gastric
cancer. Cancer Lett. 375:114–126. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang Y, Wu X, Zhou Y, Jiang H, Pan S and
Sun B: Piperlongumine suppresses growth and sensitizes pancreatic
tumors to gemcitabine in a xenograft mouse model by modulating the
NF-kappa B pathway. Cancer Prev Res (Phila). 9:234–244. 2016.
View Article : Google Scholar
|
|
37
|
Kwon MY, Park E, Lee SJ and Chung SW: Heme
oxygenase-1 accelerates erastin-induced ferroptotic cell death.
Oncotarget. 6:24393–24403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee HN, Jin HO, Park JA, Kim JH, Kim JY,
Kim B, Kim W, Hong SE, Lee YH, Chang YH, et al: Heme oxygenase-1
determines the differential response of breast cancer and normal
cells to piperlongumine. Mol Cells. 38:327–335. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kawakami K, Hattori M, Inoue T, Maruyama
Y, Ohkanda J, Kato N, Tongu M, Yamada T, Akimoto M, Takenaga K, et
al: A novel fusicoccin derivative preferentially targets hypoxic
tumor cells and inhibits tumor growth in xenografts. Anticancer
Agents Med Chem. 12:791–800. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kasukabe T, Okabe-Kado J, Haranosono Y,
Kato N and Honma Y: Inhibition of rapamycin-induced Akt
phosphorylation by cotylenin A correlates with their synergistic
growth inhibition of cancer cells. Int J Oncol. 42:767–775. 2013.
View Article : Google Scholar
|
|
41
|
Miyake T, Honma Y, Urano T, Kato N and
Suzumiya J: Combined treatment with tamoxifen and a fusicoccin
derivative (ISIR-042) to overcome resistance to therapy and to
enhance the antitumor activity of 5-fluorouracil and gemcitabine in
pancreatic cancer cells. Int J Oncol. 47:315–324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S,
Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada
K, Aguirre AJ, et al: Dependency of a therapy-resistant state of
cancer cells on a lipid peroxidase pathway. Nature. 547:453–457.
2017. View Article : Google Scholar : PubMed/NCBI
|