Introduction
Neuroblastoma, an embryonal malignancy of the
developing sympathetic nervous system, remains one of the most
difficult paediatric cancers to cure with <50% of high-risk
patients being long-term survivors despite intensive multi-modal
therapy. MYCN amplification occurs in ~50% of high-risk
cases, associated with rapid tumour progression and a poor
prognosis (1). The role of MYCN in
neuroblastoma tumourigenesis has been demonstrated by the
generation of TH-MYCN transgenic mice in 1997 through the
transfer of a construct incorporating human MYCN cDNA under
the control of the rat tyrosine hydroxylase promoter into the
nucleus of fertilised murine oocytes and subsequent integration
into genomic DNA (2,3). The tyrosine hydroxylase promoter
leads to tissue-targeted overexpression of human MYCN in
neural crest cells, and mice develop spontaneous highly penetrant
abdominal and para-spinal thoracic tumours consistent with sites of
human neuroblastoma (2). Tumour
penetrance and growth have been shown to be related to MYCN
gene dosage where homozygotes develop tumours with increased
incidence and decreased latency (2,4).
Analysis of TH-MYCN tumours has shown that they recapitulate
many histological features of human neuroblastoma with varying
degrees of neuronal differentiation and express synaptophysin and
neuron-specific enolase (2).
Moreover, tumours also exhibit chromosomal changes syntenic with
those observed in human neuroblastoma tumours, such as gain of
chromosome 17 (2,4). The TH-MYCN transgenic mouse is
now a well-established model of neuroblastoma, and a panel of
homozygous and hemizygous TH-MYCN cell lines have been
derived from tumour resections from these mice, similarly
reflecting both the genetic and biological features of human
neuroblastoma (5). Although
micrometastases can occur, one major limitation of the
TH-MYCN transgenic model in preclinical drug development
studies is the very low incidence of clinically relevant metastases
to sites such as bone marrow, thus limiting its usefulness as a
model for high-risk metastatic neuroblastoma (6). To overcome this, TH-MYCN cell
lines have been used to generate highly valuable orthotopic and
pseudometastatic syngeneic models of neuroblastoma in an
immunocompetent background (7,8).
This is of particular importance as immunotherapies, such as
anti-GD2 antibody are currently standard of care for the
treatment of children with high-risk neuroblastoma.
The tumour suppressor gene, TP53, is critical
in maintaining genomic stability and is mutationally inactivated in
>50% of all human malignancies (9). Abnormalities in the p53 pathway can
contribute to tumour resistance against ionising radiation (IR) and
cytotoxic chemotherapies (10–13).
In neuroblastoma tumours and cell lines established at diagnosis,
TP53 mutations are rare; however an increased frequency of
mutations has been reported at relapse/post-chemotherapy (14–16).
Knowledge of Trp53 genetic and functional status in
TH-MYCN cell lines is important if they are to be used in
preclinical syngeneic neuroblastoma models.
In this study, we characterised the genetic and
functional status of p53 in 6 adherent TH-MYCN transgenic
murine cell lines, and demonstrate that 3/6 cell lines have
Trp53 mutations and could be used to generate valuable
syngeneic models of p53 non-functional relapsed neuroblastoma.
Moreover, we provide evidence of the species-dependent selectivity
of some MDM2 inhibitors, which should be considered when selecting
murine models for preclinical toxicity testing of MDM2
inhibitors.
Materials and methods
Cell lines
The TH-MYCN transgenic murine cell lines used
were MYCN homozygous NHO2A, 844MYCN+/+,
3261MYCN+/+, 3394MYCN+/+,
3399MYCN+/+ and hemizygous
282MYCN+/−. The NHO2A cell line has been
previously described (5) and
together with NHO2A mouse tumour DNA, was obtained from Professor
Michelle Haber (Children's Cancer Institute, Sydney, NSW,
Australia). All other previously unreported TH-MYCN cell
lines were established from the TH-MYCN colony at the
Children's Hospital of Philadelphia (Philadelphia, PA, USA) under
an IACUC approved animal protocol. Genetically-engineered 129X1/SvJ
mice carrying the TH-MYCN transgene in one concatamer
(TH-MYCN+/− mice) or two concatamers
(TH-MYCN+/+ mice) develop tumours closely
resembling human neuroblastoma (2).
TH-MYCN+/− mice were bred and
offspring genotyped as described previously (17). TH-MYCN mice were euthanised
at the time of tumour progression according to humane approved
guidelines using 3–5% isoflurane inhalation followed by cervical
dislocation. The mice were then disinfected with 70% ethanol prior
to resecting the tumour free. Tumours were fragmented and filtered
through a 40 μm nylon mesh filter into a conical tube,
spun-pelleted, and resuspended in sterile Tris Ammonia Chloride
buffer, buffered to pH 7.65. Tumour cell pellets were incubated at
37°C for 5 min, rinsed and spun-pelleted multiple times in
phosphate-buffered saline (PBS) and then plated in RPMI tissue
culture media supplemented with 10% fetal calf serum (FCS), 1%
L-glutamine and 1% penicillin/streptamycin/gentamycin. The cells
were propagated under routine tissue culture conditions and ~50%
resulted in an established cell line over ~8 weeks. The
TH-MYCN cell lines established were validated as tyrosine
hydroxylase-positive by RT-PCR (>95% positive) and had surface
expression of the GD2 disialoganglioside, as detected by
flow cytometry. The control murine cell lines used were
Trp53 wild-type (wt) MEFPARP−/− obtained
from Dr Gilbert de Murcia (18)
and NIH3T3 cells. The control human neuroblastoma cell lines used
were TP53 mutant, MYCN-amplified SK-N-BE(2)-C, and TP53 wt
non-MYCN-amplified SHSY5Y [obtained from Professor Penny
Lovat (Newcastle University, Newcastle upon Tyne, UK) and Dr June
Biedler (Memorial Sloan Kettering Cancer Center, New York, NY, USA)
and authenticated by multiplex short tandem repeat profiling by
NewGene Ltd. (Newcastle upon Tyne, UK) using the
GenePrint® 10 system] and TP53 wt
MYCN-amplified NGP cells (19). The NHO2A cells were cultured as
previously described (5). The
844MYCN+/+, 282MYCN+/−, SHSY5Y,
SK-N-BE(2)-C and NGP cells were
cultured in RPMI-1640 (Sigma-Aldrich, Irvine, UK) supplemented with
10% fetal calf serum (FCS) (Gibco, Paisley, Scotland) and the
3261MYCN+/+, 3394MYCN+/+ and
3399MYCN+/+ cells were cultured in RPMI-1640
supplemented with 20% FCS. MEFPARP−/− and NIH3T3
cells were cultured in Dulbecco's modified Eagle's medium
(Sigma-Aldrich) supplemented with 10% FCS.
Trp53 sequencing
DNA was extracted from tumours and cell lines using
a DNeasy Blood and Tissue kit (Qiagen, Manchester, UK) according to
the manufacturer's instructions. DNA from NHO2A,
844MYCN+/+ and 282MYCN+/− cells
was amplified for Trp53 exons 2–10 and sequenced in both
directions by DBS genomics (Durham University, Durham, UK). The
primer sequences are available upon request. The
3261MYCN+/+,
3394MYCN+/+ and
3399MYCN+/+ DNA samples were amplified and
sequenced in both directions for Trp53 exons 5–10 by LGC
Genomics GmbH (Berlin, Germany).
MDM2 inhibitors, IR, western blot
analysis and flow cytometry
In brief, RG7388 was provided by Hoffman-La Roche
(Nutley, NJ, USA), Nutlin-3 was purchased from Cambridge Bioscience
Ltd. (Cambridge, UK) and MI-63 and NDD0005 were synthesised as
previously described (20,21). The cells were irradiated using a
RS320 irradiator (Gulmay Medical, Surrey, UK). For protein
analysis, whole cell lysates were harvested and proteins separated
using 4–20% Mini-Protean TGX Precast Gels (Bio-Rad Laboratories
Ltd., Hemel Hempstead, UK) and transferred onto Hybond-C Extra
membrane prior to incubation with antibodies and detection using
enhanced chemiluminescence (both from GE Healthcare Life Sciences,
Little Chalfont, UK) and X-ray film (Fujifilm, Bedford, UK). The
primary antibodies used were p53 (1:1,000; CM5; Leica Microsystems
Ltd., Buckinghamshire, UK), MYCN (1:100; NCMII00; Merck
Millipore, Billerica, MA, USA), p21WAF1 (1:1,000; SX118;
BD Biosciences, San Jose, CA, USA), Mdm2 1:500 (2A10),
p19ARF 1:500 (ab80) (both from Abcam, Cambridge, MA,
USA) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) 1:500
(FL335; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). Cell
growth inhibition was determined using the XTT cell proliferation
assay (Roche, Burgess Hill, UK). The cells were seeded in 96-well
plates (Corning, VWR International Ltd., Lutterworth, UK), allowed
to adhere overnight prior to treatment with MDM2-p53 antagonists
for 48 h. For flow cytometry analysis, cells were fixed in ice-cold
70% (v/v) ethanol, and stained with 50 μg/ml propidium
iodide and 50 μg/ml RNAse A (both from Sigma-Aldrich) at
37°C for 30 min prior to analysis on the FACSCalibur
(Becton-Dickinson, Oxford, UK). Data were analysed using Cyflogic
(CyFlo Ltd., Turku, Finland).
Statistical analyses
Two-sided unpaired t-tests were performed using
GraphPad Prism v6.0 software with a value of P<0.05 considered
as the level of significance.
Results
Trp53 status of TH-MYCN cell lines
All TH-MYCN transgenic cell lines used in the
present study were cultured as adherent monolayers (Fig. 1A). Trp53 Sanger sequencing
identified that 3/6 cell lines, the NHO2A,
844MYCN+/+ and 282MYCN+/−
cells, had missense coding region point mutations (Fig. 1B–D). The NHO2A cells were
homozygous for a p.F106S (phenylalanine to serine; c.317 C>T)
Trp53 mutation corresponding to the human p.F109S
TP53 missense mutation (Fig.
1B). The 844MYCN+/+ cells were Trp53
homozygous mutant for a p.C173W (cysteine to tryptophan; c.519
C>G) mutation corresponding to the human p.C176W missense
mutation (Fig. 1C). Of note, the
282MYCN+/− cells were heterozygously mutated for
the same Trp53 p.C173W mutant allele as the
844MYCN+/+ cells (Fig. 1D). Both p.F106S and p.C173W
mutations are within the DNA binding domain and were predicted to
affect p53-mediated transactivation using the TP53Mutload
database. The homozygous mutations detected in NHO2A and
844MYCN+/+ cells are most likely to be due to
allelic loss of one allele with mutation in the remaining
allele.
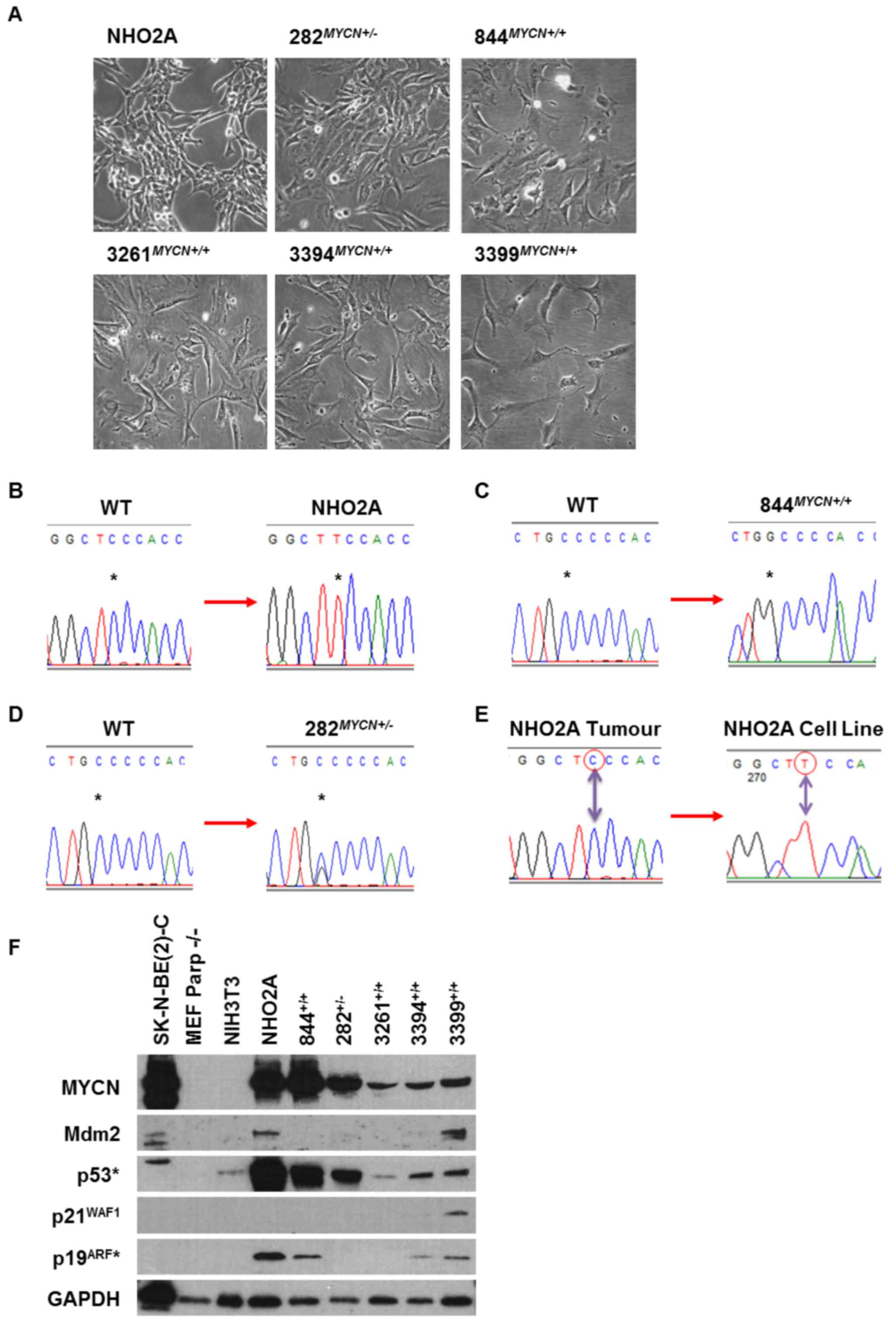 | Figure 1(A) Photomicrographs showing the
morphological appearance of NHO2A, 282MYCN+/−,
844MYCN+/+, 3261MYCN+/+,
3394MYCN+/+ and 3399MYCN+/+
TH-MYCN murine neuroblastoma cell lines. Chromatograms
showing Trp53 gene mutations identified in (B) NHO2A
(homozygous mutation, codon 106, phenylalanine to serine), (C)
844MYCN+/+ (homozygous mutation, codon 173,
cysteine to tryptophan) and (D) 282MYCN+/−
(heterozygous mutation, codon 173, cysteine to tryptophan). All
mutations are within the DNA binding domain of p53. The asterisk
(*) marks the nucleotide change. (E) Chromatograms
showing the Trp53 wild-type (wt) status of the original
tumour from which the NHO2A cell line was derived compared to the
homozygous p.F106S (c.317C>T) mutation identified in the NHO2A
cell line. (F) Western blot analysis of the basal expression of
MYCN, Mdm2, p53, p21WAF1 and p19ARF (murine
homologue of p14ARF) in the murine cell line panel in
comparison to the TP53 mutant MYCN amplified
SK-N-BE(2)-C human neuroblastoma
cell line. *p53 and p19ARF antibodies are
only reactive against murine p53 and p19ARF. |
To determine whether the mutations present in cell
lines were also present in the original tumours or selected for
during establishment of the cell line, tumour DNA was sequenced for
Trp53 exons 2–10. Only 1/6 cell lines (NHO2A) had tumour DNA
available and was found to be wt (Fig.
1E).
MYCN and the p53 pathway in TH-MYCN cell
lines
Basal expression of MYCN and p53 pathway components,
namely p53, Mdm2, p21WAF1 and p19ARF were
assessed in TH-MYCN cell lines by western blot analysis
(Fig. 1F). The Trp53 wt
murine MEFPARP−/− and NIH3T3, and human
MYCN amplified TP53 mutant SK-N-BE(2)-C cells were included as controls.
Overexpression of the MYCN transgene was observed in all
TH-MYCN cell lines, with levels comparable to those of the
MYCN-amplified SK-N-BE(2)-C
cells. NHO2A and 844MYCN+/+ cells had the highest
MYCN levels compared to the other TH-MYCN cell lines.
Accumulation of p53 was observed in NHO2A,
844MYCN+/+ and 282MYCN+/−
cells, consistent with their mutant Trp53 status. All mouse
cell lines had very low or undetectable baseline Mdm2 expression
(Fig. 1F). p21WAF1 was
expressed only in Trp53 wt 3399MYCN+/+
cell line (Fig. 1F).
p19ARF was expressed in NHO2A,
844MYCN+/+, 3394MYCN+/+ and
3399MYCN+/+ cells (Fig. 1F).
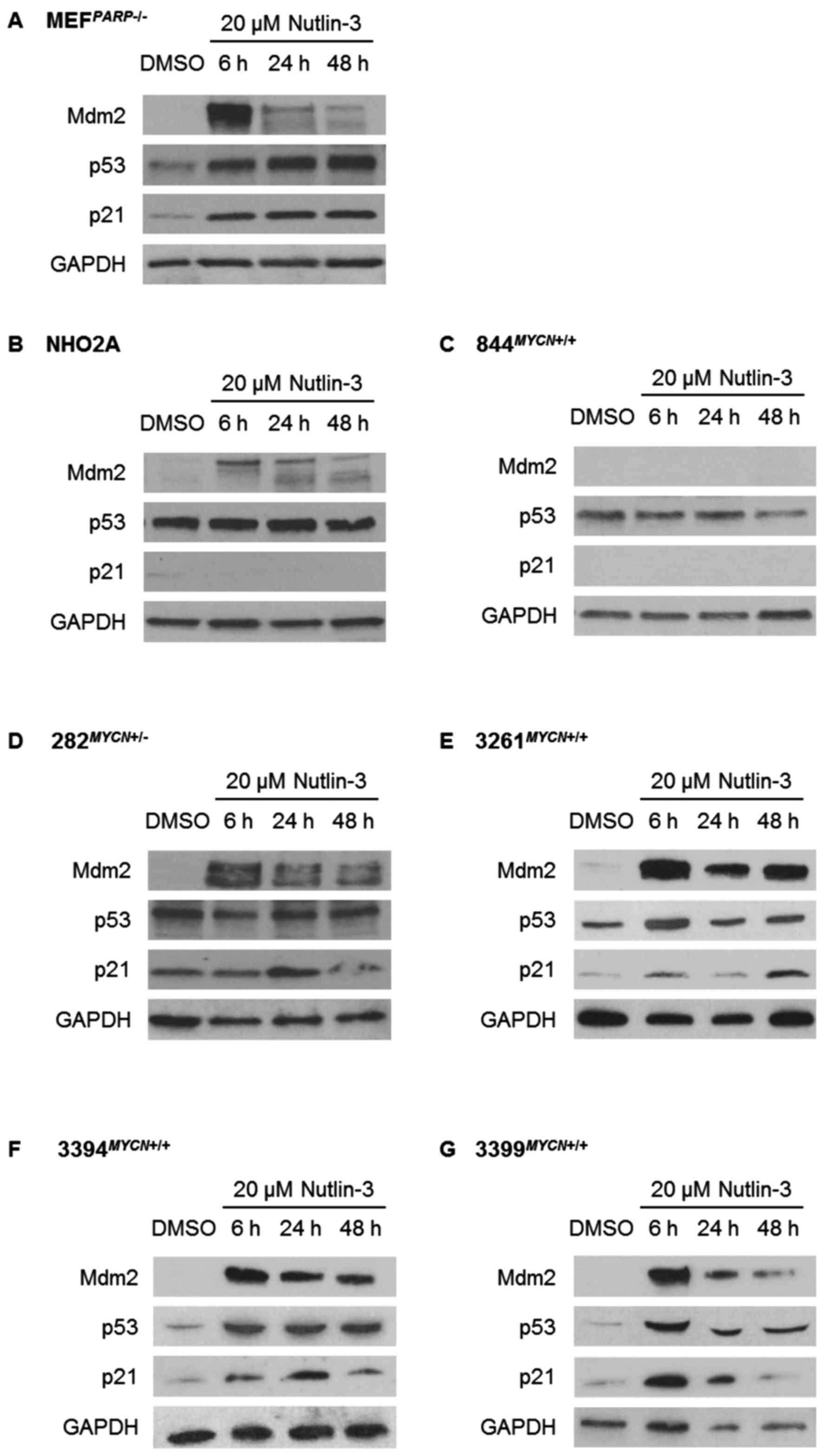
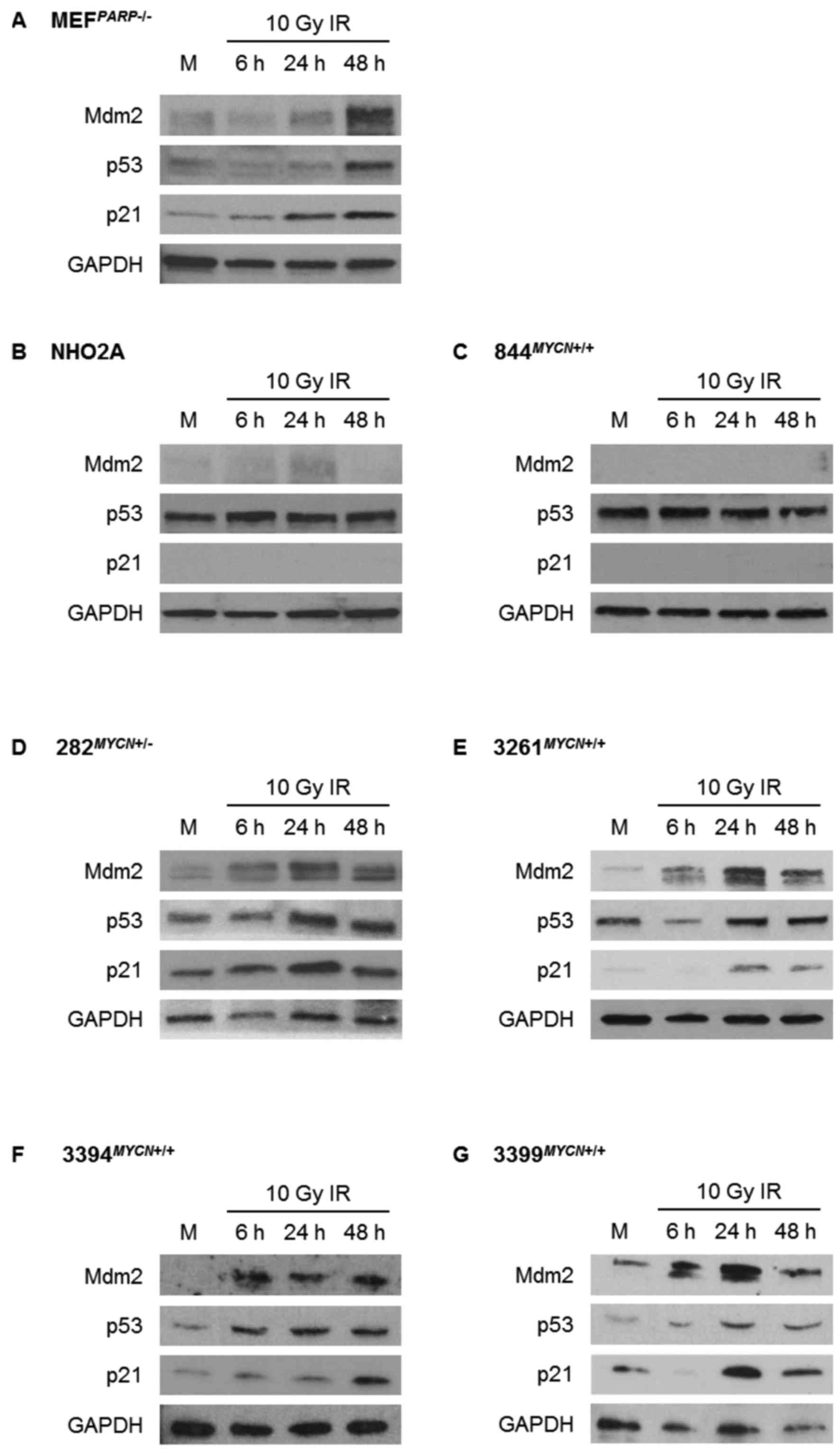
The response of the p53 pathway to
Nutlin-3 and IR
Using Nutlin-3 and IR as different methods to induce
p53 activation, activation of the p53 signalling pathway was
assessed by western blot analysis in the TH-MYCN cell lines
and control Trp53 wt MEFPARP−/− cells
(Figs. 2 and 3). Consistent with their Trp53 wt
status, MEFPARP−/− cells exhibited an intact p53
signalling pathway in response to both Nutlin-3 and IR, evident by
p53 stabilisation and Mdm2 and p21WAF1 upregulation
(Figs. 2A and 3A). Of note, the p53 pathway response of
MEFPARP−/− cells to IR was slightly diminished
and delayed in comparison to Nutlin-3 (Figs. 2A and 3A), and is most likely a consequence of
PARP-1 knockout in this cell line (22). As expected, homozygously
Trp53 mutant NHO2A and 844MYCN+/+ cells
failed to show p53 induction in response to Nutlin-3 (Fig. 2B and C) or IR (Fig. 3B and C). Consistent with this
observation, no p21WAF1 induction was observed in either
cell line, and no Mdm2 induction was observed in
844MYCN+/+ cells. Of note, despite a lack of p53
induction, weak induction of Mdm2 was observed in NHO2A cells
following both treatment with Nutlin-3 and exposure to IR (Figs. 2B and 3B). The heterozygously Trp53
mutant 282MYCN+/− cells also failed to show p53
stabilisation in response to Nutlin-3 or IR; however in spite of
this, an increase in both Mdm2 and p21WAF1 expression
was observed (Figs. 2D and
3D), most likely as a result of
the presence of one remaining wt Trp53 allele. Finally, in
line with their Trp53 wt status, the 3 remaining
TH-MYCN cell lines, 3261MYCN+/+,
3394MYCN+/+ and 3399MYCN+/+,
all showed evidence of IR- and Nutlin-3-induced p53 pathway
activation, with stabilisation of p53, and induction of p21 and
Mdm2 (Figs. 2E–G and 3E–G).
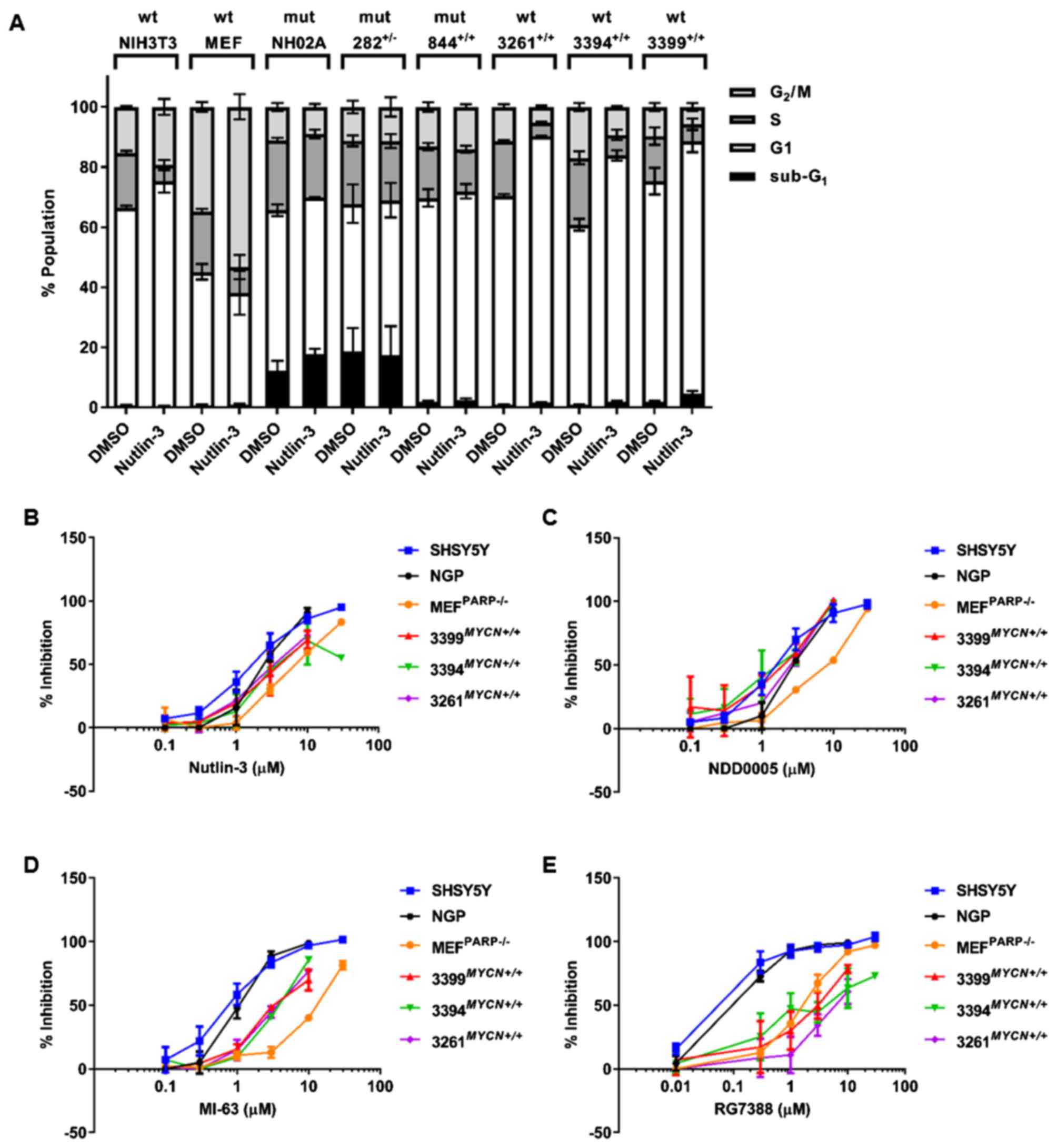
Nutlin-3-induced cell cycle
distribution
To further characterise the p53 functional response
in the TH-MYCN cell lines to Nutlin-3, the sub-G1
and cell cycle distribution of all the 6 TH-MYCN cell lines
following 24 h treatment with 20 μM Nutlin-3 were analysed
by propidium iodide-based flow cytometry. Sub-G1 events
was used as a surrogate marker of apoptosis and the G1:S
ratio calculated as an indicator of G1 cell cycle
arrest. Trp53 wt NIH3T3 and MEFPARP−/−
cells were included as positive controls. The functional assessment
of Nutlin-3-mediated cell cycle arrest and apoptosis of human
neuroblastoma cell lines have previously been reported (23,24).
In response to Nutlin-3 treatment, NIH3T3 cells underwent
G1 arrest, evident by a 4.6-fold increase in their G1/S
ratio [16.58±4.43 (Nutlin-3) vs. 3.61±0.17 (DMSO)] (Table I and Fig. 4A). Consistent with the observed
activation of the p53 pathway in Fig.
2A, the MEFPARP−/− cells demonstrated a
3.2-fold increase in their G1:S ratio [7.19±3.32
(Nutlin-3) vs. 2.23±0.23 (DMSO)], and a significant increase in the
percentage of cells in G2/M phase [53.32%±4.19
(Nutlin-3) vs. 34.75%±1.63 (DMSO); P<0.005, paired t-test]
(Table I and Fig. 4A), indicative of a Nutlin-3-induced
G1 and particularly G2 arrest in this cell
line. In contrast to control Trp53 wt murine cell lines, and
as expected, no noticeable changes in the G1/S ratio or
percentage of cells in G2/M phase were observed in any
Trp53 mutant TH-MYCN NHO2A,
844MYCN+/+ or 282MYCN+/− cells
(Table I and Fig. 4A). Of note, compared to the other
murine cell lines, NHO2A and 282MYCN+/− cells had
a high proportion of sub-G1 basal events (Table I and Fig. 4A), a surrogate marker of apoptosis,
which following Nutlin-3 treatment, increased slightly and remained
unaltered, respectively. Consistent with their Trp53 wt
status and observed p53 pathway activation, as shown in Fig. 2E–G, the 3 Trp53 wt
TH-MYCN cell lines, 3261MYCN+/+,
3394MYCN+/+ and 3399MYCN+/+,
all underwent a G1 phase arrest, as evident by a 5.2-,
5- and 3.4-fold increase in G1/S ratio compared to
control cells, respectively (Table
I and Fig. 4A). In addition,
the 3399MYCN+/+ cells also exhibited an increased
sub-G1 population (Table
I and Fig. 4A).
 | Table IFlow cytometric analysis of the
proportion of cells in Sub-G1, G1, S, and
G2/M phase, and the G1/S ratios in the panel
cell lines used in this study. |
Table I
Flow cytometric analysis of the
proportion of cells in Sub-G1, G1, S, and
G2/M phase, and the G1/S ratios in the panel
cell lines used in this study.
| Cell line and
Trp53 status | Treatment
condition | % Cell population
in each phase
|
|---|
|
Sub-G1 | G1 | S |
G2/M | G1/S
ratio |
|---|
|
MEFPARP−/− (wt) | DMSO | 0.66±0.37 | 44.46±2.57 | 20.13±0.89 | 34.75± 1.63 | 2.23±0.23 |
| Nutlin-3 | 0.86±0.31 | 37.29±7.29 | 8.52±4.03 | 53.32±4.19 | 7.19± 3.32 |
| NIH3T3 (wt) | DMSO | 0.58±0.28 | 65.84±0.7 | 18.31±0.70 | 15.27±0.26 | 3.61±0.17 |
| Nutlin-3 | 0.39±0.17 | 74.89±3.73 | 5.37±1.64 | 19.34±2.68 | 16.58±4.43 |
| NHO2A (mutant) | DMSO | 12.29± 3.2 | 53.35±1.98 | 23.28±0.79 | 11.07±1.33 | 2.29±0.01 |
| Nutlin-3 | 17.76± 1.68 | 52.06±0.23 | 21.14±1.44 | 9.04±0.94 | 2.48±0.15 |
|
282MYCN+/− (mutant) | DMSO | 18.60±7.73 | 49.16±6.4 | 20.95±1.81 | 11.28±2.12 | 2.4±0.41 |
| Nutlin-3 | 17.37±9.66 | 51.58±5.73 | 19.69±2.29 | 11.37±3.22 | 2.68±0.41 |
|
844MYCN+/+ (mutant) | DMSO | 1.91±0.23 | 67.80±2.88 | 17.09±1.16 | 13.18±1.59 | 4.03±0.45 |
| Nutlin-3 | 2.44±0.58 | 69.50±2.4 | 13.96±1.20 | 14.10±0.92 | 5.09±0.64 |
|
3261MYCN+/+ (wt) | DMSO | 0.86± 0.09 | 69.45±0.65 | 18.28±0.32 | 11.40±0.87 | 3.8±0.03 |
| Nutlin-3 | 1.58± 0.05 | 88.61±0.28 | 4.57±0.41 | 5.24±0.41 | 19.67±1.68 |
|
3394MYCN+/+ (wt) | DMSO | 0.91±0.16 | 59.88±1.98 | 22.28±2.12 | 16.94± 1.31 | 2.79±0.38 |
| Nutlin-3 | 1.86±0.36 | 82.01±1.72 | 6.82±1.68 | 9.31± 1.39 | 14.03±2.82 |
|
3399MYCN+/+ (wt) | DMSO | 1.80±0.37 | 73.46±4.43 | 14.98±2.88 | 9.77±1.39 | 5.55±1.69 |
| Nutlin-3 | 4.63±0.97 | 83.93±3.61 | 5.76±1.79 | 5.69±1.26 | 18.67±7.01 |
Species-dependent MDM2 inhibitor
selectivity
MDM2 inhibitors are currently under preclinical and
clinical evaluation as a novel therapeutic for neuroblastoma. To
further evaluate the p53 pathway status of cell lines studied and
establish their response to MDM2 inhibitors, sensitivity to
Nutlin-3 and additional structurally unrelated MDM2 inhibitors,
NDD0005, MI-63 and RG7388, and their effects on growth inhibition
were assessed (Table II). Human
TP53 wt non-MYCN amplified SHSY5Y and MYCN
amplified NGP cells, which have previously been shown to be
sensitive to the tested MDM2 inhibitors (20,21,23),
were included as positive controls. The concentrations of Nutlin-3,
MI63, NDD0005 and RG7388, which led to a 50% growth inhibition
(GI50) after 48 h of treatment, are shown in Table II. In comparison to TP53 wt
human neuroblastoma cells, the Trp53 wt control and
TH-MYCN murine cell lines were less sensitive to
Nutlin-3-mediated growth inhibition, as evidenced by higher
GI50 concentrations (1.2–7.4-fold less sensitive)
(Table II and Fig. 4B). As expected, Trp53 mutant
TH-MYCN cell lines NHO2A, 844MYCN+/+ and
282MYCN+/− had the highest GI50 values
(14.1–21.8-fold less sensitive) (Table II).
| Table IISummary of 48-h GI50
values for MDM2 antagonists in human and murine cells. |
Table II
Summary of 48-h GI50
values for MDM2 antagonists in human and murine cells.
| Cell line | 48 h | Antagonist
(μM)
|
|---|
| Nutlin-3 | NDD0005 | MI-63 | RG7388 |
|---|
| SHSY5Y |
GI50 | 1.95±0.56 | 1.93±0.69 | 0.78±0.16 | 0.11±0.05 |
| NGP |
GI50 | 2.51±0.3 | 2.78±0.02 | 1.06±0.12 | 0.14±0.03 |
|
MEFPARP−/− |
GI50 | 7.10±0.38 | 6.98±0.06 | 12.05±0.63 | 1.61±0.11 |
| Rel Fold/P-value
vs. SHSY5Y | 3.6/b | 3.6/b | 15.4/d | 15.0/d |
| Rel Fold/P-value
vs. NGP | 2.8/b | 2.5/c | 11.3/c | 11.9/c |
| NIH3T3 |
GI50 | 14.1±2.2 | ND | ND | ND |
| Rel Fold/P-value
vs. SHSY5Y | 7.4/b | | | |
| Rel Fold/P-value
vs. NGP | 5.6/b | | | |
| NHO2A |
GI50 | 41.4±2.7 | ND | ND | ND |
| Rel Fold/P-value
vs. SHSY5Y | 21.8/d | | | |
| Rel Fold/P-value
vs. NGP | 16.5/d | | | |
|
282MYCN+/− |
GI50 | 35.3±1.8 | ND | ND | ND |
| Rel Fold/P-value
vs. SHSY5Y | 18.6/d | | | |
| Rel Fold/P-value
vs. NGP | 14.1/d | | | |
|
844MYCN+/+ |
GI50 | 36.5±1.4 | ND | ND | ND |
| Rel Fold/P-value
vs. SHSY5Y | 19.2/d | | | |
| Rel Fold/P-value
vs. NGP | 14.5/d | | | |
|
3261MYCN+/+ |
GI50 | 3.53±0.54 | 2.38±0.39 | 3.71±0.3 | 6.36±1.65 |
| Rel Fold/P-value
vs. SHSY5Y | 1.8/ns | 1.2/ns | 4.7/c | 59.1/b |
| Rel Fold/P-value
vs. NGP | 1.4/ns | 0.9/ns | 3.5/b | 47.0/a |
|
3394MYCN+/+ |
GI50 | 3.09±0.19 | 1.57±0.63 | 4.0±0.41 | 1.84±0.50 |
| Rel Fold/P-value
vs. SHSY5Y | 1.6/ns | 0.8/ns | 5.1/c | 17.1/b |
| Rel Fold/P-value
vs. NGP | 1.2/ns | 0.6/ns | 3.8/b | 13.6/a |
|
3399MYCN+/+ |
GI50 | 4.3±1.2 | 1.58±0.65 | 3.94±0.58 | 3.49±0.37 |
| Rel Fold/P-value
vs. SHSY5Y | 2.2/ns | 0.8/ns | 5/b | 32.4/c |
| Rel Fold/P-value
vs. NGP | 1.7/ns | 0.6/ns | 3.7/b | 25.8/b |
Further evaluation and comparison of Trp53
wt TH-MYCN and MEFPARP−/− cells to
NDD0005, MI-63 and RG7388, demonstrated that although the
MEFPARP−/− cells were significantly
(2.5–3.6-fold) less sensitive to NDD0005, there was no difference
in the sensitivity of Trp53 wt TH-MYCN cell lines to
NDD0005 compared with human SHSY5Y and NGP neuroblastoma cells
(Table II and Fig. 4C). Of the tested MDM2 inhibitors,
MI-63 and RG7388 were the most potent against human neuroblastoma
cell lines, consistent with the findings of our previous studies
(20,23). The data also revealed that compared
with SHSY5Y and NGP cells, Trp53 wt
MEFPARP−/− cells were significantly (11.3–15.4-
and 11.9–15.0-fold) less sensitive to both MI-63 and RG7388,
respectively (Table II and
Fig. 4D and E). Furthermore, the
data also indicated that Trp53 wt TH-MYCN cell lines
were significantly (3.5–5.1-fold) less sensitive to MI-63 (Table II and Fig. 4D), and even less so
(13.6–59.1-fold) to RG7388 (Table
II and Fig. 4E). This
highlights an inverse association between potency in TP53 wt
human neuroblastoma cells and potency in Trp53 wt murine
cells.
Discussion
Cell lines established from primary tumours
resected from TH-MYCN mice have been used to develop
valuable preclinical immunocompetent, syngeneic models of
neuroblastoma (7,8), for which knowledge of their p53
pathway status is important. In this study, we demonstrated that
2/6 TH-MYCN cell lines were Trp53 homozygous mutant
(NHO2A and 844MYCN+/+) and 1/6 was heterozygous
mutant (282MYCN+/−). In the only case where DNA
from the original tumour was available (NHO2A), the original tumour
was Trp53 wt. This is consistent with our previous analysis
of 13 primary TH-MYCN tumours, which did not show any
Trp53 mutations (exons 4–8) (25). It is possible that Trp53
mutant subpopulations existed within the primary tumour, but were
below the level of detection of Sanger sequencing, or that the
mutation spontaneously developed and was selected for during ex
vivo culturing pressures and cell line establishment. A
MYCN oncogenic drive may be a strong contributing factor for
the positive selection of Trp53 mutations and may account
for why 3/6 TH-MYCN cell lines tested in the present study
were either homozygous or heterozygous Trp53 mutant.
Certainly, MYCN is known to play a dual role in driving both
proliferation and apoptosis, and there are several lines of
evidence, including studies using TH-MYCN models, which
suggest that MYCN driven p53-dependent apoptosis is an
important mechanism for tumour suppression in neuroblastoma and
that MYCN amplified neuroblastoma cells may circumvent
MYCN driven p53-dependent apoptosis by selecting for cells
with aberrations in the p53/MDM2/p14ARF pathway, as has
been observed for MYCC-driven lymphoma (3,19,26–29).
Specifically, as previously demonstrated, tumours formed with
greater penetrance and reduced latency in TH-MYCN mice
heterozygous for an inactivated germline p53 allele (27). The analysis of human neuroblastoma
cell lines reported to date with aberrations in the
p53/MDM2/p14ARF pathway demonstrates that 31/40 (78%)
are MYCN-amplified (19).
More recently, a study of the role of p53 function in neuroblastoma
pathogenesis using TH-MYCN murine models observed that loss
of p53 function led to reduced survival (30).
Nutlin-3 is a potent selective inhibitor of the
MDM2-p53 interaction (31),
previously shown to be highly effective against TP53 wt
neuroblastoma cell lines, inducing cell cycle arrest and/or
apoptosis, and used to functionally screen large neuroblastoma cell
line panels for p53 pathway aberrations (23,24,32).
In this study, we found that in all cell lines tested, the presence
of a mutation was consistent with high basal levels of p53 and
aberrant p53 signalling in response to Nutlin-3 and IR, and failure
to growth arrest in response to Nutlin-3. Consistent with the
mechanisms of action of MDM2 inhibitors and existing data from
human neuroblastoma cell lines, overall, Nutlin-3 was found to
induce cell cycle arrest and/or apoptosis of Trp53 wt
control and TH-MYCN murine cell lines (23,24).
Of note, in the current study, in response to Nutlin-3 and IR,
although there was no induction of p53, an increase in both Mdm2
and p21WAF1 expression was observed in heterozygously
mutant 282MYCN+/− cells, suggesting there is some
residual p53 function from the remaining wt allele (33); however this did not lead to growth
arrest or apoptosis in response to Nutlin-3.
MDM2 inhibitors are currently under preclinical and
clinical development as a novel therapeutic, both alone and in
combination, for human malignancies including neuroblastoma. Of
particular interest in view of the latter, data from the present
study demonstrated that in comparison to human TP53 wt
neuroblastoma cells, murine control (MEFPARP−/−)
and TH-MYCN cell lines were significantly less sensitive to
MI-63 and RG7388 induced growth inhibition. Although human and
murine MDM2 show a high degree of amino acid sequence homology,
with only 2 non-identical amino acids within the p53 binding domain
(34), these subtle differences
could account for a weaker binding affinity to murine Mdm2 which is
believed to contribute to the increased resistance and higher
GI50 concentrations of murine cells to some MDM2
inhibitors that have been designed with high potency against human
MDM2 (35). In support of this,
the present data showed that the more potent the MDM2 inhibitor was
against human neuroblastoma cells, the less sensitive the murine
cells were, and the greater the fold difference between
GI50 values of Trp53 wt murine cells vs.
TP53 wt human cells. These observations are consistent with
the inter-species selectivity of spiro-oxindole-based MDM2
inhibitors (36) and
dihydroisoquinolinone NVP-CGM097 (37), but not pyrazolo-pyrrolidinone
NVP-HDM201 (38,39), and should be taken into account
when designing studies of MDM2 inhibitors either alone or in
combination using either preclinical transgenic or human tumour
xenograft models as p53-dependent normal tissue toxicity will not
be adequately modelled.
Currently, murine neuroblastoma models include
genetically engineered mouse models, syngeneic models, and
subcutaneous, orthotopic, pseudometastatic and patient-derived
xenografts (3,40–43).
All models have associated advantages and disadvantages, and it is
likely that the most comprehensive preclinical assessment of
efficacy will include a combination of existing models. Several of
the models predominantly use immunocompromised mice and thus are
unsuitable for assessment of immunotherapies, which are emerging as
effective targeted therapies in patients with neuroblastoma. To
overcome this, cell lines established from the TH-MYCN
transgenic model/or other murine neuroblastoma cell lines can be
used to generate orthotopic or pseudometastatic syngeneic models of
neuroblastoma in an immunocompetent background (7,8). For
this, the genetic and functional characterisation of murine cell
lines, including Trp53 status and pathway function, are very
important and highly warranted, as existing data are limited.
In conclusion, the Trp53 wt and mutant
TH-MYCN cell lines characterised in this study can be used
in syngeneic models of neuroblastoma, the former to test
p53-dependent therapies in combination with immunotherapies, such
as anti-GD2 antibody, and the latter as models of
immunocompetent, chemoresistant relapsed neuroblastoma in which
aberrations in the p53 pathway are more common (14–16).
Glossary
Abbreviations
Abbreviations:
|
IR
|
ionising radiation
|
|
wt
|
wild-type
|
|
PBS
|
phosphate-buffered saline
|
|
FCS
|
fetal calf serum
|
Acknowledgments
This study was supported by The Dubois Child Cancer
Fund, SPARKS, the North of England Children's Cancer Research Fund
(NECCRF), Neuroblastoma UK and Niamh's Next Step. We would like to
thank Professor Michelle Haber for the NHO2A cell line and tumour
DNA, Dr Gilbert de Murcia for providing the
MEFPARP−/− cells, Dr Fabio Del Bello and Dr
Alessandro Piergentili for providing MI-63, Dr Steven Middleton
(Hoffmann-La Roche, Nutley, NJ, USA) for providing RG7388, and Dr
Anna Watson, Dr Karen Haggerty and Dr Ian Hardcastle (Newcastle
University) for synthesising NDD0005. We disclose that Dr L. Chen
and Professor D.A. Tweddle are part of an international
collaborative research consortium with Hoffmann-La Roche Ltd.
Professor J. Lunec is a collaborative co-investigator of the CRUK
funded Drug Discovery Programme at Newcastle University which
developed NDD0005. Newcastle University, Cancer Research Technology
and Astex Pharmaceuticals Inc. are part of an alliance agreement
since 2012.
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Cohn SL and Tweddle DA: MYCN amplification
remains prognostically strong 20 years after its 'clinical debut'.
Eur J Cancer. 40:2639–2642. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weiss WA, Aldape K, Mohapatra G,
Feuerstein BG and Bishop JM: Targeted expression of MYCN causes
neuroblastoma in transgenic mice. EMBO J. 16:2985–2995. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chesler L and Weiss WA: Genetically
engineered murine models - contribution to our understanding of the
genetics, molecular pathology and therapeutic targeting of
neuroblastoma. Semin Cancer Biol. 21:245–255. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rasmuson A, Segerström L, Nethander M,
Finnman J, Elfman LH, Javanmardi N, Nilsson S, Johnsen JI,
Martinsson T and Kogner P: Tumor development, growth
characteristics and spectrum of genetic aberrations in the TH-MYCN
mouse model of neuroblastoma. PLoS One. 7:e512972012. View Article : Google Scholar
|
|
5
|
Cheng AJ, Cheng NC, Ford J, Smith J,
Murray JE, Flemming C, Lastowska M, Jackson MS, Hackett CS, Weiss
WA, et al: Cell lines from MYCN transgenic murine tumours reflect
the molecular and biological characteristics of human
neuroblastoma. Eur J Cancer. 43:1467–1475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lehembre F and Regenass U: Metastatic
disease: A drug discovery perspective. Semin Cancer Biol.
22:261–271. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stauffer JK, Orentas RJ, Lincoln E, Khan
T, Salcedo R, Hixon JA, Back TC, Wei JS, Patidar R, Song Y, et al:
High-throughput molecular and histopathologic profiling of tumor
tissue in a novel transplantable model of murine neuroblastoma: New
tools for pediatric drug discovery. Cancer Invest. 30:343–363.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kroesen M, Nierkens S, Ansems M, Wassink
M, Orentas RJ, Boon L, den Brok MH, Hoogerbrugge PM and Adema GJ: A
transplantable TH-MYCN transgenic tumor model in C57Bl/6 mice for
preclinical immunological studies in neuroblastoma. Int J Cancer.
134:1335–1345. 2014. View Article : Google Scholar
|
|
9
|
Brown CJ, Lain S, Verma CS, Fersht AR and
Lane DP: Awakening guardian angels: drugging the p53 pathway.
Nature reviews. 9:pp. 862–873. 2009, https://doi.org/10.1038/nrc2763.
|
|
10
|
Carr J, Bell E, Pearson ADJ, Kees UR,
Beris H, Lunec J and Tweddle DA: Increased frequency of aberrations
in the p53/MDM2/p14(ARF) pathway in neuroblastoma cell lines
established at relapse. Cancer Res. 66:2138–2145. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Keshelava N, Zuo JJ, Chen P, Waidyaratne
SN, Luna MC, Gomer CJ, Triche TJ and Reynolds CP: Loss of p53
function confers high-level multidrug resistance in neuroblastoma
cell lines. Cancer Res. 61:6185–6193. 2001.PubMed/NCBI
|
|
12
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Soussi T and Béroud C: Assessing TP53
status in human tumours to evaluate clinical outcome. Nat Rev
Cancer. 1:233–240. 2001. View Article : Google Scholar
|
|
14
|
Carr-Wilkinson J, O'Toole K, Wood KM,
Challen CC, Baker AG, Board JR, Evans L, Cole M, Cheung NK, Boos J,
et al: High Frequency of p53/MDM2/p14ARF Pathway
Abnormalities in Relapsed Neuroblastoma. Clin Cancer Res.
16:1108–1118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tweddle DA, Malcolm AJ, Bown N, Pearson AD
and Lunec J: Evidence for the development of p53 mutations after
cytotoxic therapy in a neuroblastoma cell line. Cancer Res.
61:8–13. 2001.PubMed/NCBI
|
|
16
|
Padovan-Merhar OM, Raman P, Ostrovnaya I,
Kalletla K, Rubnitz KR, Sanford EM, Ali SM, Miller VA, Mossé YP,
Granger MP, et al: Enrichment of targetable mutations in the
relapsed neuroblastoma genome. PLoS Genet. 12:e10065012016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Evageliou NF, Haber M, Vu A, Laetsch TW,
Murray J, Gamble LD, Cheng NC, Liu K, Reese M, Corrigan KA, et al:
Polyamine antagonist therapies inhibit neuroblastoma initiation and
progression. Clin Cancer Res. 22:4391–4404. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
de Murcia JM, Niedergang C, Trucco C,
Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A,
LeMeur M, et al: Requirement of poly(ADP-ribose) polymerase in
recovery from DNA damage in mice and in cells. Proc Natl Acad Sci
USA. 94:7303–7307. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen L and Tweddle DA: p53, SKP2, and DKK3
as MYCN target genes and their potential therapeutic significance.
Front Oncol. 2:1732012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen L, Zhao Y, Halliday GC, Berry P,
Rousseau RF, Middleton SA, Nichols GL, Del Bello F, Piergentili A,
Newell DR, et al: Structurally diverse MDM2-p53 antagonists act as
modulators of MDR-1 function in neuroblastoma. Br J Cancer.
111:716–725. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen L, Rousseau RF, Middleton SA, Nichols
GL, Newell DR, Lunec J and Tweddle DA: Pre-clinical evaluation of
the MDM2-p53 antagonist RG7388 alone and in combination with
chemotherapy in neuroblastoma. Oncotarget. 6:10207–10221.
2015.PubMed/NCBI
|
|
22
|
Valenzuela MT, Guerrero R, Núñez MI, De
Ruiz Almodóvar JM, Sarker M, de Murcia G and Oliver FJ: PARP-1
modifies the effectiveness of p53-mediated DNA damage response.
Oncogene. 21:1108–1116. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gamble LD, Kees UR, Tweddle DA and Lunec
J: MYCN sensitizes neuroblastoma to the MDM2-p53 antagonists
Nutlin-3 and MI-63. Oncogene. 31:752–763. 2012. View Article : Google Scholar
|
|
24
|
Van Maerken T, Rihani A, Dreidax D, De
Clercq S, Yigit N, Marine JC, Westermann F, De Paepe A,
Vandesompele J and Speleman F: Functional analysis of the p53
pathway in neuroblastoma cells using the small-molecule MDM2
antagonist nutlin-3. Mol Cancer Ther. 10:983–993. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mazanek P, Dam V, Morgan BT, Liu X, Pawar
N and Hogarty MD: Assessment of p19/ARF-MDM-p53 and RAS pathways
for alterations in neuroblastomas arising in the transgenic TH-MYCN
mouse model. In: Advances in Neuroblastoma Research; June 16–19th
2004; Genoa, Italy. 2004
|
|
26
|
Chen L, Iraci N, Gherardi S, Gamble LD,
Wood KM, Perini G, Lunec J and Tweddle DA: p53 is a direct
transcriptional target of MYCN in neuroblastoma. Cancer Res.
70:1377–1388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chesler L, Goldenberg DD, Collins R,
Grimmer M, Kim GE, Tihan T, Nguyen K, Yakovenko S, Matthay K and
Weiss WA: Chemotherapy-induced apoptosis in a transgenic model of
neuroblastoma proceeds through p53 induction. Neoplasia.
10:1268–1274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen Z, Lin Y, Barbieri E, Burlingame S,
Hicks J, Ludwig A and Shohet JM: Mdm2 deficiency suppresses
MYCN-Driven neuroblastoma tumorigenesis in vivo. Neoplasia.
11:753–762. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eischen CM, Weber JD, Roussel MF, Sherr CJ
and Cleveland JL: Disruption of the ARF-Mdm2-p53 tumor suppressor
pathway in Myc-induced lymphomagenesis. Genes Dev. 13:2658–2669.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yogev O, Barker K, Sikka A, Almeida GS,
Hallsworth A, Smith LM, Jamin Y, Ruddle R, Koers A, Webber HT, et
al: p53 loss in MYC-driven neuroblastoma leads to metabolic
adaptations supporting radioresistance. Cancer Res. 76:3025–3035.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vassilev LT, Vu BT, Graves B, Carvajal D,
Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et
al: In vivo activation of the p53 pathway by small-molecule
antagonists of MDM2. Science. 303:844–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen L, Malcolm AJ, Wood KM, Cole M,
Variend S, Cullinane C, Pearson AD, Lunec J and Tweddle DA: p53 is
nuclear and functional in both undifferentiated and differentiated
neuroblastoma. Cell Cycle. 6:2685–2696. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun Y, Yi H, Yang Y, Yu Y, Ouyang Y, Yang
F, Xiao Z and Chen Z: Functional characterization of p53 in
nasopharyngeal carcinoma by stable shRNA expression. Int J Oncol.
34:1017–1027. 2009.PubMed/NCBI
|
|
34
|
Vassilev LT: Small-molecule antagonists of
p53-MDM2 binding: research tools and potential therapeutics. Cell
Cycle. 3:419–421. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Khoo KH, Verma CS and Lane DP: Drugging
the p53 pathway: Understanding the route to clinical efficacy. Nat
Rev Drug Discov. 13:217–236. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Delaisi C, Meaux I, Dos-Santos O, Barrière
C, Duffieux F, Hoffmann D, Rak A, Wolfrom M, Flèche, Zhou-Liu Q, et
al: In vitro characterization of spiro-oxindole-based modulators of
the MDM2-53 interaction and their interspecies selectivity. 103rd
Annual Meeting of the American Association for Cancer Research, Mar
31–Apr 4, 2012. Cancer Res. 72. Abstract nr 4648. Chicago, IL:
2012
|
|
37
|
Holzer P, Masuya K, Furet P, Kallen J,
Valat-Stachyra T, Ferretti S, Berghausen J, Bouisset-Leonard M,
Buschmann N, Pissot-Soldermann C, et al: Discovery of a
dihydroisoquinolinone derivative (NVP-CGM097): A highly potent and
selective MDM2 inhibitor undergoing phase 1 clinical trials in
p53wt tumors. J Med Chem. 58:6348–6358. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stachyra-Valat T, Baysang F, D'Alessandro
A-C, Dirk E, Furet P, Guagnano V, Kallen J, Leder L, Mah R, Masuya
K, et al: Abstract 1239: NVP-HDM201: Biochemical and biophysical
profile of a novel highly potent and selective PPI inhibitor of
p53-Mdm2. Cancer Res. 76(Suppl 14): 1239. 2016. View Article : Google Scholar
|
|
39
|
Furet P, Masuya K, Kallen J,
Stachyra-Valat T, Ruetz S, Guagnano V, Holzer P, Mah R, Stutz S,
Vaupel A, et al: Discovery of a novel class of highly potent
inhibitors of the p53-MDM2 interaction by structure-based design
starting from a conformational argument. Bioorg Med Chem Lett.
26:4837–4841. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Heukamp LC, Thor T, Schramm A, De Preter
K, Kumps C, De Wilde B, Odersky A, Peifer M, Lindner S, Spruessel
A, et al: Targeted expression of mutated ALK induces neuroblastoma
in transgenic mice. Sci Transl Med. 4:141ra912012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Khanna C and Hunter K: Modeling metastasis
in vivo. Carcinogenesis. 26:513–523. 2005. View Article : Google Scholar
|
|
42
|
Khanna C, Jaboin JJ, Drakos E, Tsokos M
and Thiele CJ: Biologically relevant orthotopic neuroblastoma
xenograft models: Primary adrenal tumor growth and spontaneous
distant metastasis. In Vivo. 16:77–85. 2002.PubMed/NCBI
|
|
43
|
Beltinger C and Debatin KM: Murine models
for experimental therapy of pediatric solid tumors with poor
prognosis. Int J Cancer. 92:313–318. 2001. View Article : Google Scholar : PubMed/NCBI
|