Introduction
At present, non-small cell lung cancer (NSCLC) is
one of the leading causes of cancer-associated mortality in elderly
patients. Although the early treatment for NSCLC has improved in
recent years, including platinum-based chemotherapy and
radiotherapy, NSCLC remains an aggressive type of cancer, and the
majority of patients have a poor prognosis, since NSCLC is
typically detected at the late stages of the disease (1). Excessive cancer cell migration and
survival are known to serve a crucial role in the development and
progression of NSCLC (2).
Accordingly, suppressing cancer cell migration and survival is
vital for slowing or preventing NSCLC progression.
Mammalian STE20-like kinase 1 (Mst1) is an element
of the Hippo pathway, which was initially identified as a major
growth suppressor that interrupts stem cell growth, proliferation
and apoptosis (3). Subsequent
studies have illustrated that Mst1 is also involved in sustaining
cardiomyocyte survival in diabetic cardiomyopathy (4,5),
suppressing endometrial stromal cell migration in endometriosis
(6) and promoting cancer cell
apoptosis in colorectal carcinoma (7). These findings indicated that Mst1
functions as a tumor suppressor by modulating cancer cell
apoptosis, migration and proliferation. However, the role of Mst1
in NSCLC A549 cells remains to be elucidated.
Mitochondria are vital for cancer development
(8). Mitochondria are at the
center of energy production, and damage to mitochondria reduces
cancer metabolism and therefore limits cancer growth (9). In addition, mitochondria are
messengers of cellular apoptotic signals; poorly structured
mitochondria release the pro-apoptotic factor cytochrome c
(cyt-c) into the nucleus, where it cooperates with the caspase
family to initiate the cellular death program. Furthermore,
mitochondria are calcium pumps that help the endoplasmic reticulum
(ER) to regulate cellular calcium homeostasis (10), thus critically regulating cancer
migration. Therefore, the roles of mitochondria in the regulation
of cancer migration, apoptosis and metabolism have been well
established. However, whether Mst1 can reduce NSCLC A549 cell
viability by restricting mitochondrial function has yet to be fully
elucidated.
F-actin is an important structural protein that is
required for cellular cytoskeleton organization and cellular
movement, and is also involved in processes including the
regulation of cellular division, mitochondrial fission and
filopodia formation (11). This
affords F-actin a central position within cellular response
networks. Based on previous studies, F-actin dysregulation is
associated with gastric cancer migration inhibition via sirtuin
1/mitofusin 2-mediated mitophagy (12,13).
Furthermore, F-actin downregulation contributes to rectal cancer
mitochondrial apoptosis via activation of the c-Jun N-terminal
kinase (JNK)-dynamin-related protein 1-mitochondrial fission-HtrA
serine peptidase 2/Omi axis (14).
In cardiovascular disease, F-actin degradation promotes cardiac
microvascular ischemia-reperfusion injury (11). Collectively, these findings
confirmed that functional F-actin signaling is imperative to normal
cell function. Notably, a relationship between Mst1 and F-actin has
previously been established (6).
Activated Mst1 has the ability to induce F-actin degradation, thus
promoting apoptosis in endometriosis, colorectal cancer cell death
and arrested liver cancer invasion. However, whether Mst1 has a
critical role in NSCLC A549 cell survival via regulating F-actin
homeostasis, invasion and metastasis remains to be elucidated.
At the molecular level, F-actin homeostasis is
governed by Rho-associated coiled-coil containing protein kinase 1
(ROCK1) (15), which depolymerizes
F-actin into G-actin. Furthermore, ample evidence has suggested the
possibility of ROCK1 acting as a tumor suppressor in several types
of cancer. Activated ROCK1 signaling promotes prostate cancer
apoptosis by inducing cofilin-1 translocation onto the surface of
mitochondria (16), whereas ROCK1
suppression accounts for renal cell carcinoma aggressiveness
(17). Furthermore, overexpression
of ROCK1 enhances myeloid leukemia apoptosis (18), inhibits osteosarcoma cell
metastasis (19) and increases
radiosensitization in pancreatic cancer (20). Taken together, these findings have
established a central role for ROCK1 in suppressing cancer
development and progression. However, whether ROCK1-mediated
F-actin inactivation is regulated by Mst1 and is involved in NSCLC
A549 cell migration, proliferation and apoptosis remains unclear.
Therefore, the present study aimed to explore the role of Mst1 in
the NSCLC A549 cell stress response, involving cancer cell
mobility, death and growth, with a focus on ROCK1-mediated F-actin
degradation and mitochondrial injury signaling.
Materials and methods
Cell culture and treatments
The normal pulmonary epithelial cell line BEAS-2B
(American Type Culture Collection (ATCC)® no. CRL-9609™) and the
NSCLC cell line A549 (ATCC® no. CCL-185EMT™) were purchased from
ATCC (Manassas, VA, USA). The cells were cultured in Low
Glucose-Dulbecco's modified Eagle's medium (L-DMEM; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) containing low glucose,
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.)
and 1% streptomycin and penicillin at 37°C in an atmosphere
containing 5% CO2. To inhibit ROCK1 activity, Y-27632 (5
mM; cat. no. S1049; Selleck Chemicals, Houston, TX, USA) was added
to the medium for 4 h (21).
Mst1 overexpression assay
The pDC315-Mst1 vector (1,336 bp;
pDC315-Mst1-NheI-forward, 5′-CTAATGCGT
TGCAATACGTGCGTCCTATATG-3′ and pDC315-Mst1-HindIII-reverse,
5′-TTGTCCATTGCAAGGCCTCTGATT GAGTCTG-3′) was designed and purchased
from Shanghai GenePharma Co., Ltd. (Shanghai, China). Briefly, the
plasmid (3.0 μg per 1×104 cells/well) was
transfected into 293 cells using Lipofectamine 2000® (Invitrogen;
Thermo Fisher Scientific, Inc.). When the cells detached from the
plates, the medium supernatant was collected (22). Subsequently, the viral supernatant
was identified and was amplified by transfection into 293 cells
three times, in order to obtain adenovirus (Ad)-Mst1, after which,
Ad-Mst1 was transduced into the cells to induce overexpression of
Mst1. A total of 1×105 cells/well were infected with 100
multiplicity of infection Ad-Mst1 or empty vector (Ad-ctrl) in
Opti-MEM media (Gibco; Thermo Fisher Scientific, Inc.) for 6 h at
37°C, according to the manufacturer's protocol. Cells transduced
with the Ad-ctrl were used as the control group (23).
Reactive oxygen species (ROS) detection
via flow cytometry and immunofluorescence
Cellular ROS production was analyzed by flow
cytometry, according to a previous study (24). Cells (1×106) were washed
with cold PBS and cultured with the ROS probe dihydroethidium (1
mg/ml; Molecular Probes; Thermo Fisher Scientific, Inc.) at 37°C in
the dark for 15 min. After the cells were washed three times with
cold PBS, the cells were collected using 0.25% pancreatin. After
resuspension in cold PBS, the cells were analyzed using a flow
cytometer (BD FACSVerse; BD Biosciences, San Jose, CA, USA) and the
data were analyzed with Flowmax software (Version 2.3; Sysmex
Partec GmbH, Görlitz, Germany) (25). In addition, ROS was observed using
a ROS probe. Cells (1×106) were loaded with the ROS
probe dihydroethidium (5 mg/ml; Molecular Probes; Thermo Fisher
Scientific, Inc.) in the dark for 10 min at room temperature. After
washing with PBS, ROS production was monitored under an Olympus
IX81 microscope (Olympus Corporation, Tokyo, Japan).
ATP production and mitochondrial
potential
Cellular ATP generation was measured to reflect
mitochondrial function. Firstly, A549 cells were washed three times
with cold PBS at room temperature. Subsequently, a luciferase-based
ATP assay kit (CellTiter-Glo® Luminescent Cell Viability Assay;
cat. no. G7570; Promega Corporation, Madison, WI, USA) was used to
analyze ATP content, according to the manufacturer's protocols
(10). ATP production was measured
using a microplate reader at a wavelength of 570 nm (Epoch 2;
BioTek Instruments, Inc., Winooski, VT, USA) (26). To observe the mitochondrial
potential, JC-1 staining (cat no. M34152; Thermo Fisher Scientific
Inc.) was used. Briefly, 10 mg/ml JC-1 was added to the medium for
10 min at 37°C in the dark, in order to label mitochondria. Images
were captured under an Olympus IX81 microscope (Olympus
Corporation). Normal mitochondrial potential was indicated by red
fluorescence, whereas damaged mitochondrial potential was indicated
by green fluorescence (27).
Cell migration assay
For the cell migration assay, Transwell units
(Corning Incorporation, Corning, NY, USA) with an 8-μm pore
size polycarbonate filter were used. A549 cells were initially
transduced with Ad-Mst1 and were then isolated using 0.25% trypsin.
Trypsin-mediated digestion and isolation was conducted to remove
apoptotic cells. To further exclude the influence of cell apoptosis
on the Transwell assay, a lactate dehydrogenase (LDH) release assay
was used to evaluate cell viability. Subsequently,
~1×105 cells were seeded in the upper chamber of the
Transwell units; the lower chamber was filled with 600 μl
L-DMEM supplemented with 1% FBS (Gibco; Thermo Fisher Scientific,
Inc.). Following a 12-h incubation at 37°C, the medium was removed
and the cells were fixed with 3.7% paraformaldehyde for ~10 min at
room temperature (28). The cells
in the upper chamber were removed using a cotton swab, and the
migrated cells were stained with 0.1% crystal violet for 15 min at
room temperature. Subsequently, the samples were observed under a
digital microscope system (IX81; Olympus Corporation). The images
were captured and the number of migrated cells was recorded in at
least five fields.
LDH assay and caspase-9 activity
detection
LDH is released into the medium when cellular
membranes rupture. To evaluate the levels of LDH in the medium, an
LDH Release Detection kit (Beyotime Institute of Biotechnology,
Haimen, China) was used according to the manufacturer's protocol.
To analyze alterations in caspase-9, a caspase-9 activity kit (cat
no. C1158; Beyotime Institute of Biotechnology) was conducted,
according to the manufacturer's protocol (29). Briefly, to measure caspase-9
activity, 5 μl LEHD-p-NA substrate (4 mM, 200 μM
final concentration) was added to the samples for 1 h at 37°C.
Subsequently, the absorbance was recorded at 400 nm using a
microplate reader, in order to reflect caspase-9 activities
(30).
Glutathione (GSH), GSH peroxidase (GPX)
and superoxide dismutase (SOD) detection, mitochondrial respiratory
complex activity estimation, lactate production measurement and
glucose uptake evaluation
GSH (cat. no. T10095; Thermo Fisher Scientific
Inc.), GPX (cat. no. S0056; Beyotime Institute of Biotechnology)
and SOD (cat. no. BMS222TEN; Thermo Fisher Scientific Inc.) levels
were measured, according to the manufacturers' protocols, using a
microplate reader (Epoch 2; BioTek Instruments, Inc.) (31). Mitochondrial respiratory complex
activity was estimated using commercially available kits (cat. nos.
ab109721 and ab109878; Abcam, Cambridge, MA, USA) according to the
manufacturer's protocols. To analyze lactate production, a lactate
assay kit (cat. no. K607-100; BioVision, Inc.) was used, according
to a previous study (32). In
addition, cellular glucose uptake was evaluated using a glucose
absorption assay kit (cat. no. K606-100; BioVision, Inc.),
according to the manufacturer's protocol (33).
Western blotting
Cells were lysed in radioimmunoprecipitation assay
lysis buffer (Beyotime Institute of Biotechnology). Total protein
was analyzed using the bicinchoninic acid assay (Beyotime Institute
of Biotechnology) and 70 μg lysates were separated by 10-15%
SDS-PAGE, and proteins were electrotransferred onto Pure
Nitrocellulose Blotting membranes (EMD Millipore, Billerica, MA,
USA). The membranes were then blocked with 5% non-fat milk for 2 h
at room temperature (34) and were
washed with Tris-buffered saline-0.1% Tween (TBST). The membranes
were then incubated with the following primary antibodies:
Caspase-9 (1:1,000; cat. no. ab32539; Abcam), pro-caspase-3
(1:1,000; cat. no. ab13847; Abcam), cleaved caspase-3 (1:1,000;
cat. no. ab49822; Abcam), B-cell lymphoma 2 (Bcl2; 1:1,000; cat.
no. 3498; Cell Signaling Technology, Inc.), Bcl2-associated death
promoter (Bad; 1:1,000; cat. no. 9292; Cell Signaling Technology,
Inc.), cellular inhibitor of apoptosis protein (c-IAP; 1:1,000;
cat. no. 4952; Cell Signaling Technology, Inc.), Mst1 (1:1,000;
cat. no. 3682; Cell Signaling Technology, Inc.), F-actin (1:1,000;
cat. no. ab205; Abcam), complex III subunit core 2 (CIII-core2;
1:1,000; cat. no. 459220; Invitrogen; Thermo Fisher Scientific,
Inc.), complex II (CII-30; 1:1,000; cat. no. ab110410; Abcam),
C-X-C chemokine receptor type (CXCR)4 (1:1,000; cat. no. ab1670;
Abcam), CXCR7 (1:1,000; cat. no. ab38089; Abcam), cyclin D1
(1:1,000; cat. no. ab16663; Abcam), cyclin E (1:1,000; cat. no.
ab33911; Abcam), cyt-c (1:1,000; cat. no. ab90529; Abcam) and ROCK1
(1:1,000; cat. no. ab45171; Abcam) at 4°C overnight. After washing
with TBST, the membranes were incubated with horseradish
peroxidase-coupled secondary antibodies (1:2,000; cat. nos. 7074
and 7076; Cell Signaling Technology, Inc.) for 1 h at room
temperature. The proteins were visualized using Pierce enhanced
chemiluminescence western blotting substrate (Pierce; Thermo Fisher
Scientific, Inc.) and autoradiography. Subsequently, the blots were
analyzed using Quantity One 4.6 software (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). GAPDH (1:1,000; cat. no. 5174; Cell
Signaling Technology, Inc.), β-actin (1:1,000; cat. no. 4970; Cell
Signaling Technology, Inc.) and translocase of the outer membrane
20 (1:1,000; cat. no. ab56783; Abcam) were used as internal
controls (35).
Isolation of mitochondria-enriched
fraction
Mitochondrial and cytoplasmic cyt-c expression was
measured via isolation of the mitochondrial-enriched fraction,
followed by western blotting. After isolation of
mitochondria-enriched fraction, the remaining fraction was used to
analyze cytoplasmic protein expression. In order to isolate the
mitochondria-enriched fraction, cells were washed with cold PBS and
were scraped; the homogenates were then centrifuged at 800 x g for
5 min at 4°C. The supernatants were centrifuged at 10,000 x g for
20 min at 4°C to acquire pellets, which were spun again. The final
pellets were suspended in lysis buffer (cat. no. P0013E; Beyotime
Institute of Biotechnology) containing 1% Triton X-100 and were
noted as mitochondrial-rich lysate fractions.
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL)
Apoptotic cells were detected using an In Situ Cell
Death Detection kit (cat. no. C10245; Thermo Fisher Scientific
Inc.), according to the manufacturer's protocol. Briefly, cells
(1×106) were fixed with 4% paraformaldehyde at 37°C for
15 min. Blocking buffer (3% H2O2 in
CH3OH) was added to the wells, and cells were then
permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2
min on ice. Finally, the cells were incubated with TUNEL reaction
mixture for 1 h at 37°C. DAPI (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was used to counterstain the nuclei, and the
numbers of TUNEL-positive cells were recorded (36) under a digital microscope system
(IX81; Olympus Corporation).
5-ethynyl-2′-deoxyuridine (EdU)
incorporation assay and MTT experiments
The EdU incorporation assay was performed using an
EdU kit (cat. no. A10044; Thermo Fisher Scientific Inc.) (37). Briefly, EdU (2 nM/well) was diluted
in complete culture medium, and the cells (1×106) were
incubated with the dilution for 2 h at 37°C. Subsequently, the
cells were fixed with 4% paraformaldehyde for 15 min at 37°C and
were incubated with Apollo Staining reaction liquid for 30 min.
DAPI was used to counterstain the nuclei for 15 min at room
temperature under a digital microscope system (IX81; Olympus
Corporation).
MTT was used to analyze cellular viability. A549
cells (1×106 cells/well) were cultured on a 96-well
plate at 37°C in an atmosphere containing 5% CO2, with
or without Ad-Mst1 transduction. Subsequently, 40 μl MTT
solution (2 mg/ml; Sigma-Aldrich; Merck KGaA) was added to the
medium for 4 h at 37°C in an atmosphere containing 5%
CO2. Subsequently, the cell medium was discarded, and 80
μl dimethyl sulfoxide was added to the wells for 1 h at 37°C
in an atmosphere containing 5% CO2 in the dark. The
optical density (OD) of each well was observed at an absorbance of
490 nm via a spectrophotometer (Epoch 2; BioTek Instruments, Inc.)
(38).
Immunofluorescence confocal
microscopy
The cells (1×106) were washed twice with
PBS and permeabilized in 0.1% Triton X-100 overnight at 4°C.
Subsequently, 10% goat serum albumin (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to block the samples for 1 h at room
temperature. The sections were then cryoprotected in a PBS solution
supplemented with 0.9 mol/l sucrose overnight at 4°C. Following
neutralization with NH4Cl buffer, the sections were
permeabilized for 45 min with 0.05% saponin/PBS (pH 7.4) and
incubated with H2O2 (3%) for 10 min
Subsequently, samples were treated overnight at 4°C with the
following primary antibodies: Cyt-c (1:500; cat. no. ab90529;
Abcam), Mst1 (1:200; cat. no. 3682; Cell Signaling Technology,
Inc.), F-actin (1:1,000; cat. no. ab205; Abcam), cyclin D1 (1:500;
cat. no. ab16663; Abcam) and cyclin E (1:500; cat. no. ab33911;
Abcam). After three rinses in PBS, secondary antibodies (Alexa
Fluor® 488 donkey anti-rabbit antibody; 1:1,000; cat. no. A-21206
and Alexa Fluor® Plus 647 goat anti-rabbit antibody; 1:1,000; cat.
no. A-32733; Invitrogen; Thermo Fisher Scientific, Inc.) were added
to the samples for 1 h at room temperature. The samples were
stained with DAPI (10 nM) for 5 min. Confocal immunofluorescence
images were captured using FV10-ASW 1.7 software and an Olympus
IX81 microscope (Olympus Corporation) (39). The length of filopodia formation
was measured using Image-Pro Plus 6.0 (Media Cybernetics,
Rockville, MD, USA).
Statistical analysis
Experiments were repeated three times and data are
expressed as the means ± standard error of the mean. Statistical
analyses were performed using one-way analysis of variance followed
by a Bonferroni post hoc test. P<0.05 was considered to indicate
a statistically significant difference. Statistical analysis was
performed using GraphPad Prism 5.0 (GraphPad Software, Inc., La
Jolla, CA, USA).
Results
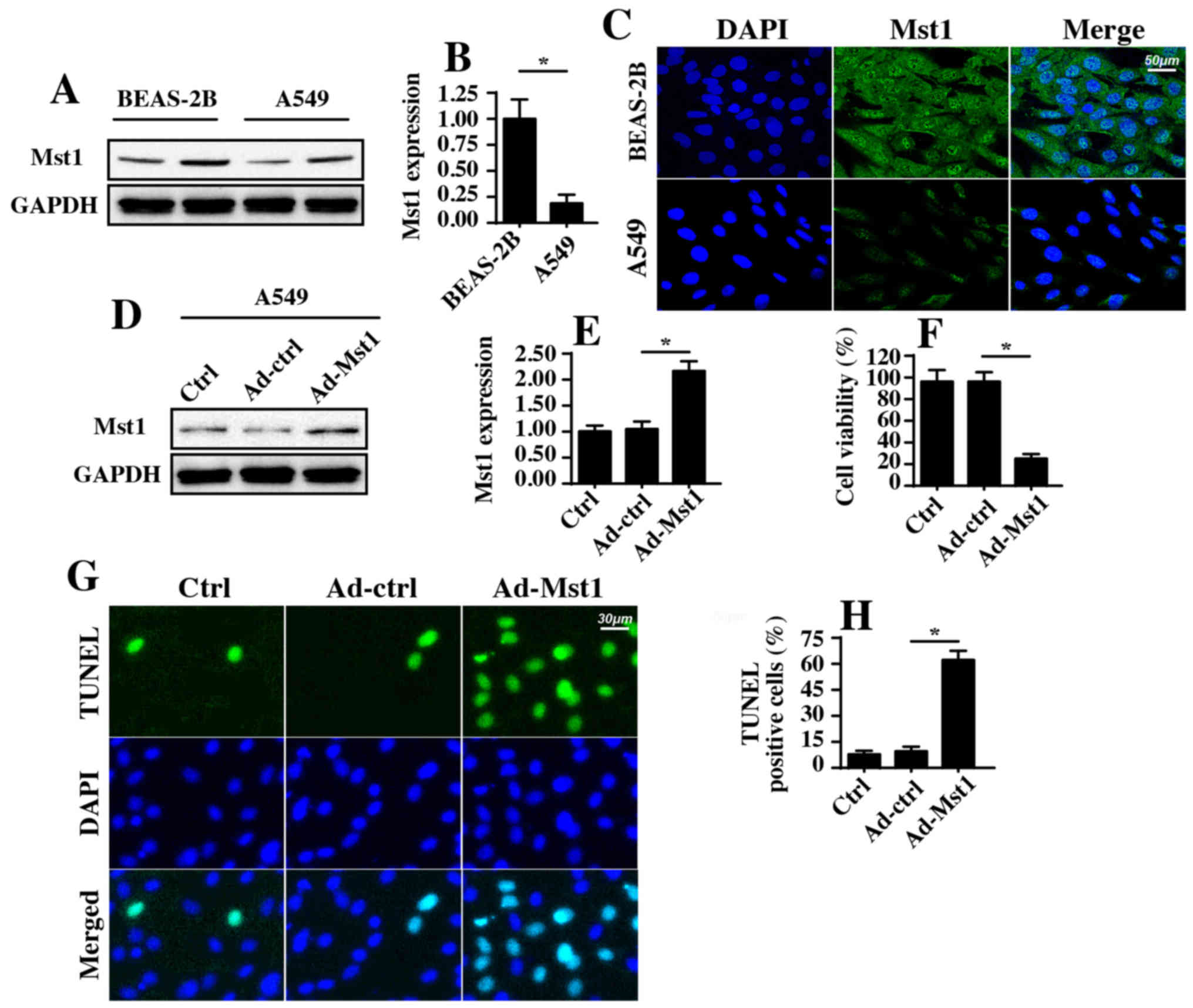
Mst1 is downregulated in NSCLC A549
cells, and overexpression of Mst1 promotes A549 cell apoptosis
To address the functional role of Mst1 in the
phenotypic alterations of A549 cells, western blotting was used to
examine the protein expression levels of Mst1 in A549 cells. As
shown in Fig. 1A and B, abundant
Mst1 expression was observed in the normal pulmonary epithelial
cell line BEAS-2B. In comparison, Mst1 expression was markedly
reduced in A549 cells; these findings were further supported by
immunofluorescence staining (Fig.
1C). These results indicated that Mst1 expression may be
downregulated in A549 cells. Subsequently, Mst1 was overexpressed
in A549 cells via an adenovirus-based Mst1 overexpression method.
Transduction efficiency is shown in Fig. 1D and E, as determined by western
blotting. Following overexpression of Mst1, cellular viability and
apoptosis were analyzed. MTT assays revealed that Mst1
overexpression in A549 cells significantly reduced cellular
viability (Fig. 1F). In agreement
with these results, the cellular apoptotic rate, as measured by
TUNEL staining (Fig. 1G and H),
revealed that the number of TUNEL-positive apoptotic cells was
significantly increased in response to Ad-Mst1 transduction
compared with in the control group. These results suggested that
Mst1 may function as a tumor suppressor in A549 cells by promoting
cancer cell apoptosis.
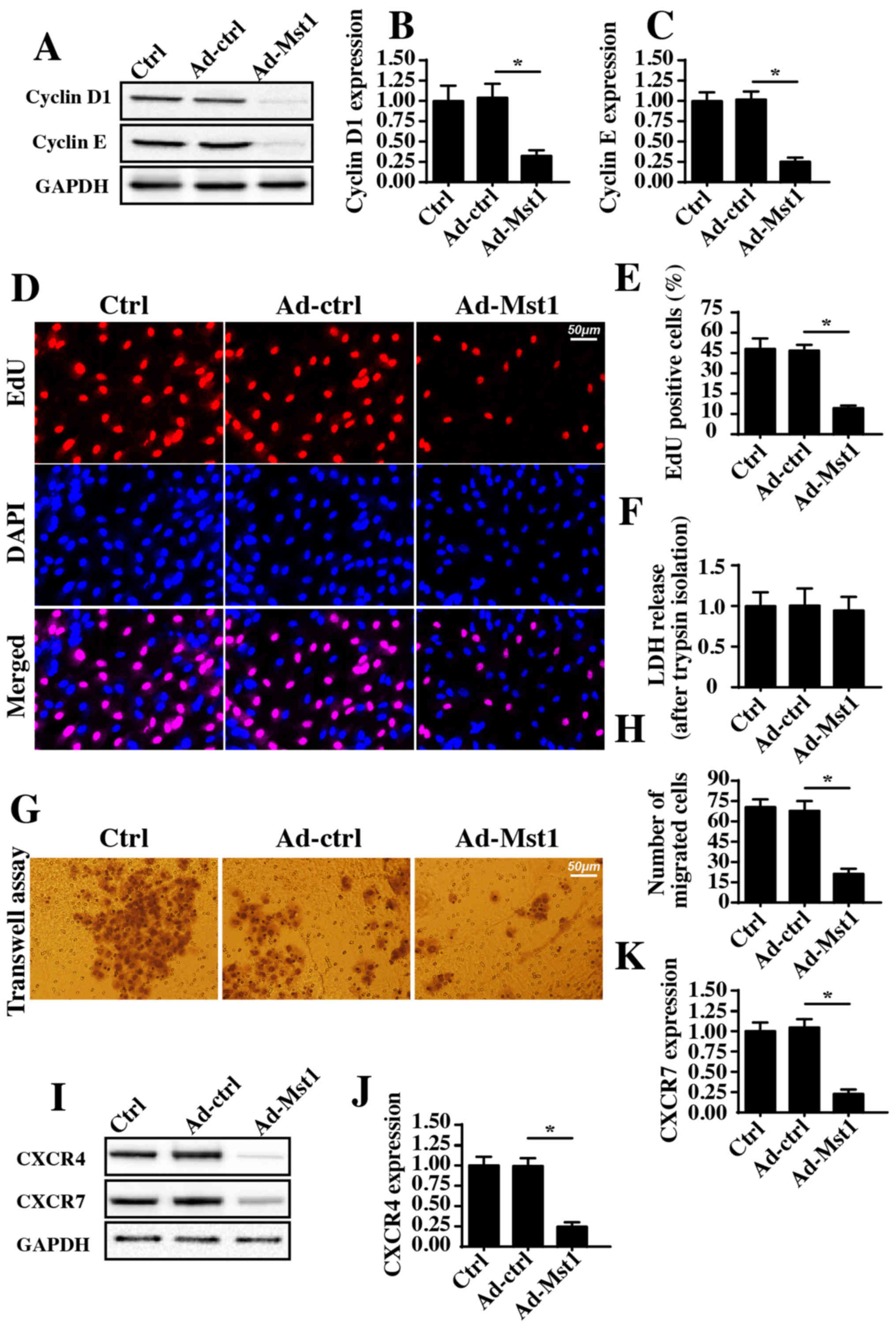
Overexpression of Mst1 reduces A549 cell
proliferation and migration
The present study further investigated the effects
of Mst1 overexpression on the growth and metastasis of A549 cells.
Initially, cyclin D1 and cyclin E expression levels, which were
detected by western blotting, were significantly decreased in
response to Mst1 overexpression compared with in the control group
(Fig. 2A–C). As cyclin D1 and
cyclin E interact with each other and generate cyclin-dependent
kinase (Cdk)4/6-cyclin D and/or Cdk2-cyclin E complexes, which
accelerate transition from the G0/G1 to S stage, the present study
aimed to determine whether Mst1 overexpression augmented the number
of cancer cells at S stage. Through EdU staining, which labels
cells at S stage, it was demonstrated that Ad-Mst1 transduction
markedly decreased the ratio of EdU-positive cells compared with in
the control group (Fig. 2D and E).
Therefore, these findings indicated that Mst1 affects A549 cell
proliferation.
The present study also observed cellular migration
in response to Mst1 overexpression. To exclude the influence of
cell viability on the Transwell assay, A549 cells transduced with
Ad-Mst1 were isolated using 0.25% trypsin. Subsequently, a LDH
release assay was used to evaluate cell viability following trypsin
isolation. The present results demonstrated that there was no
significant difference in cell viability in the Ad-Mst1 and Ad-ctrl
groups (Fig. 2F). Subsequently, a
Transwell assay was conducted. The number of migrated cells was
decreased in response to Mst1 overexpression (Fig. 2G and H). Furthermore, the
expression levels of the chemotactic factors CXCR4 and CXCR7 were
detected. Notably, the expression levels of CXCR4 and CXCR7 were
downregulated following Ad-Mst1 transduction (Fig. 2I–K), thus suggesting that Mst1
overexpression suppressed A549 cell migration. Collectively, these
results demonstrated that overexpression of Mst1 inhibited A549
cell growth and metastasis in vitro.
Overexpression of Mst1 activates the
mitochondrial apoptotic pathways
Recent studies (40,41)
have indicated that mitochondrial dysfunction is associated with
cancer cell apoptosis, inhibition of migration and proliferation
arrest via numerous mechanisms. Therefore, the present study
measured mitochondrial function in A549 cells with or without Mst1
overexpression. Initially, mitochondrial potential was evaluated by
JC-1 staining. Compared with in the control group, Mst1
overexpression reduced mitochondrial potential (Fig. 3A and B), as evidenced by reduced
red fluorescence and increased green fluorescence. In response to
mitochondrial potential collapse, excessive electrons would be
released from the mitochondrial respiratory complex, thus promoting
ROS production, which induces oxida-tive stress in cancer cells.
Based on this information, cellular ROS was measured by flow
cytometry. Compared with in the control group, Mst1 overexpression
enhanced ROS production (Fig. 3C and
D). In response to increased ROS, cellular antioxidant factors,
including GSH, SOD and GPX, were markedly downregulated in response
to Mst1 overexpression (Fig.
3E–G). Furthermore, excessive ROS has the ability to promote
mitochondrial pro-apoptotic factor translocation into the nucleus.
Through immunofluorescence assays, it was revealed that Mst1
overexpression induced cyt-c release from the mitochondria into the
nucleus compared with in the control group (Fig. 3H and I). These findings were
further supported by western blotting (Fig. 3J–L). Furthermore, Mst1
overexpression upregulated the expression levels of mitochondrial
pro-apoptotic proteins (Bad, caspase-9 and caspase-3) and
downregulated the expression levels of anti-apoptotic factors
(c-IAP and Bcl2) (Fig. 3J–Q).
Collectively, these findings indicated that Mst1 overexpression may
activate mitochondrial apoptosis in A549 cells.
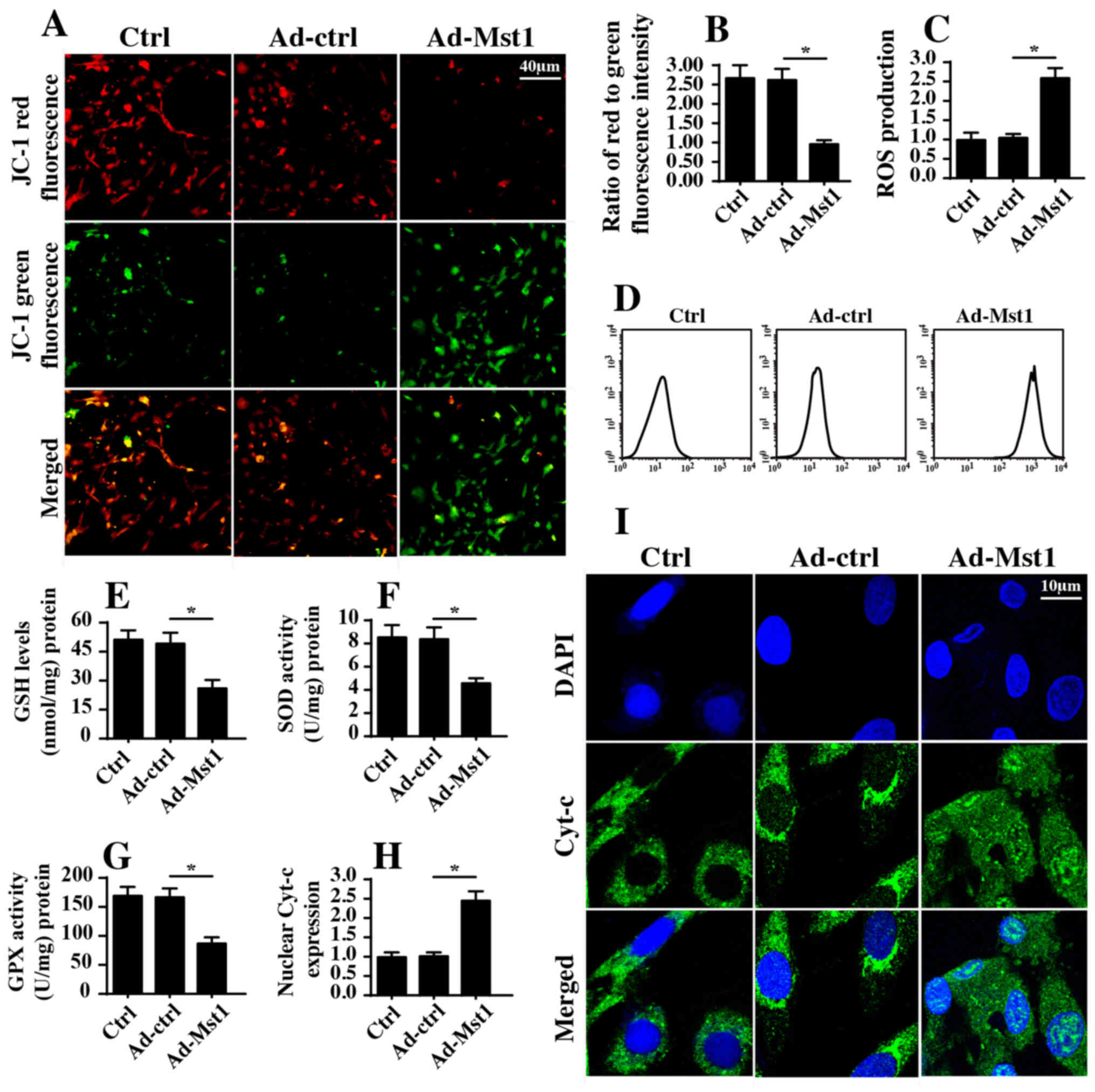 | Figure 3Mst1 overexpression is associated
with mitochondrial damage. (A and B) Mitochondrial potential was
observed via JC-1 staining. Red fluorescence indicated normal
mitochondrial potential, whereas green fluorescence indicated
damage to mitochondrial potential. (C and D) ROS production was
measured by flow cytometry. Ad-Mst1 transduction increased the ROS
content in A549 cells. (E-G) Antioxidants were detected by ELISA.
(H and I). Cyt-c translocation into the nucleus was observed by
co-staining cells with cyt-c and DAPI. (J-Q) Western blotting of
proteins associated with mitochondrial apoptosis. The expression
levels of pro-apoptotic and anti-apoptotic proteins were analyzed
by western blotting. TOM20 was used as a loading control for mito
cyt-c expression. *P<0.05. Ad, adenovirus; Bad,
Bcl2-associated death promoter; Bcl2, B-cell lymphoma 2; c-IAP,
cellular inhibitor of apoptosis protein; cyt-c, cytochrome
c; GPX, GSH peroxidase; GSH, glutathione; mito,
mitochondrial; Mst1, mammalian STE20-like kinase 1; ROS, reactive
oxygen species; SOD, superoxide dismutase; TOM20, translocase of
the outer membrane 20. |
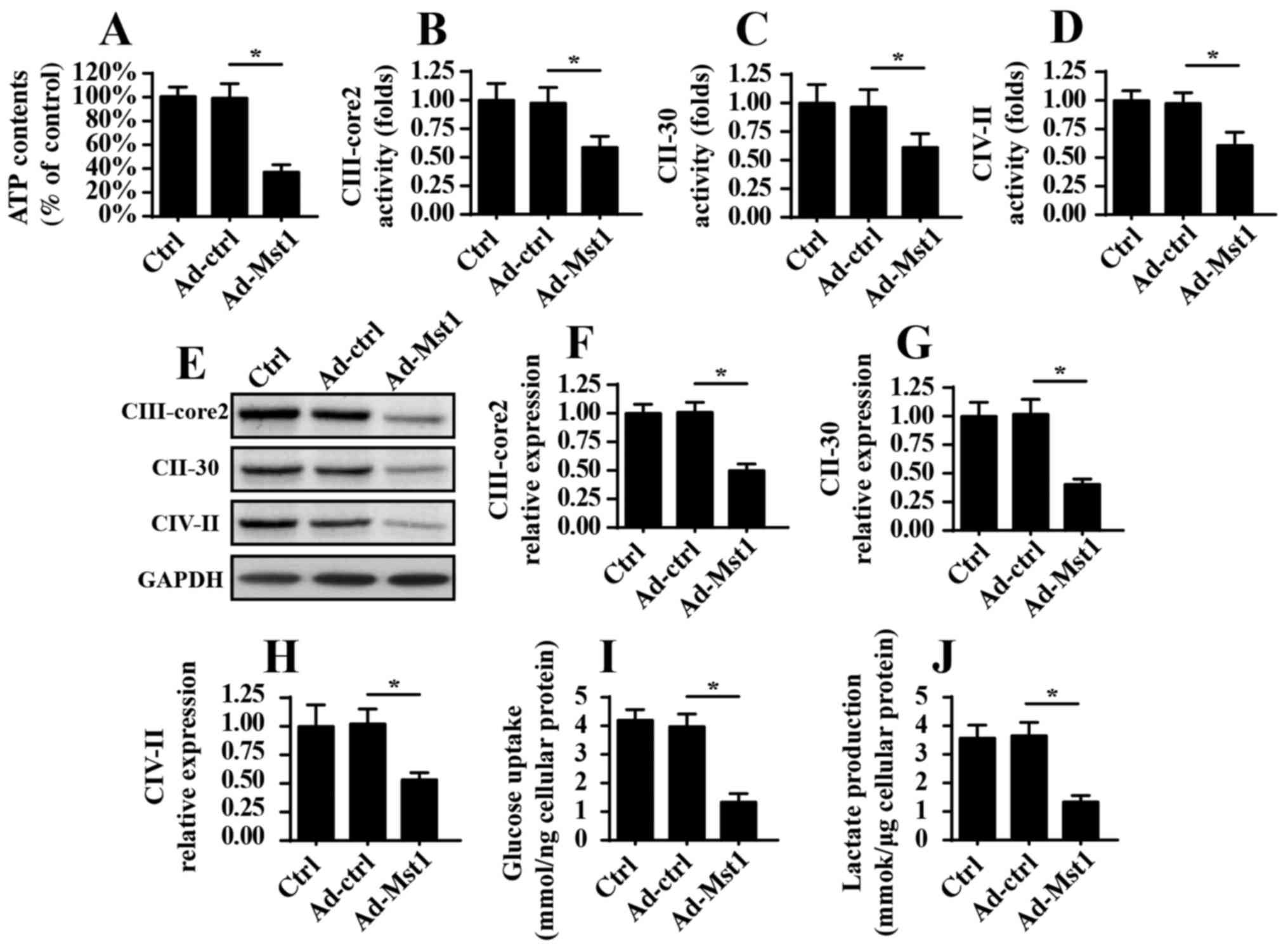
Overexpression of Mst1 induces
mitochondrial energy disorder
As well as their involvement in mitochondrial
apop-tosis, mitochondria are the center of cellular ATP production,
which provides enough energy to sustain cancer cell growth and
mobilization. The present study aimed to investigate the role of
Mst1 in mitochondrial metabolism. Initially, ATP production was
measured in A549 cells; the results indicated that Mst1
overexpression suppressed ATP production (Fig. 4A). Since ATP production is
dependent on the activity and expression of the mitochondrial
respiratory complex, this study aimed to determine whether Mst1
affected the activity and expression of the mitochondrial
respiratory complex. Through ELISA analyses, it was revealed that
mitochondrial respiratory complex activity was decreased in
response to Ad-Mst1 transduction (Fig.
4B–D). Furthermore, the protein expression levels of the
mitochondrial respiratory complexes, as measured by western
blotting, were predominantly suppressed by Mst1 overexpression
(Fig. 4E–H). Collectively, these
data indicated that Mst1 impaired mitochondrial respiration and
induced energy disorder. To provide more solid evidence for the
role of Mst1 in cellular energy metabolism, the present study
analyzed glucose and lactate content in the medium following
Ad-Mst1 transduction. As shown in Fig.
4I and J, Mst1 overexpression interrupted glucose consumption
and thus reduced the production of lactate, which is indicative of
the termination of glycometabolism. Therefore, these data indicated
that Mst1 overexpression may suppress mitochondrial energy
metabolism in A549 cells.
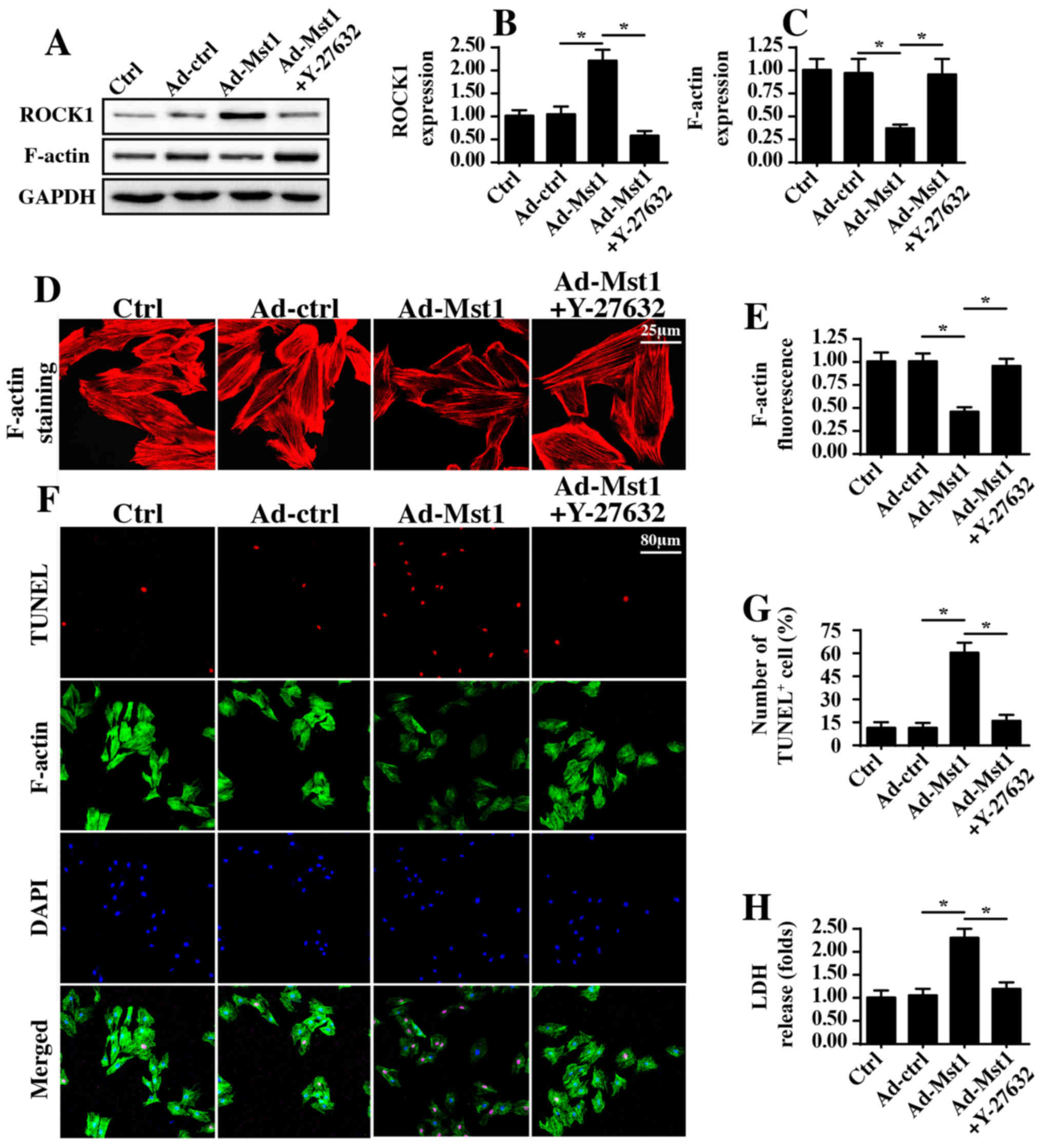
ROCK1/F-actin signaling pathways are
activated by Mst1
To determine the underlying mechanism by which Mst1
regulates mitochondrial damage and energy disorder, this study
focused on the ROCK1/F-actin axis, since ROCK1/F-actin pathways
have been revealed to be the upstream regulatory molecules for
mitochondrial damage in several types of cancer cell (42,43).
Through western blotting, it was demonstrated that Mst1
overexpression enhanced the protein expression levels of ROCK1,
which was followed by a decrease in F-actin expression (Fig. 5A–C). Subsequently, a ROCK1
loss-of-function assay was conducted via administration of Y-27632
after Ad-Mst1 transduction, which is an inhibitor of ROCK1.
Following application of Y-27632 in Mst1-overexpressed cells, ROCK1
expression was reduced (Fig.
5A–C), whereas F-actin expression was increased. These findings
were further supported by immunofluorescence assays, which
indicated that Mst1 overexpression induced the degradation of
F-actin, whereas this effect was reversed by Y-27632 administration
(Fig. 5D and E). Based on these
results, it may be suggested that Mst1 overexpression has the
ability to activate ROCK1 expression and thus reduce F-actin
stabilization.
In order to observe whether ROCK1/F-actin was
involved in Mst1-mediated A549 cell apoptosis, F-actin staining and
TUNEL assays were performed. Compared with in the control group,
Mst1 overexpression augmented the number of TUNEL-positive cells,
which was accompanied by a drop in the fluorescence intensity of
F-actin (Fig. 5F and G). However,
Mst1-induced cell apoptosis and F-actin degradation were reversed
by Y-27632 (Fig. 5F and G). This
finding was further supported by an LDH release assay (Fig. 5H), which indicated that
Mst1-mediated LDH release could be reversed by Y-27632 treatment.
Collectively, these observations indicated that Mst1 may regulate
A549 cell apoptosis via the ROCK1/F-actin pathway.
The Mst1/ROCK/F-actin axis regulates
mitochondrial injury
Since mitochondria are potential targets of Mst1 and
impact A549 cell viability, the present study aimed to determine
whether mitochondrial homeostasis was regulated by the
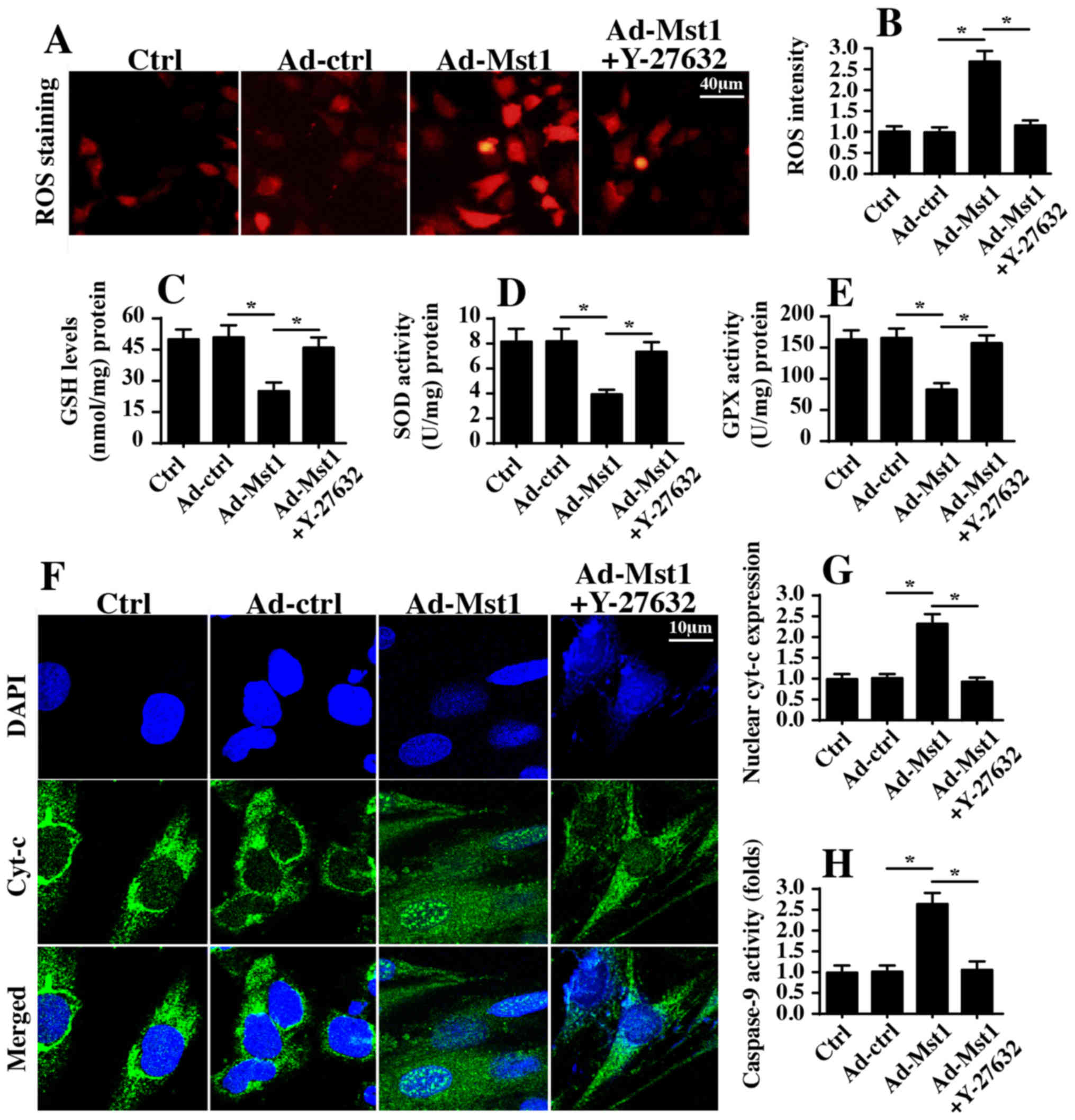
Mst1/ROCK/F-actin axis. To verify the hypothesis, ROS production
was initially detected, which is an early indicator of
mitochondrial damage. Consistent with the aforementioned findings,
Mst1 overexpression augmented ROS generation, as assessed by ROS
staining (Fig. 6A and B). However,
inhibition of ROCK1 pathways via Y-27632 significantly suppressed
ROS production despite transduction with Ad-Mst1 (Fig. 6A and B). Furthermore, inhibition of
ROCK1 blocked the pro-oxidant effects of Mst1, and the contents of
SOD, GSH and GPX were recovered in A549 cells (Fig. 6C–E). In addition, Mst1-mediated
cyt-c translocation into the nucleus was disrupted by Y-27632
administration (Fig. 6F and G).
These findings indicated that the ROCK1/F-actin axis may be
required for Mst1-associated mitochondrial apoptosis. To provide
more solid evidence to explain the role of the ROCK1/F-actin axis
in mitochondrial damage, caspase-9 activity, which is a hallmark of
mitochondrial death, was measured via ELISA. Compared with in the
control group, Mst1 overexpression elevated caspase-9 activity
(Fig. 6H); however, this effect
was reversed by Y-27632. Taken together, these data suggested that
the ROCK1/F-actin axis may be activated by Mst1 and could
contribute to mitochondrial injury in A549 cells.
Mst1/ROCK1/F-actin pathways are involved
in A549 cell migration inhibition and proliferation arrest
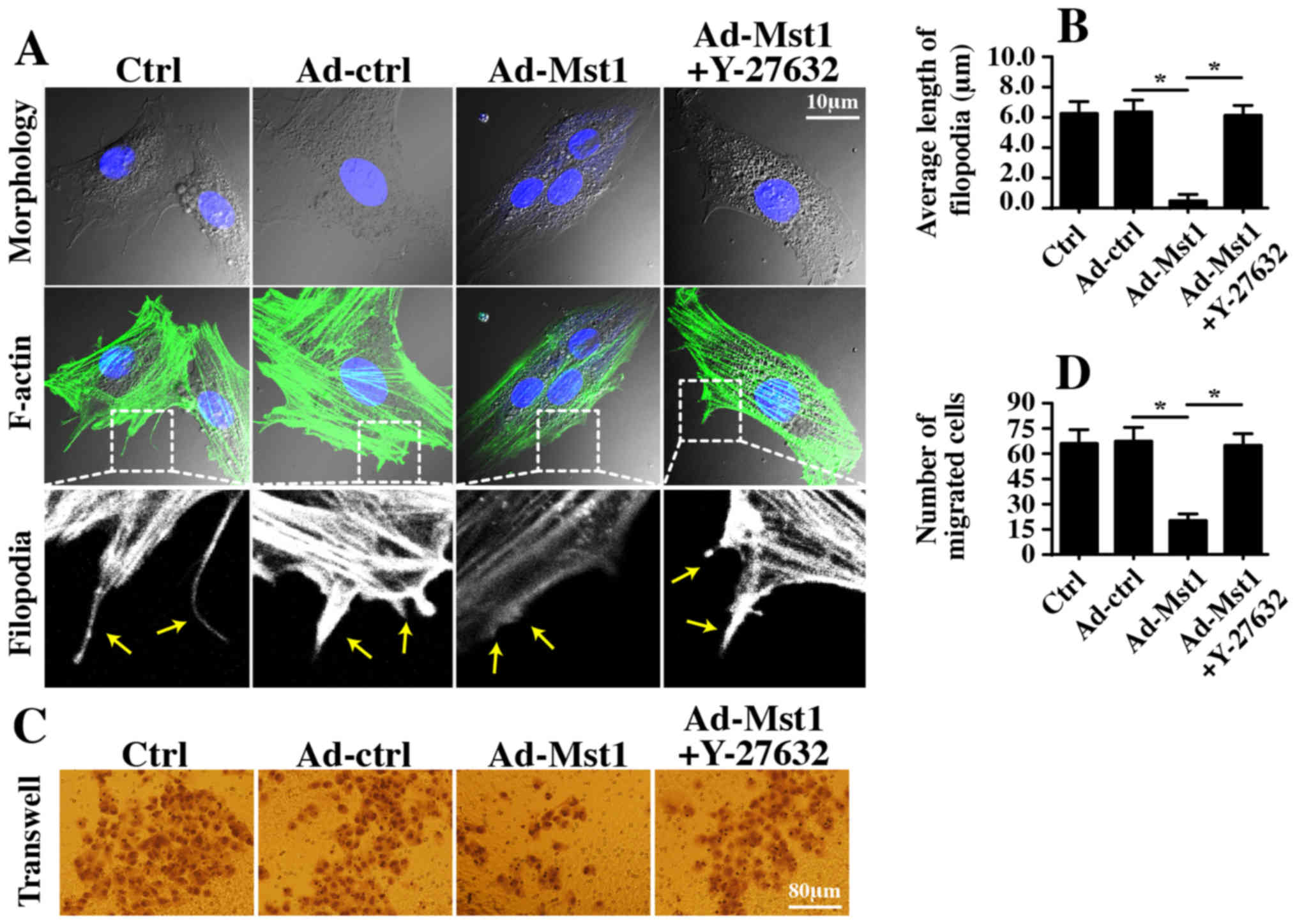
Finally, the present study aimed to determine
whether the ROCK/F-actin pathway is necessary for Mst1-mediated
A549 cell migration inhibition and proliferation arrest. Therefore,
filopodia formation was observed; filopodia is the major vehicle
for cellular migration. Mst1 overexpression hampered filopodia
formation, as evidenced by shorter and fewer filopodia outside of
the cellular membrane (Fig. 7A and
B). Conversely, this conformational alteration was reversed by
Y-27632 (Fig. 7A and B).
Furthermore, Transwell assays revealed that the number of migrated
cells was reduced in response to Mst1 overexpression but reverted
to normal levels following ROCK1 suppression (Fig. 7C and D). These findings indicated
that the ROCK1/F-actin pathway was responsible for Mst1-mediated
A549 cell migration inhibition.
The EdU assay was used to observe cell growth.
Similar to the aforementioned results, Mst1 overexpression reduced
the number of EdU-positive cells, whereas this effect was reversed
by Y-27632 (Fig. 7E and F).
Furthermore, through immunofluorescence analysis, it was revealed
that Mst1-inhibited cyclin D1/E expression was rescued by Y-27632
(Fig. 7G–I). These findings
indicated that Mst1 suppressed A549 cell proliferation by
regulating the ROCK1/F-actin pathway.
Discussion
Lung cancer is currently one of the leading causes
of cancer-associated mortality for older patients. Despite advances
in its early diagnosis and treatment, additional investigations
elucidating the mechanism underlying tumor progression are
clinically required due to the high prevalence, invasiveness and
metastatic potential of lung cancer. The present study aimed to
determine the potential molecular events regulating NSCLC A549 cell
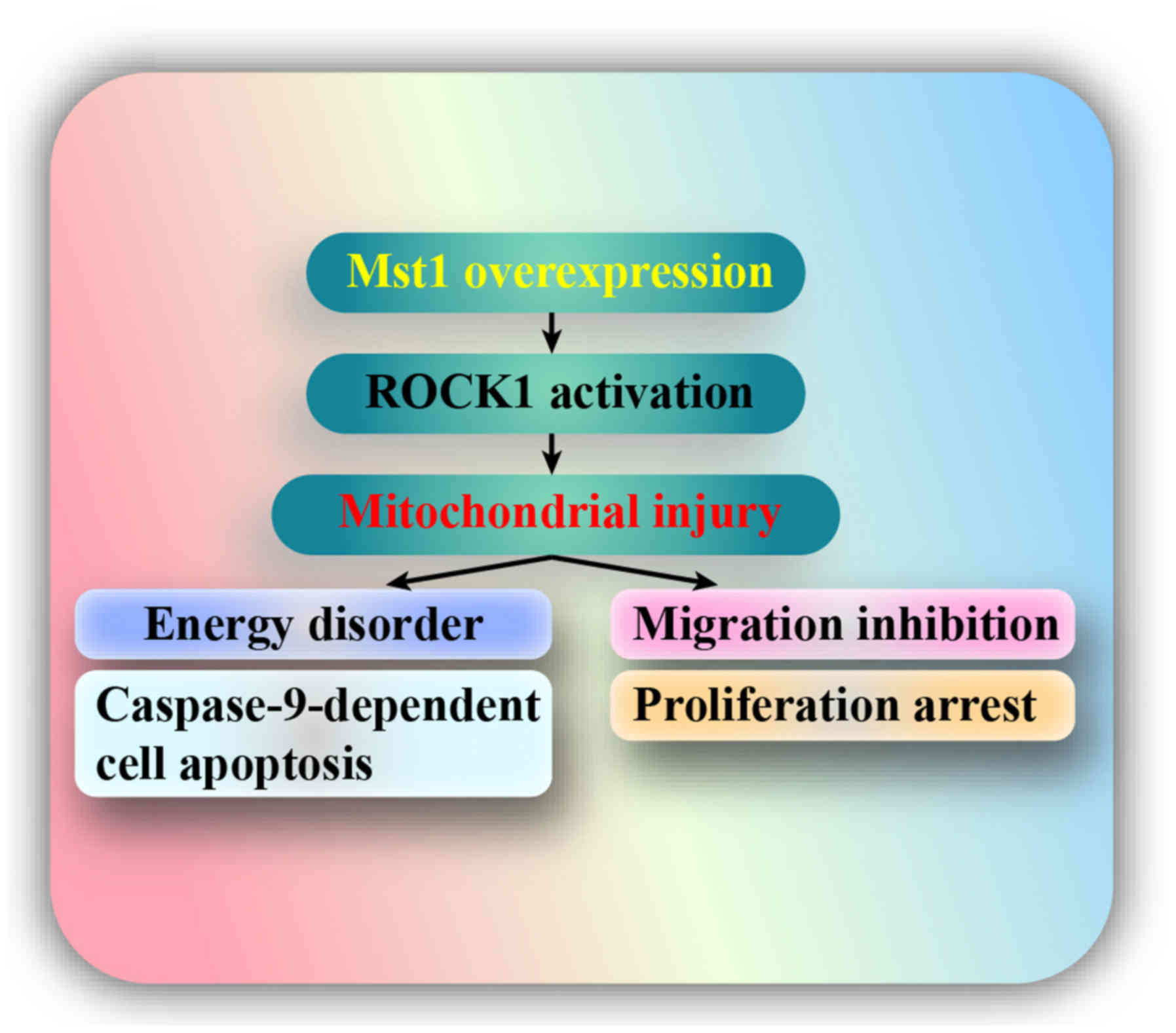
migration, survival and proliferation. The results indicated that:
i) Mst1 was downregulated in A549 cells; ii) overexpression of Mst1
promoted A549 cell apoptosis, inhibited migration and arrested
proliferation; iii) mechanistically, Mst1 activation was closely
associated with mitochondrial injury, as evidenced by reduced
mitochondrial potential, uncontrolled oxidative stress,
pro-apoptotic protein translocation, reduced mitochondrial energy
metabolism and activated caspase-9 signals; iv) at the molecular
level, the ROCK1/F-actin pathway was triggered by Mst1
overexpression; and v) activated ROCK1 signaling promoted F-actin
degradation and finally triggered mitochondrial apoptosis,
migration impairment and proliferation delay in A549 cells. Taken
together, these findings regarding A549 cell stress response lay
the foundation for a detailed study into the molecular mechanism
underlying cancer progression (Fig.
8). To the best of our knowledge, this is the first study to
investigate the role of the Mst1-mediated ROCK1/F-actin pathway in
A549 cell migration, apoptosis and proliferation, with a focus on
their roles in modulating mitochondrial damage.
The association between mitochondrial homeostasis
and tumor progression is an area of research that has rapidly
evolved over the past decade (1).
Several biological processes, including apoptosis, proliferation,
metabolic reprogramming and autophagy, are tightly linked to
mitochondrial homeo-stasis (44),
and each of these processes can be affected by alterations in
mitochondrial function and structure (6). These findings have indicated that
alterations in mitochondrial homeostasis may underlie many of the
phenotypes that drive tumorigenic growth. In addition,
mitochondrial damage has been acknowledged to have an unfavorable
effect on cancer development. It has previously been reported that
mitochondrial damage impairs liver cancer migration via
JNK/Bcl2-interacting protein 3/sarcoendoplasmic reticulum calcium
transport ATPase/Ca2+/calmodulin-dependent protein
kinase II pathways (8).
Furthermore, alterations in mitochondrial morphology, such as
mitochondrial fission, promote rectal cancer cell apoptosis
(7). The mitochondrial autophagy
repair system, which is also known as mitophagy, provides a
survival advantage to gastric cancer (13). These data confirmed that
mitochondria may be considered a potential target for regulating
cancer development. In agreement with these findings, the present
study demonstrated a strong association between mitochondrial
injury and cancer cell migration inhibition, apoptosis and
proliferation arrest. Therefore, it may be suggested that
disturbance of mitochondrial integrity is of utmost importance when
designing antitumor therapies in combination with chemotherapy.
The present study revealed that Mst1 affected
mitochondrial injury via the ROCK1/F-actin pathway. Mst1 is a
downstream factor of the Hippo pathway, which has been implicated
in several pathological processes, including diabetic
cardiomyopathy, osteoblast differentiation (45) and milk fat metabolism disorder
(46). Furthermore, Mst1
downregulation has been identified as a potential early detection
biomarker for numerous types of cancer, including colorectal cancer
(47), breast cancer (48), pancreatic cancer (49), lung cancer (50) and liver cancer. At the molecular
level, Mst1 has been reported to be associated with the ER stress
response (51), cellular oxidative
stress (52),
epithelial-to-mesenchymal transition (53), transforming growth factor
β-mediated tissue fibrosis (54)
and mitophagy (7). The present
study reported that Mst1 was downregulated in A549 cells, which was
similar to previous conclusions (55). Furthermore, the present study
revealed that Mst1 activation was associated with mitochondrial
damage. Damaged mitochondria are particularly prone to activating
the apoptotic program and mediating cellular energy disorder
(11), thus leading to cancer cell
death, mobilization impairment and growth arrest. This finding
highlights that Mst1 dysregulation may be involved in the ability
of tumor cells to escape programmed cell death. Notably, to the
best of our knowledge, the present data established a novel role
for Mst1 in initiating mitochondrial damage for the first time.
Furthermore, it was confirmed that the ROCK1/F-actin pathway was
necessary for Mst1-induced mitochondrial dysfunction and cancer
cell damage. It is well documented that ROCK1 activation is
strongly associated with cancer suppression. ROCK1 activation
reduces the development of osteosarcoma (56), alleviates chemotherapeutic
resistance in colorectal cancer (57), suppresses prostate cancer survival
(16), attenuates the
aggressiveness of renal cell carcinoma (17) and impairs epithelial-to mesenchymal
transition in patients with lung cancer (58). Similar to these findings, the
present data indicated that Mst1 overexpression-induced ROCK1
activation hampered NSCLC A549 cell migration, proliferation and
survival. These findings, combined with evidence that ROCK1 is a
potentially actionable therapeutic target for tumor suppression,
provided novel information regarding cancer development and may
provide a novel therapeutic approach for cancer inhibition via the
modulation of mitochondrial homeostasis. At present, since Mst1
lung-specific transgenic mice are currently unavailable, further
in vivo studies using Mst1 adenoviral injection into the
lung tissue of rats and/or mice to induce Mst1 overexpression are
required to verify these findings.
Acknowledgments
Not applicable.
Funding
The present study was supported by The Wu Jie Ping
Medical Research Fund (2016 grant no. 320.6750.14136)
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
WQZ designed the study. JBM, KQL, JT, YXP and JZ
performed the experiments. All authors participated in the
discussion and revision of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors have declared that they have no
competing interests.
References
|
1
|
Ilie M, Benzaquen J, Hofman V, Lassalle S,
Yazbeck N, Leroy S, Heeke S, Bence C, Mograbi B, Glaichenhaus N, et
al: Immunotherapy in non-small cell lung cancer: Biological
principles and future opportunities. Curr Mol Med. 17:527–540.
2017. View Article : Google Scholar
|
|
2
|
Zhang Y, Lian J and Wang X: Actein
inhibits cell proliferation and migration and promotes cell
apoptosis in human non-small cell lung cancer cells. Oncol Lett.
15:3155–3160. 2018.PubMed/NCBI
|
|
3
|
Patel SH, Camargo FD and Yimlamai D: Hippo
signaling in the liver regulates organ size, cell fate, and
carcinogenesis. Gastroenterology. 152:533–545. 2017. View Article : Google Scholar :
|
|
4
|
Liu M, Liu S, Tan W, Tang F, Long J, Li Z,
Liang B, Chu C and Yang J: Gaseous signalling molecule
SO2 via Hippo-MST pathway to improve myocardial fibrosis
of diabetic rats. Mol Med Rep. 16:8953–8963. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang M, Lin J, Wang S, Cheng Z, Hu J,
Wang T, Man W, Yin T, Guo W, Gao E, et al: Melatonin protects
against diabetic cardiomyopathy through Mst1/Sirt3 signaling. J
Pineal Res. Jun 9–2017.Epub ahead of print. View Article : Google Scholar
|
|
6
|
Zhao Q, Ye M, Yang W, Wang M, Li M, Gu C,
Zhao L, Zhang Z, Han W, Fan W, et al: Effect of Mst1 on
endometriosis apoptosis and migration: Role of Drp1-related
mitochondrial fission and Parkin-required mitophagy. Cell Physiol
Biochem. 45:1172–1190. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Q, Qi F, Meng X, Zhu C and Gao Y: Mst1
regulates colorectal cancer stress response via inhibiting
Bnip3-related mitophagy by activation of JNK/p53 pathway. Cell Biol
Toxicol. 34:263–277. 2018. View Article : Google Scholar
|
|
8
|
Shi C, Cai Y, Li Y, Li Y, Hu N, Ma S, Hu
S, Zhu P, Wang W and Zhou H: Yap promotes hepatocellular carcinoma
metastasis and mobilization via governing
cofilin/F-actin/lamellipodium axis by regulation of
JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 14:59–71. 2018.
View Article : Google Scholar
|
|
9
|
Lee MS, Yin TC, Sung PH, Chiang JY, Sun CK
and Yip HK: Melatonin enhances survival and preserves functional
integrity of stem cells: A review. J Pineal Res. Jan 23–2017.Epub
ahead of print. View Article : Google Scholar
|
|
10
|
Zhu H, Jin Q, Li Y, Ma Q, Wang J, Li D,
Zhou H and Chen Y: Melatonin protected cardiac microvascular
endothelial cells against oxidative stress injury via suppression
of IP3R-[Ca2+] c/VDAC-[Ca2+]m axis by
activation of MAPK/ERK signaling pathway. Cell Stress Chaperones.
23:101–113. 2018. View Article : Google Scholar
|
|
11
|
Zhou H, Wang J, Zhu P, Hu S and Ren J:
Ripk3 regulates cardiac microvascular reperfusion injury: The role
of IP3R-dependent calcium overload, XO-mediated oxidative stress
and F-action/filopodia-based cellular migration. Cell Signal.
45:12–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yan H, Xiao F, Zou J, Qiu C, Sun W, Gu M
and Zhang L: NR4A1-induced increase in the sensitivity of a human
gastric cancer line to TNFα-mediated apoptosis is associated with
the inhibition of JNK/Parkin-dependent mitophagy. Int J Oncol.
52:367–378. 2018.
|
|
13
|
Yan H, Qiu C, Sun W, Gu M, Xiao F, Zou J
and Zhang L: Yap regulates gastric cancer survival and migration
via SIRT1/Mfn2/mitophagy. Oncol Rep. 39:1671–1681. 2018.PubMed/NCBI
|
|
14
|
Li H, He F, Zhao X, Zhang Y, Chu X, Hua C,
Qu Y, Duan Y and Ming L: YAP inhibits the apoptosis and migration
of human rectal cancer cells via suppression of
JNK-Drp1-mitochondrial fission-HtrA2/Omi pathways. Cell Physiol
Biochem. 44:2073–2089. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang X, Guo D, Lin C, Shi Z, Qian R, Fu W,
Liu J, Li X and Fan L: hCLOCK causes Rho-kinase-mediated
endothelial dysfunction and NF-κB-mediated inflammatory responses.
Oxid Med Cell Longev. 2015:6718392015. View Article : Google Scholar
|
|
16
|
Mu D, Zhou G, Li J, Su B and Guo H:
Ursolic acid activates the apoptosis of prostate cancer via
ROCK/PTEN mediated mitochondrial translocation of cofilin-1. Oncol
Lett. 15:3202–3206. 2018.PubMed/NCBI
|
|
17
|
Zhang X, Li P, Ding Z, Wang H, Wang J, Han
L and Ding S: The putative tumor suppressor, miR-199a, regulated by
Snail, modulates clear cell renal cell carcinoma aggressiveness by
repressing ROCK1. OncoTargets Ther. 11:103–112. 2017. View Article : Google Scholar
|
|
18
|
Zhao WH, Huang BT, Zhang JY and Zeng QC:
Distinct EphB4-mediated mechanisms of apoptotic and resistance to
dasatinib in human chronic myeloid leukemia and K562 cell lines.
Leuk Res. 63:28–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Zhang Y, Yang T, Zhao W, Wang N,
Li P, Zeng X and Zhang W: Long non-coding RNA MALAT1 for promoting
metastasis and proliferation by acting as a ceRNA of miR-1443pin
osteosarcoma cells. Oncotarget. 8:59417–59434. 2017.PubMed/NCBI
|
|
20
|
Vennin C, Rath N, Pajic M, Olson MF and
Timpson P: Targeting ROCK activity to disrupt and prime pancreatic
cancer for chemotherapy. Small GTPases. Oct 3–2017.Epub ahead of
print. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Le Cras TD, Mobberley-Schuman PS, Broering
M, Fei L, Trenor CC III and Adams DM: Angiopoietins as serum
biomarkers for lymphatic anomalies. Angiogenesis. 20:163–173. 2017.
View Article : Google Scholar
|
|
22
|
Zhang W, Tao A, Lan T, Cepinskas G, Kao R,
Martin CM and Rui T: Carbon monoxide releasing molecule-3 improves
myocardial function in mice with sepsis by inhibiting NLRP3
inflammasome activation in cardiac fibroblasts. Basic Res Cardiol.
112:162017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou H, Shi C, Hu S, Zhu H, Ren J and Chen
Y: BI1 is associated with microvascular protection in cardiac
ischemia reperfusion injury via repressing
Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis.
21:599–615. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
García-Niño WR, Correa F,
Rodríguez-Barrena JI, León-Contreras JC, Buelna-Chontal M,
Soria-Castro E, Hernández-Pando R, Pedraza-Chaverri J and Zazueta
C: Cardioprotective kinase signaling to subsarcolemmal and
inter-fibrillar mitochondria is mediated by caveolar structures.
Basic Res Cardiol. 112:152017. View Article : Google Scholar
|
|
25
|
Ackermann M, Kim YO, Wagner WL, Schuppan
D, Valenzuela CD, Mentzer SJ, Kreuz S, Stiller D, Wollin L and
Konerding MA: Effects of nintedanib on the microvascular
architecture in a lung fibrosis model. Angiogenesis. 20:359–372.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou H, Yang J, Xin T, Zhang T, Hu S, Zhou
S, Chen G and Chen Y: Exendin-4 enhances the migration of
adipose-derived stem cells to neonatal rat ventricular
cardiomyocyte-derived conditioned medium via the phosphoinositide
3-kinase/Akt-stromal cell-derived factor-1α/CXC chemokine receptor
4 pathway. Mol Med Rep. 11:4063–4072. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kalyanaraman B: Teaching the basics of
cancer metabolism: Developing antitumor strategies by exploiting
the differences between normal and cancer cell metabolism. Redox
Biol. 12:833–842. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Das N, Mandala A, Naaz S, Giri S, Jain M,
Bandyopadhyay D, Reiter RJ and Roy SS: Melatonin protects against
lipid-induced mitochondrial dysfunction in hepatocytes and inhibits
stellate cell activation during hepatic fibrosis in mice. J Pineal
Res. Mar 27–2017.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alghanem AF, Wilkinson EL, Emmett MS,
Aljasir MA, Holmes K, Rothermel BA, Simms VA, Heath VL and Cross
MJ: RCAN1.4 regulates VEGFR-2 internalisation, cell polarity and
migration in human microvascular endothelial cells. Angiogenesis.
20:341–358. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vargas LA, Velasquez FC and Alvarez BV:
Compensatory role of the NBCn1 sodium/bicarbonate cotransporter on
Ca2+-induced mitochondrial swelling in hypertrophic
hearts. Basic Res Cardiol. 112:142017. View Article : Google Scholar
|
|
31
|
Jovancevic N, Dendorfer A, Matzkies M,
Kovarova M, Heckmann JC, Osterloh M, Boehm M, Weber L, Nguemo F,
Semmler J, et al: Medium-chain fatty acids modulate myocardial
function via a cardiac odorant receptor. Basic Res Cardiol.
112:132017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou H, Wang S, Hu S, Chen Y and Ren J:
ER-mitochondria microdomains in cardiac ischemia-reperfusion
injury: A fresh perspective. Front Physiol. 9:7552018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ligeza J, Marona P, Gach N, Lipert B,
Miekus K, Wilk W, Jaszczynski J, Stelmach A, Loboda A, Dulak J, et
al: MCPIP1 contributes to clear cell renal cell carcinomas
development. Angiogenesis. 20:325–340. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ronchi C, Torre E, Rizzetto R, Bernardi J,
Rocchetti M and Zaza A: Late sodium current and intracellular ionic
homeostasis in acute ischemia. Basic Res Cardiol. 112:122017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Couto JA, Ayturk UM, Konczyk DJ, Goss JA,
Huang AY, Hann S, Reeve JL, Liang MG, Bischoff J, Warman ML, et al:
A somatic GNA11 mutation is associated with extremity capillary
malformation and overgrowth. Angiogenesis. 20:303–306. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang
D, Wang J, Qin Y, Liu Y, Tang C, et al: The mitochondria-targeted
antioxidant MitoQ ameliorated tubular injury mediated by mitophagy
in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 11:297–311.
2017. View Article : Google Scholar :
|
|
37
|
Pickard JMJ, Burke N, Davidson SM and
Yellon DM: Intrinsic cardiac ganglia and acetylcholine are
important in the mechanism of ischaemic preconditioning. Basic Res
Cardiol. 112:112017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Torres-Estay V, Carreño DV, Fuenzalida P,
Watts A, San Francisco IF, Montecinos VP, Sotomayor PC, Ebos J,
Smith GJ and Godoy AS: Androgens modulate male-derived endothelial
cell homeostasis using androgen receptor-dependent and
receptor-independent mechanisms. Angiogenesis. 20:25–38. 2017.
View Article : Google Scholar
|
|
39
|
Randriamboavonjy V, Kyselova A, Elgheznawy
A, Zukunft S, Wittig I and Fleming I: Calpain 1 cleaves and
inactivates prostacyclin synthase in mesenteric arteries from
diabetic mice. Basic Res Cardiol. 112:102017. View Article : Google Scholar
|
|
40
|
Zhou H, Yue Y, Wang J, Ma Q and Chen Y:
Melatonin therapy for diabetic cardiomyopathy: A mechanism
involving Syk-mitochondrial complex I-SERCA pathway. Cell Signal.
47:88–100. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li R, Xin T, Li D, Wang C, Zhu H and Zhou
H: Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic
fatty liver disease: The role of the ERK-CREB pathway and
Bnip3-mediated mitophagy. Redox Biol. 18:229–243. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tamura H, Kawamoto M, Sato S, Tamura I,
Maekawa R, Taketani T, Aasada H, Takaki E, Nakai A, Reiter RJ, et
al: Long-term melatonin treatment delays ovarian aging. J Pineal
Res. Jan 23–2017.Epub ahead of print. View Article : Google Scholar
|
|
43
|
Zhou H, Zhu P, Wang J, Zhu H, Ren J and
Chen Y: Pathogenesis of cardiac ischemia reperfusion injury is
associated with CK2α-disturbed mitochondrial homeostasis via
suppression of FUNDC1-related mitophagy. Cell Death Differ.
25:1080–1093. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Van Nostrand JL, Bowen ME, Vogel H, Barna
M and Attardi LD: The p53 family members have distinct roles during
mammalian embryonic development. Cell Death Differ. 24:575–579.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li W, Deng Y, Feng B and Mak KK: Mst1/2
kinases modulate glucose uptake for osteoblast differentiation and
bone formation. J Bone Miner Res. 33:1183–1195. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen Z, Shi H, Sun S, Luo J, Zhang W, Hou
Y and Loor JJ: miR-183 regulates milk fat metabolism via MST1 in
goat mammary epithelial cells. Gene. 646:12–19. 2018. View Article : Google Scholar
|
|
47
|
Yu J, Zhai X, Li X, Zhong C, Guo C, Yang
F, Yuan Y and Zheng S: Identification of MST1 as a potential early
detection biomarker for colorectal cancer through a proteomic
approach. Sci Rep. 7:142652017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ruiz-Torres SJ, Benight NM, Karns RA,
Lower EE, Guan JL and Waltz SE: HGFL-mediated RON signaling
supports breast cancer stem cell phenotypes via activation of
non-canonical β-catenin signaling. Oncotarget. 8:58918–58933. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gao ZQ, Wang JF, Chen DH, Ma XS, Yang W,
Zhe T and Dang XW: Long non-coding RNA GAS5 antagonizes the
chemo-resistance of pancreatic cancer cells through down-regulation
of miR-181c-5p. Biomed Pharmacother. 97:809–817. 2018. View Article : Google Scholar
|
|
50
|
Zheng X, Dong Q, Zhang X, Han Q, Han X,
Han Y, Wu J, Rong X and Wang E: The coiled-coil domain of oncogene
RASSF 7 inhibits hippo signaling and promotes non-small cell lung
cancer. Oncotarget. 8:78734–78748. 2017.PubMed/NCBI
|
|
51
|
Ashokkumar R, Jamuna S, Sakeena Sadullah
MS and Niranjali Devaraj S: Vitexin protects isoproterenol induced
post myocardial injury by modulating hipposignaling and ER stress
responses. Biochem Biophys Res Commun. 496:731–737. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yu L, Liu Y, Jin Y, Cao X, Chen J, Jin J,
Gu Y, Bao X, Ren Z, Xu Y, et al: Lentivirus-mediated HDAC3
inhibition attenuates oxidative stress in APPswe/PS1dE9 mice. J
Alzheimers Dis. 61:1411–1424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li X, Zhao X, Song W, Tian Z, Yang L, Niu
Q, Zhang Q, Xie M, Zhou B, Xu Y, et al: Pseudolaric acid B inhibits
proliferation, invasion and epithelial-to-mesenchymal transition in
human pancreatic cancer cell. Yonsei Med J. 59:20–27. 2018.
View Article : Google Scholar
|
|
54
|
Wang S, Fan Y, Feng X, Sun C, Shi Z, Li T,
Lv J, Yang Z, Zhao Z and Sun D: Nicorandil alleviates myocardial
injury and post-infarction cardiac remodeling by inhibiting Mst1.
Biochem Biophys Res Commun. 495:292–299. 2018. View Article : Google Scholar
|
|
55
|
Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu
S, Ren J and Chen Y: NR4A1 aggravates the cardiac microvascular
ischemia reperfusion injury through suppressing FUNDC1-mediated
mitophagy and promoting Mff-required mitochondrial fission by CK2α.
Basic Res Cardiol. 113:232018. View Article : Google Scholar
|
|
56
|
Su S and Nie X: miR-139 prompts the
development of osteosarcomas mainly through targeting ROCK1.
Pharmazie. 72:759–763. 2017.
|
|
57
|
Wang X, Sun D, Tai J, Chen S, Yu M, Ren D
and Wang L: TFAP2C promotes stemness and chemotherapeutic
resistance in colorectal cancer via inactivating hippo signaling
pathway. J Exp Clin Cancer Res. 37:272018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lin L, Li M, Lin L, Xu X, Jiang G and Wu
L: FPPS mediates TGF-β1-induced non-small cell lung cancer cell
invasion and the EMT process via the RhoA/Rock1 pathway. Biochem
Biophys Res Commun. 496:536–541. 2018. View Article : Google Scholar : PubMed/NCBI
|