Introduction
Liver cancer accounts for ~500,000 deaths annually,
and is ranked as the second leading cause of cancer-associated
mortality globally (1). The
development of liver cancer is not completely understood; hepatitis
B virus (HBV) infection is considered to be the most common cause
of this disease (2). Individuals
chronically infected with HBV are at major risk for developing
liver cancer, with the risk 10-25-fold greater compared with
non-infected individuals (3,4). The
pathogenesis of this disease is complex and requires further
investigation. HBV induces liver cancer via distinct mechanisms.
For example, the HBV X protein (HBx) produced by HBV is one of the
most important factors that induce the development of liver cancer;
a variety of metabolic pathways in hepatocytes are involved, and
lead to disorders of liver function. HBx is critical for viral
replication, pathogenesis and oncogenesis (5,6). It
is the most frequently integrated viral gene in liver cancer,
altering various signal transduction pathways in hepatocytes by
interacting with Bcl-2 and Bcl-xl, and regulating cellular
microRNAs (miRNAs/miRs), which in turn promotes cell metastasis and
hepatocellular proliferation (7-9). The
surface proteins of HBV, including HBV surface antigen (HBsAg), are
also crucial factors in HBV-associated hepatocellular
carcinogenesis; they interact with enoyl coenzyme A hydratase 1,
jumping translocation breakpoint protein and aldolase A (10-12),
thereby affecting cell proliferation and apoptosis. Another
mechanism underlying the development of HBV-associated liver cancer
is the integration of HBV DNA into the host genome, which not only
disrupts the normal expression of endogenous genes and induces the
mutagenesis of numerous cancer-associated genes, but also leads to
chromosomal instability (13,14).
Of note, mutations in the HBV genome have been linked to liver
cancer (15). An additional
mechanism of HBV-associated liver cancer development is
inflammation induced by HBV, which leads to persistent damage
mediated by the immune response against HBV-infected cells
(16). Subsequently, this will
induce the proliferation of bipotential hepatobiliary progenitors
and differentiated hepatocytes rather than stem cells present in
bile canaliculi, resulting in epigenetic and genetic lesions
(17). Furthermore, chronic
inflammation, which induces injury and the regeneration of
hepatocytes, significantly increases the risk of
hepatocarcinogenesis (16). Liver
injury is not caused by HBV infection alone; the host immune
response has been proposed to serve a vital role in this process
(16). The infiltration of immune
cells, and release of inflammatory cytokines and chemokines does
not clear the virus effectively, instead causing damage to the
hepatocytes (18).
Members of the apolipoprotein B mRNA-editing enzyme
catalytic subunit (APOBEC) family have been reported to serve
pivotal roles in the mutagenesis of various human genes. The APOBEC
family comprises 11 members, including APOBEC1, -2, -3A, -3B, -3C,
-3DE, -3F, -3G, -3H and -4, and activation-induced cytidine
deaminase (AID) (19,20). These enzymes possess a distinct
range of biological functions and substrate specificities. APOBEC1
is the first and the best characterized APOBEC member; it is
primarily expressed in gastrointestinal tissues and encodes a
truncated apolipoprotein B polypeptide (21,22).
The APOBEC3 proteins, particularly members 3G and 3F, induce the
hypermutation of viral DNA, which then act as host defense factors
against viruses (23-25). APOBEC4 is mainly detectable in
mammal testes, and may be involved in spermatogenesis in humans
(19). AID is primarily expressed
in B lymphocytes, where it deaminates chromosomal DNA and triggers
antibody gene diversification (26,27).
Conversely, the function of APOBEC2 is not well characterized.
APOBEC2 was reported to be specifically expressed in cardiac and
skeletal muscles (28); however,
certain studies have suggested that APOBEC2 transcripts are
ubiquitous in human tissues, including the liver (29,30).
In addition, the aberrant expression of APOBEC2 may
contribute to the development of human liver cancer (31). APOBEC2 has been associated
with nucleotide alterations in the transcripts of its target genes,
including eukaryotic translation initiation factor 4 γ 2 and PTEN,
thereby promoting the development of liver cancer (31). In addition, APOBEC2 expression was
reported to be affected by proinflammatory cytokines via the
nuclear factor (NF)-κB pathway, which suggests a possible role for
APOBEC2 in hepatic inflammation (30). In addition, it has been reported
that HBV can inhibit miR-122 expression in hepatocytes (32), In the present study, it was
revealed that APOBEC2 expression in hepatocytes was significantly
elevated by HBV. Additionally, miR-122 was revealed to target the
3′-untranslated region (3′UTR) of APOBEC2 mRNA and inhibit
its expression. Furthermore, based on the aforementioned findings,
it was hypothesized that HBV induced the expression of APOBEC2 via
downregulation of cellular miR-122 to promote the development of
liver cancer.
Materials and methods
Cell culture and treatment
Liver cancer cell lines Huh7 and HepG2 (obtained
from Cell Bank of the Chinese Academy of Sciences; cell lines were
characterized by DNA-fingerprinting and isozyme detection) were
cultured in Dulbecco's Modified Eagle's medium (DMEM; Biological
Industries) supplemented with 10% fetal bovine serum (FBS;
Biological Industries). Cells were cultured with 100 U/ml of
penicillin/streptomycin and incubated under humidified conditions
at 37̊C with 5% CO2.
miRNA target prediction
The potential target sequences of miR-122 in
APOBEC2 mRNA were predicted using microRNA. org (microrna.org/microrna/home.do) and
RNAhybrid 2.2 (bibiserv.techfak.unibielefeld.de/rnahybrid) based on
complementary sequences and minimum free energy (mfe).
Nucleotides and plasmids
Sense and antisense miR-122 mimic (5′-TGG AGT GTG
ACA ATG GTG TTT G-3′ and 5′-CAA ACA CCA TTG TCA CAC TCC A-3′),
2′-O-methylated anti-miR-122 oligonucleotide (AMO-122; 5′-CAA ACA
CCA UUG UCA CAC UCC A-3′) and respective control miRNAs
[(miR-negative control (NC; 5′-UGG AGU GUG ACA AUG GUG UUU G-3′ and
5′-AAC ACC AUU GUC ACA CUC AAU U-3′) and AMO-NC (5′-CAG UAC UUU UGU
GUA GUA CAA-3′)] were synthesized by Shanghai GenePharma Co., Ltd.
The plasmid pGEMHBV, which contains a greater-than-unit-length cDNA
of the HBV genome (payw1.2) (33)
in a pGEM-72f(+) vector and expresses all HBV genes, was obtained
from Harvard Medical School and is stored in our laboratory. For
the pAPOBEC2 overexpres-sion plasmid, which expresses APOBEC2 and
enhanced green fluorescence protein (EGFP), APOBEC cDNA was
inserted into the EcoRI and BamHI sites of the
pEGFP-C1 vector (Clontech Laboratories, Inc.). For short hairpin
RNA targeting APOBEC2 (shAPOBEC2), used to inhibit the expression
of intracellular APOBEC2, shAPOBEC2 and shNC sequences were
inserted into pGPU6 vectors (Shanghai GenePharma Co., Ltd.); the
clone processes were supported by Genscript. Four shRNAs of APOBEC2
were constructed (shAPOBEC2-1, 2-2, 2-3 and 2-4), targeting
different regions of the APOBEC2 sequence; shAPOBEC2-4 (5′-GGA GCA
AGA AGA GGG TGA ATC TCA AGA GGA TTC ACC CTC TTC TTG CTC C-3′) was
seelcted for subsequent knockdown experiments after evaluation of
interference efficiency, and was vector subsequently referred to as
shAPOBEC2. shNC was used as control for shAPOBEC2 (5′-TTC TCC GAA
CGT GTC ACG TCA AGA GAT TAC GTG ACA CGT TCG GAG AA-3′).
RNA and plasmid transfections
Huh7 and HepG2 cells, cultured in antibiotic-free
DMEM with 10% FBS, were seeded in 6-, 24- or 96-well plates
(2×105 cells/ml) and incubated at 37°C with 5%
CO2 for 18-24 h. All transfections were performed in
Huh7 cells only, except for pGEMHBV, which was also transfected
into HepG2 cells. Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) was used for transfection. The
concentration of miRNA was 120 pmol (6-well), 40 pmol (24-well) or
10 pmol (96-well), whereas that of plasmids was 4 µg
(6-well), 1 µg (24-well) or 0.25 µg (96-well),
respectively. pEGFP-C1, pAPOBEC2 and pGEMHBV contain EGFP as a
transfection marker. Additionally, the expression of HBsAg of each
group was detected using an ELISA kit (cat. no. YBS00192010; Kehua
Biotech., Co., Ltd.) according to the manufacturer's protocols, and
the absorbance was detected at 450 nm. The cells were harvested at
24 h post-transfection for mRNA analysis, or at 48 h
post-transfection for protein and cell viability analyses.
Reverse transcription-quantitative PCR
(RT-qPCR) and gene expression analysis
According to the manufacturer's instructions, total
RNA was extracted from treated cells using TRIzol®
reagents (Thermo Fisher Scientific, Inc.) and 1 µg RNA was
reverse transcribed into cDNA using a PrimeScript™ RT reagent kit
with gDNA Eraser (cat. no. RR047A; Takara Bio, Inc.) at 42°C for 2
min, 37°C for 15 min and 85°C for 5 sec prior to storage at 4°C.
qPCR was performed using SYBR PrimeScript Ex Taq II (Takara Bio,
Inc.) in a LightCycler® 96 System (Roche Diagnostics).
qPCR was conducted as follows: 95°C for 5 min, then 45 cycles of
95°C for 15 sec, 60°C for 15 sec and 72°C for 20 sec, with a final
step at 72°C for 5 sec. Fold variations in expression between RNA
samples were calculated after normalization with U6 RNA or
GAPDH Mrna (34). The
primer sequences used in the present study were as follows:
APOBEC2, forward 5′-CCA GGC TGC TCT GAA GAA GC-3′, reverse
5′-AGG CCT TGG ATT CAC CCT CT-3′; IL-6, forward 5′-CAT TCT
GCC CTC GAG CCC ACC-3′, reverse 5′-GGC AGC AGG CAA CAC CAG GA-3′;
IKKe, forward 5′-TGC GTG CAG AAG TAT CAA GC-3′, reverse
5′-TAC AGG CAG CCA CAG AAC AG-3′; TNF-α, forward 5′-AGC CTG
TAG CCC ATG TTG TAG-3′, reverse 5′-CTC TCA GCT CCA CGC CAT TG-3′.
GAPDH, forward 5′-ATC ACT GCC ACC CAG AAG AC-3′, reverse
5′-TTT CTA GAC GGC AGG TCA GG-3′; U6, forward 5′-GCT TCG GCA
GCA CAT ATA CTA AAA T-3′, reverse 5′-CGC TTC ACG AAT TTG CGT GTC
AT-3′; miR-122, forward 5′-GGG TGG AGT GTG ACA ATG G-3′, reverse
5′-TGC GTG TCG TGG AGT C-3′.
Western blotting
Protein was extracted using
radioimmuno-precipitation assay buffer (Pierce; Thermo Fisher
Scientific, Inc.) with PMSF cocktail. An enhanced Bicinchoninic
Acid Protein Assay kit (cat. no. P0010S; Beyotime Institute of
Biotechnology) was used to determine the protein concentration, and
35 µg/lane protein was resolved via 12% SDS-PAGE, followed
by electrotransfer onto nitrocellulose membranes. Polyclonal
APOBEC2 (1:500; cat. no. 20121-1-AP; ProteinTech Group, Inc.) and
cleaved-caspase-3 antibodies (1:500; cat. no. WL01857; Wanleibio
Co., Ltd.), and monoclonal antibodies against β-actin (cat. no.
TA-09; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.;
OriGene Technologies, Inc.) were used for immunoblotting overnight
at 4°C. After a standard washing with TBS-Tween 20 (Sigma-Aldrich;
Merck KGaA), membranes were incubated with horseradish
peroxidase-conjugated goat anti-mouse and anti-rabbit (1:2,500;
cat. nos. ZB-2305 and ZB-2301, respectively; Beijing Zhongshan
Golden Bridge Biotechnology Co., Ltd.; OriGene Technologies, Inc.)
for 2 h at room temperature. The signal was visualized using an
enhanced chemiluminescence western blot substrate (Thermo Fisher
Scientific Inc.); bands were imaged with a charge-coupled camera
LAS4000 (Fujifilm) and quantified with Image J (v1.51k; National
Institutes of Health).
Dual-luciferase assay
PCR was used to amplify the 3′UTR sequence of
APOBEC2, and the amplification primers were as follows:
Forward 5′-GCG CTC GAG GCA ACT GGG CTT TGC CTC-3′ (XhoI
restriction site underlined), reverse 5′-AAT GCG GCC GCC TGC CAT
TAC CAT CAA ACA C-3′ (NotI restriction site underlined). The
PCR products were purified and cloned downstream of the
Renilla luciferase gene of the dual-luciferase vector,
pmiR-RB-REPORT™ [APOBEC2-wild type (WT); Guangzhou RiboBio Co.,
Ltd.]. To investigate the potential interaction between miR-122 and
APOBEC2, site-directed mutagenesis was performed to mutate the
miR-122 target sites in the APOBEC2 3′UTR region, replacing
ACACTCC with TGTGAGG; thus, the mutant (MUT) APOBEC2 luciferase
reporter vector was constructed (APOBEC2-MUT). For the luciferase
reporter assay, Huh7 cells (4×104 cells) were seeded in
96-well plates for 24 h. Luciferase reporter vectors (0.25
µg) were co-transfected with miR-122 mimic or miR-NC (10
pmol) into cells using Lipofectamine 2000. After 48 h of
incubation, the treated cells were harvested and analyzed using a
Dual-Luciferase Assay kit (Promega Corporation). The data were
presented as the relative luciferase activity (Renilla
lucif-erase/firefly luciferase). Luminescence was quantified using
a luminometer (Turner BioSystems). All experiments were performed
in triplicate.
MTT assay
Huh7 cells were seeded in 96-well plates and
incubated overnight; pAPOBEC2, shAPOBEC2 or corresponding control
vectors were then transfected into cells using Lipofectamine 2000.
At 48 h following treatment, sterile MTT was added to the culture
supernatant at 20 ìl/well (5 mg/ml). After incubation at 37°C with
5% CO2 for 4 h, 100 ìl dimethyl sulfoxide was added to
the supernatant and vortexed for 10 min to dissolve the formazan
crystals. The absorbance was measured at 490 nm using a microplate
reader.
Statistical analysis
All results were presented as the mean ± standard
error of the mean. A two-tailed Student's t-test was used to
evaluate the differences between two groups. For multiple
comparisons one-way ANOVA followed by Tukey's or Dunnett's test was
performed. P<0.05 was considered a statistically significant
difference. Statistical analysis was performed using SigmaPlot 11.0
(Systat Software, Inc.) and GraphPad Prism 5 (GraphPad Software,
Inc.). All experiments were repeated at least three times
independently.
Results
HBV transfection induces APOBEC2
expression in liver cells
It was previously reported that aberrant APOBEC2
expression was involved in the development of liver cancer
(31). To investigate the role of
HBV infection in modulating APOBEC2 expression and the subsequent
induction of carcinogenesis, Huh7 and HepG2 cells were transfected
with plasmids expressing HBV proteins (pGEMHBV) or the control
(pEGFP-C1). The expression of HBsAg in the pGEMHBV group increased
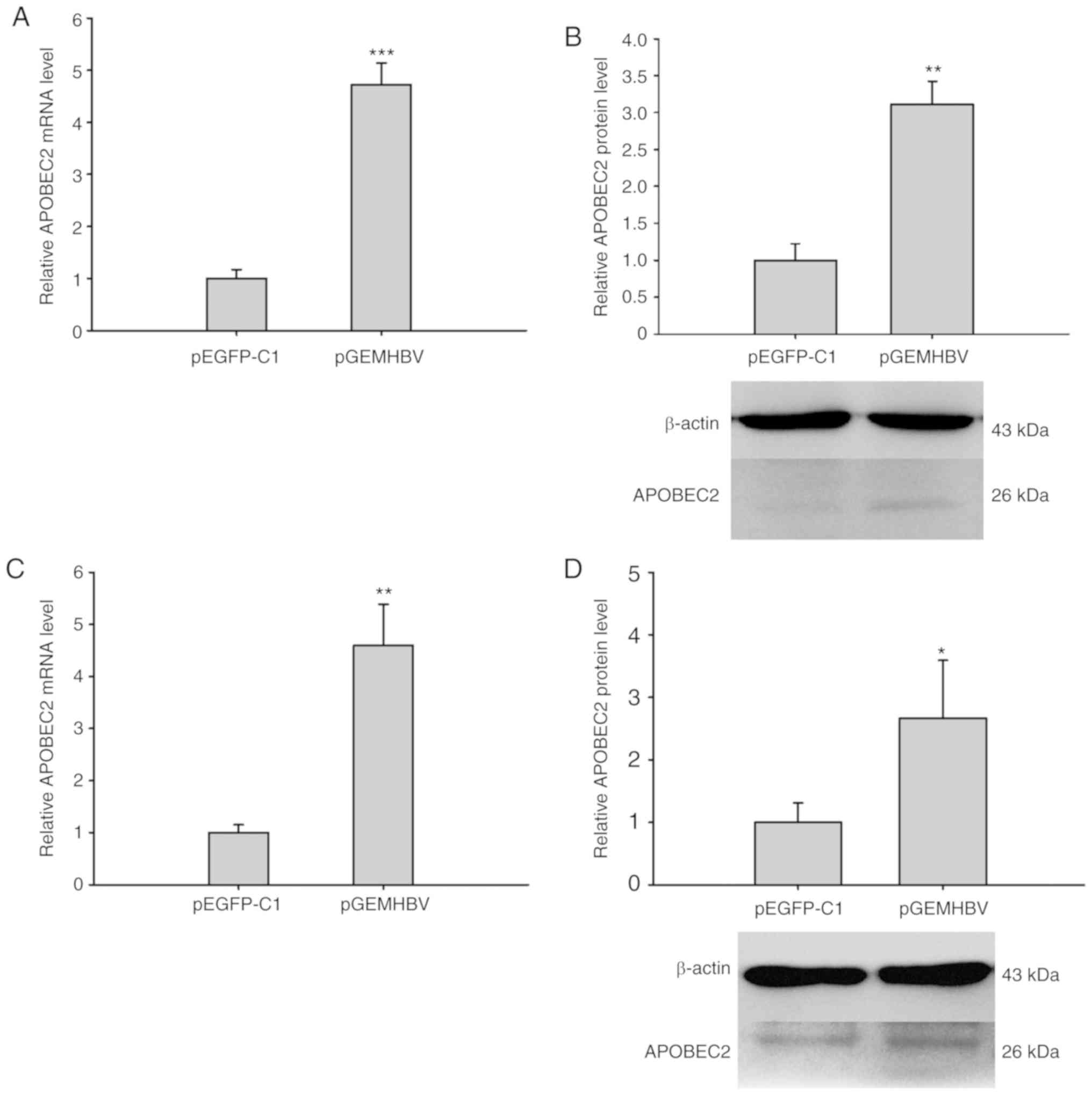
significantly compared with the control group (Fig. S1). RT-qPCR analysis revealed that the
relative mRNA expression levels of APOBEC2 were significantly
increased in pGEMHBV-transfected cells compared with
pEGFP-C1-trans-fected cells in both cell lines (Fig. 1A and C). Similarly, the protein
expression levels of APOBEC2 were upregulated in cells transfected
with pGEMHBV compared with those trans-fected with pEGFP-C1
(Fig. 1B and D). Therefore, HBV
was proposed to promote APOBEC2 expression at the mRNA and protein
levels in liver cancer cells.
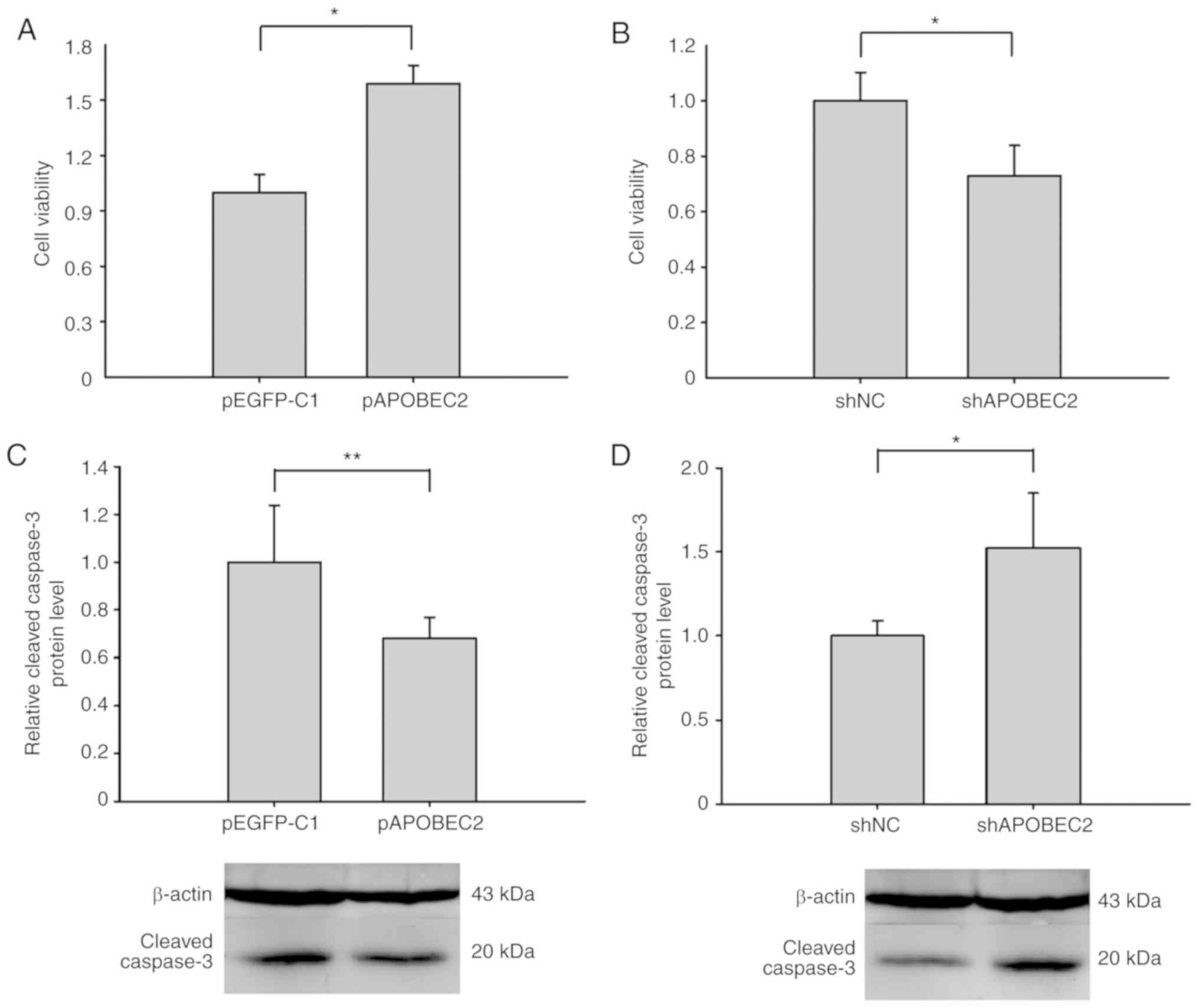
Modulation of APOBEC2 expression affects
cell viability and apoptosis
To study the role of APOBEC2 in cell proliferation,
APOBEC2 overexpression (pAPOBEC2) and APOBEC2 interference plasmid
(shAPOBEC2) vectors were constructed, which were respectively
transfected into Huh7 cells to regulate cellular APOBEC2
expression. As presented in Fig.
2, four shAPOBEC2 vectors were constructed; shAPOBEC2-4
exhibited the highest interference efficiency at the mRNA and
protein levels. Therefore, shAPOBEC2-4 was used in subsequent
experiments. RT-qPCR and western blot analyses revealed that
transfection with pAPOBEC2 significantly upregulated the expression
of APOBEC2 in Huh7 cells (Fig. S2).
An MTT assay revealed that pAPOBEC2-trans-fected cells exhibited
significantly increased viability compared with those transfected
with pEGFP-C1 (Fig. 3A).
Conversely, shAPOBEC2-transfected cells exhibited significantly
reduced viability compared with those transfected with shNC
(Fig. 3B). In addition, as
determined via western blotting, the expression of
cleaved-caspase-3 (a terminal shear enzyme in apoptosis) was
reduced following overexpression of APOBEC2, but was enhanced in
response to APOBEC2 knockdown (Fig. 3C
and D). In conclusion, APOBEC2 over-expression increased cell
viability and decreased apoptosis, whereas APOBEC2 downregulation
induced opposing effects in Huh7 cells.
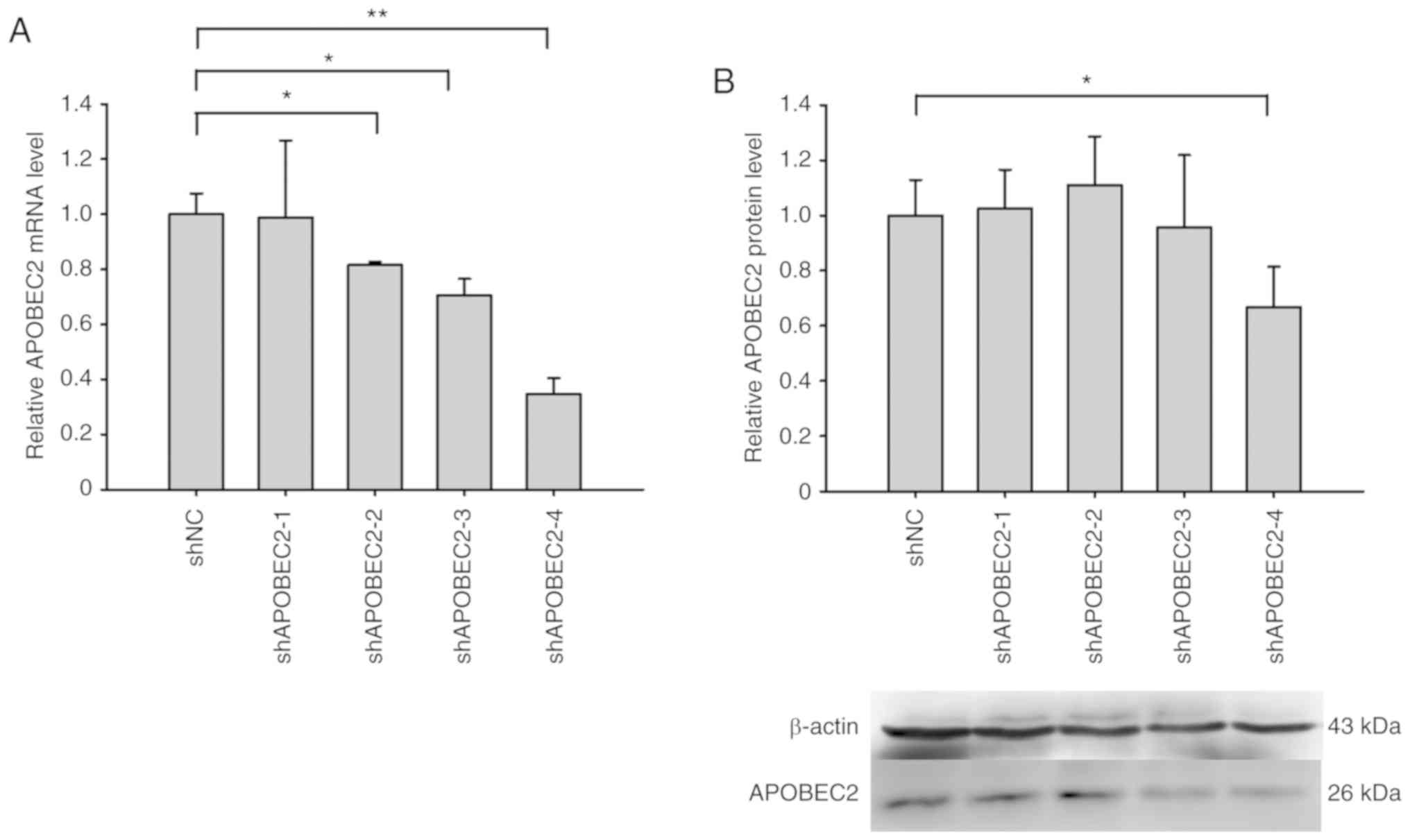 | Figure 2Verification of the inhibition
efficiency of shRNAs on APOBEC2 expression. Four shRNAs specific
for APOBEC2 were designed and transfected into Huh7 cells, and
total RNA and protein were extracted at 24 and 48 h
post-transfection, respectively. Then, reverse
transcription-quantitative PCR and western blot analyses were
conducted to detect the inhibition efficiency. Compared with shNC,
shAPOBEC2-4 effectively inhibited APOBEC2 (A) mRNA and (B) protein
expression; APOBEC2 mRNA and protein expression decreased by ~70
and ~34%, respectively, following APOBEC2 knockdown. Therefore,
shAPOBEC2-4 was selected for further experimentation in the present
study. shNC served as a control. Data are presented as the mean ±
standard error of the mean (n=3). *P<0.05,
**P<0.01. APOBEC2, apolipoprotein B mRNA-editing
enzyme catalytic subunit 2; NC, negative control; shRNAs, short
hairpin RNA. |
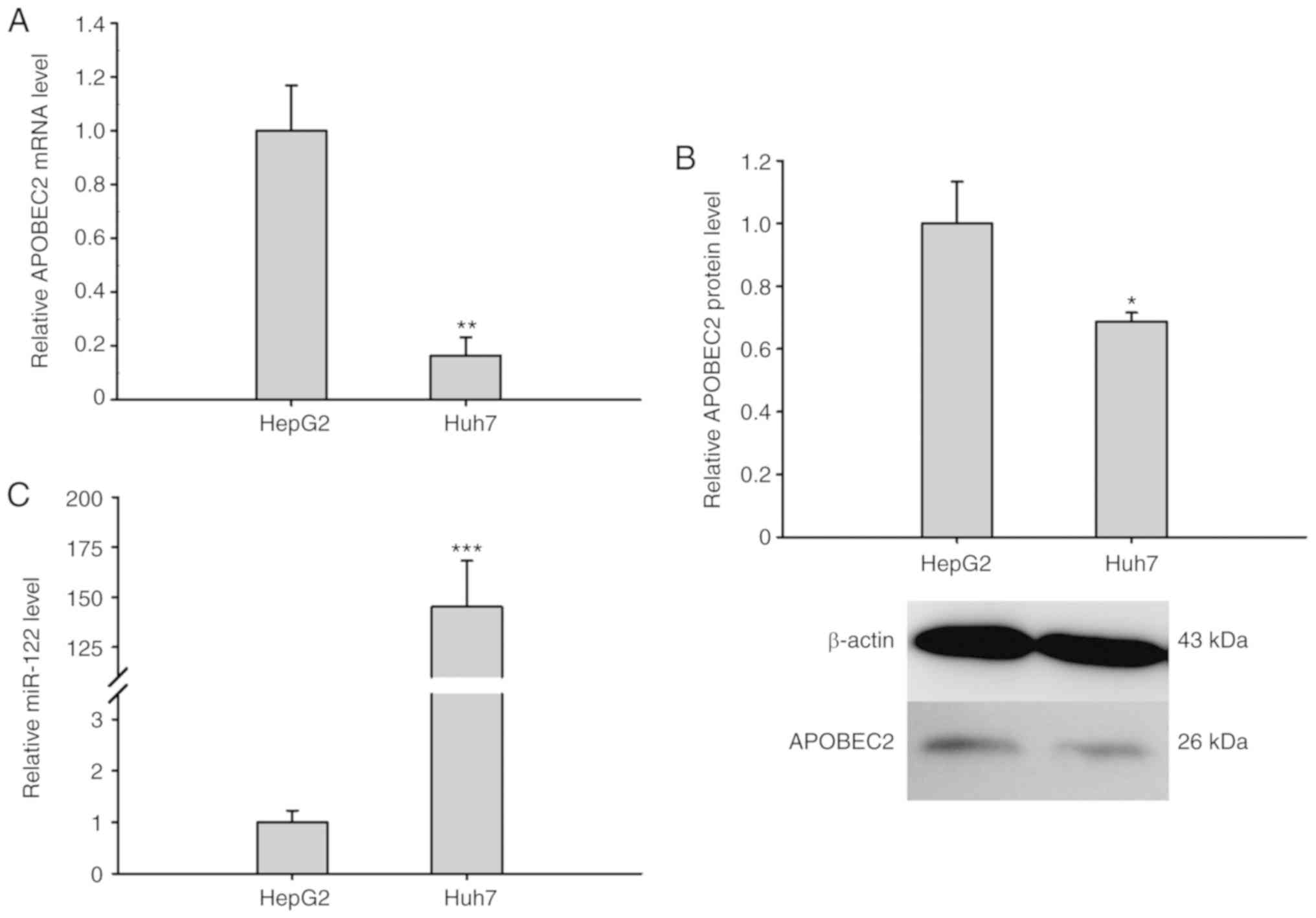
miR-122 expression opposes APOBEC2
expression
To further explore the association between APOBEC2
and liver cancer, the expression of APOBEC2 was analyzed in
different liver cancer cell lines (HepG2 and the hepatoma cell
Huh7). RT-qPCR analysis and western blotting revealed that the
expression of APOBEC2 was upregulated in HepG2 cells compared with
in Huh7 cells (Fig. 4A and B). Of
note, the expression of miR-122 exhibited opposing expression
trends to that of APOBEC2 in the two cell lines (Fig. 4C). Collectively, the results
suggested that the expression of miR-122 was negatively associated
with that of APOBEC2 in liver cells.
APOBEC2 is a target of miR-122 in liver
cells
To analyze the association between miR-122 and
APOBEC2, miR-122 was overexpressed via transfection with miR-122
mimic or knocked down via transfection with AMO-122 in Huh7 cells.
The expression levels of miR-122 in miR-122 mimic-treated cells
were significantly increased compared with miR-NC treated cells,
whereas in AMO-122 treated cells, the expression levels of miR-122
were significantly suppressed (Fig. 5A
and B). RT-qPCR and western blotting demonstrated that miR-122
overexpression resulted in a significant suppression of APOBEC2
expression. Conversely, miR-122 knockdown resulted in an increase
in APOBEC2 expression (Fig. 5A-D),
which further indicated that there may potential interaction
between miR-122 and APOBEC2 mRNA.
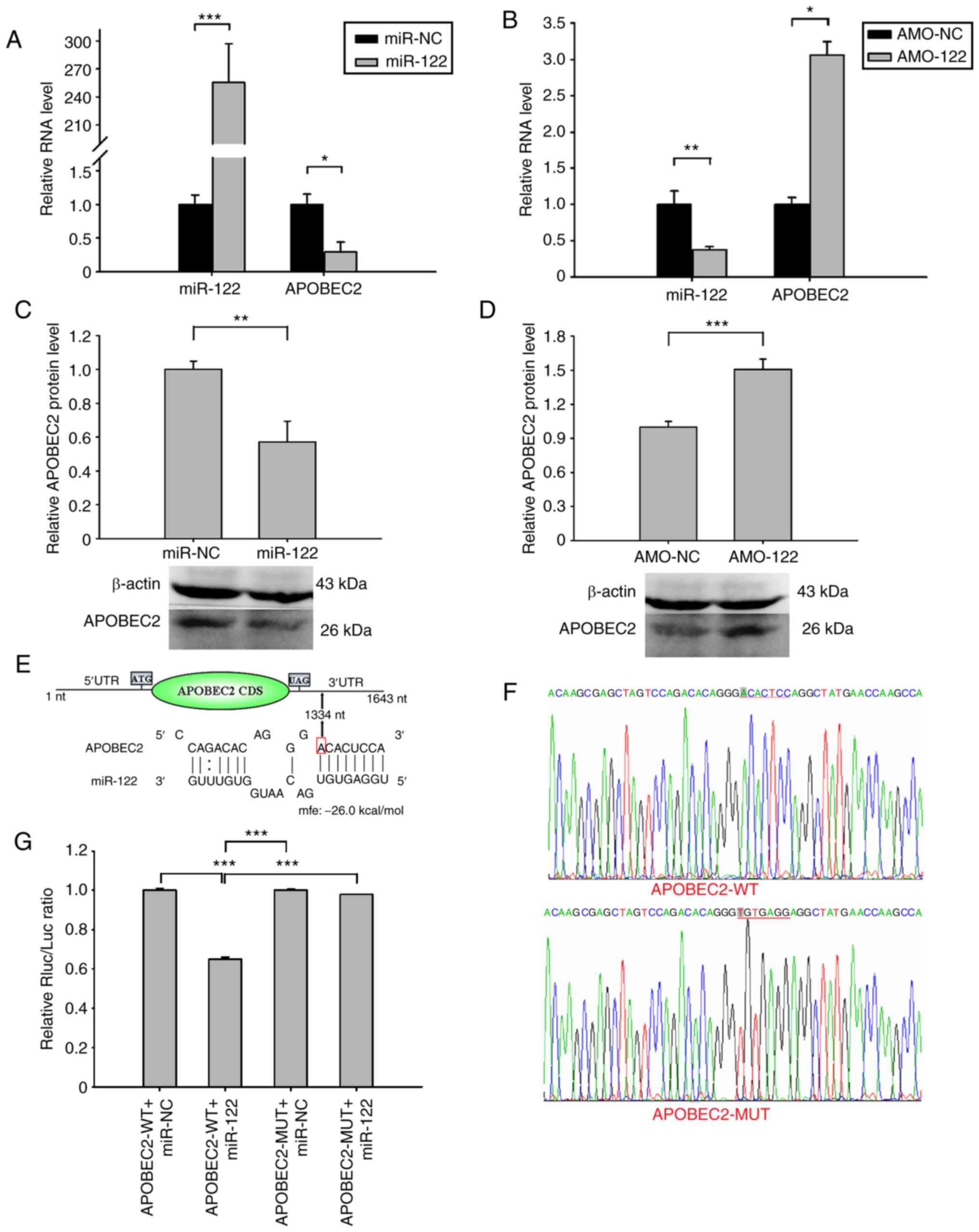 | Figure 5Validation of miR-122 targeting
APOBEC2 mRNA. The expression levels of miR-122 and APOBEC2 in Huh7
cells transfected with miR-NC, miR-122, AMO-NC or AMO-122 were
determined. Analysis of (A and B) miR-122 and APOBEC2 mRNA, and (C
and D) APOBEC2 protein expression 24 and 48 h post-transfection by
reverse transcription-quantitative PCR and western blotting,
respectively. β-actin served as an internal control. (E) Schematic
diagram of potential binding sites for miR-122 in the 3′UTR of
APOBEC2 mRNA. The locations of putative miR-122 binding sites in
the 3′UTR of APOBEC2 mRNA are marked by arrows at 1,334 nt. (F) WT
and MUT nucleotides in the putative target sequence for miR-122 in
the APOBEC2 mRNA. The MUT region indicated by the red line was
generated by over-lapping PCR to minimize complementarity between
miR-122 and the 3′UTR of APOBEC2 mRNA. (G) Relative luciferase
activity of Huh7 cells transfected with miR-122 mimics + APOBEC2-WT
or APOBEC2-MUT. Huh7 cells were co-transfected with miR-122 mimics
or miR-NC and a luciferase reporter plasmid harboring WT or MUT miR
binding sites. The effects of miR-122 on luciferase activity were
determined by luciferase reporter assays. The activity of Renilla
luciferase was normalized to that of firefly luciferase. Data are
presented as the mean ± standard error of the mean (n=3).
*P<0.05, **P<0.01,
***P<0.001. APOBEC2, apolipoprotein B mRNA-editing
enzyme catalytic subunit 2; AMO, anti-miR oligonucleotide; mfe,
minimum free energy; miR, microRNA; MUT, mutated; NC, negative
control; nt, nucleotide, the number counts from the first
nucleotide at the 5′ end of APOBEC2 mRNA; UTR, untranslated region;
WT, wild type. |
To explore this hypothesis, microRNA.org and RNAhybrid 2.2, web-based RNA analysis
tools, were employed to predict the potential target sites of
miR-122 within APOBEC2 mRNA. It was revealed that there was
a putative miR-122 target sequence in the 3′UTR of APOBEC2
mRNA. The target sequences perfectly matched the seed sequence of
miR-122, and the mfe was -26.0 kcal/mol (Fig. 5E). To further determine the
potential interaction between miR-122 and APOBEC2 mRNA, the
WT and MUT 3′UTR of APOBEC2 mRNA were inserted into
pmiR-RB-REPORT vectors, generating APOBEC2-WT and APOBEC2-MUT
vectors (Fig. 5F). As presented in
Fig. 5G, the fluorescence of
miR-122 and APOBEC2-WT co-transfection group was ~35% lower than
that of the miR-NC and APOBEC2-WT co-transfection group; however,
in the group co-transfected with APOBEC2-MUT, the decrease in
fluorescence was eliminated. Collectively, the results of the
present study indicated that miR-122 targeted the 3′UTR of
APOBEC2 mRNA and downregulated its expression.
HBV infection suppresses miR-122
expression
Furthermore, to confirm whether HBV regulates
APOBEC2 expression by directly suppressing that of miR-122,
miR-122, AMO-122, and their corresponding controls were introduced
separately into Huh7 cells. In addition, pGEMHBV was co-transfected
with the aforementioned miRNAs. The expression levels of miR-122
and APOBEC2 were subsequently measured. The results demonstrated
that pGEMHBV co-transfection significantly suppressed cellular
miR-122 expression in the control groups (miR-NC and AMO-NC)
compared with the pEGFP-C1 group (Fig.
6A), which was consistent with previous findings (32). In addition, miR-122 was
significantly suppressed following co-transfection of pGEMHBV +
AMO-122 compared with all other groups, whereas in the pGEMHBV +
miR-122 co-transfection group, the expression of miR-122 didn't
change significantly compared with the empty vector control
(Fig. 6A). Conversely, the
expression levels of APOBEC2 were increased in the pGEMHBV + miR-NC
or pGEMHBV + AMO-NC co-transfection groups, and further increased
following co-transfection with AMO-122, whereas APOBEC2 was
downregulated in the pGEMHBV + miR-122 co-transfection group
(Fig. 6B). Overall, transfection
of HBV genes suppressed the expression of miR-122, which
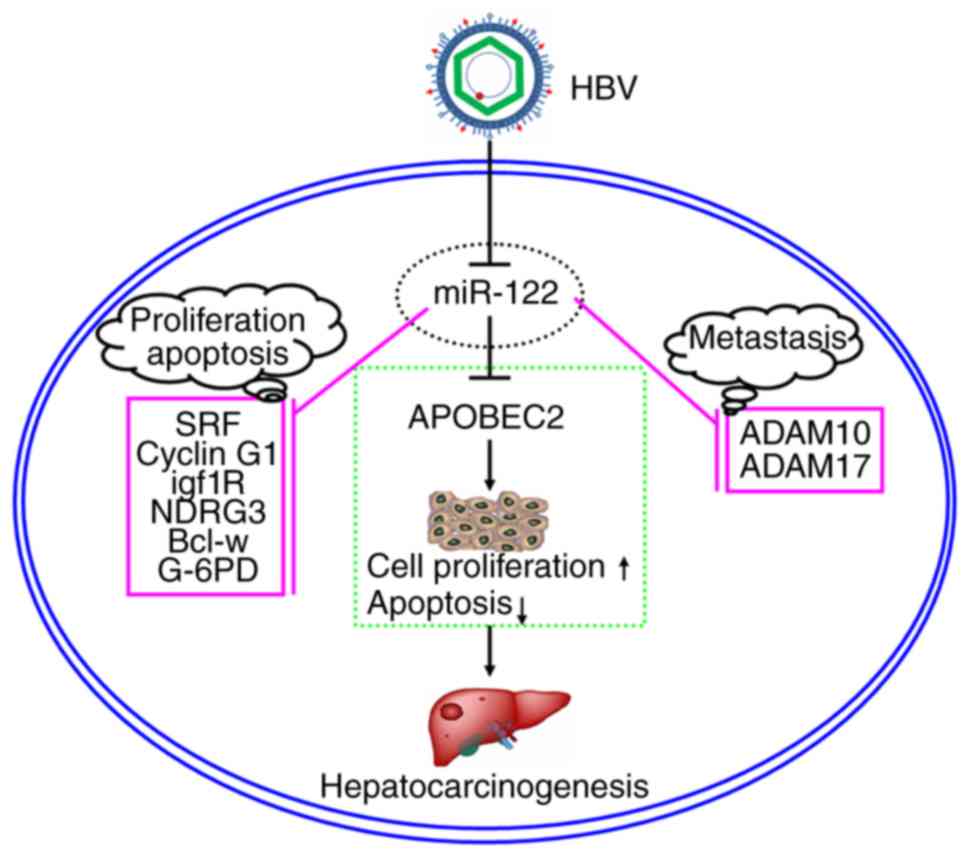
subsequently induced the expression of APOBEC2. These findings
further suggested that the effects of HBV on APOBEC2 occur via the
downregulation of cellular miR-122 expression, which may contribute
to the tumorigenesis of liver cells (Fig. 7).
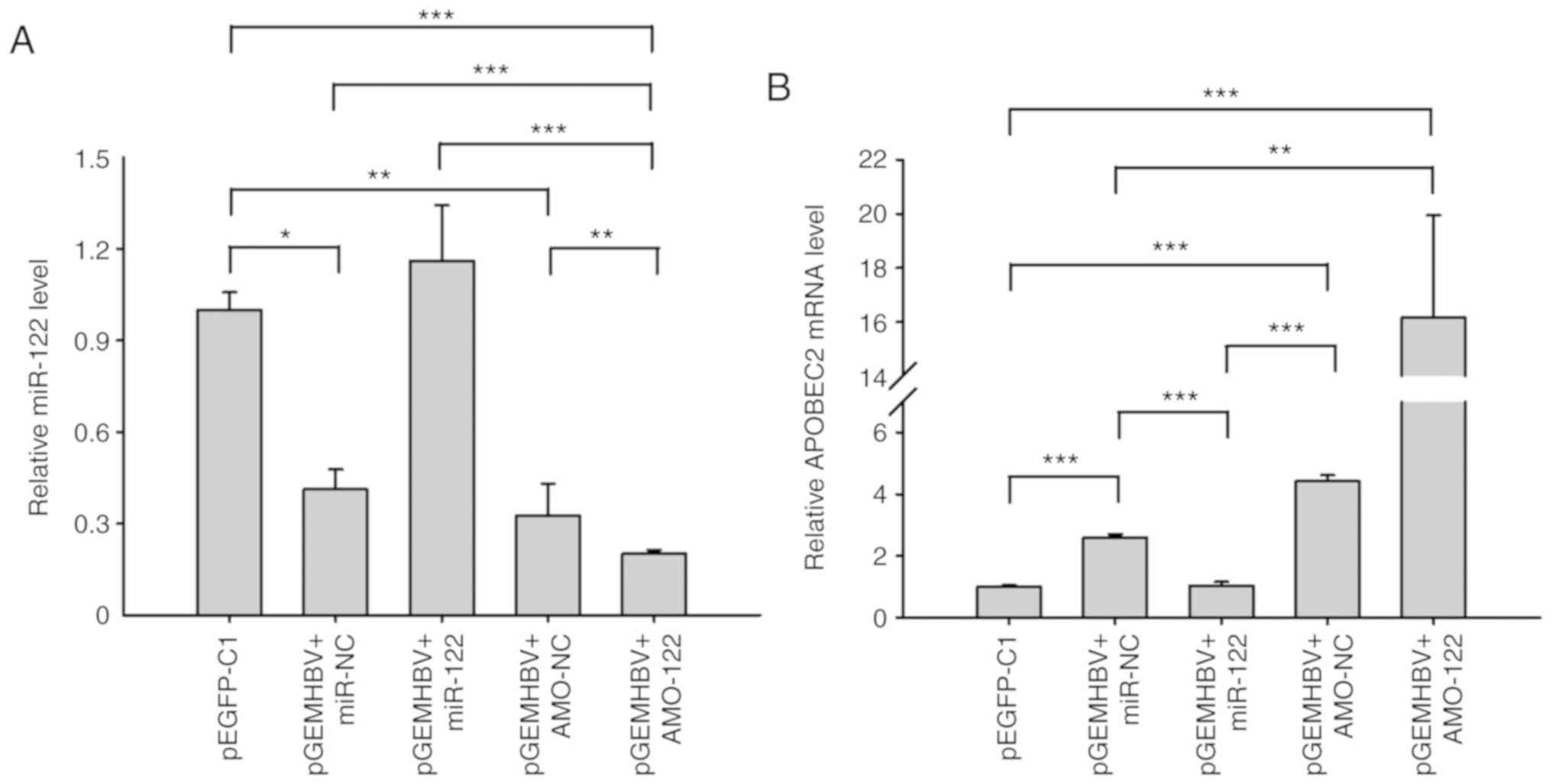 | Figure 6HBV and miR-122 regulate APOBEC2
expression. Huh7 cells were respectively transfected with miR-122,
AMO-122 or the corresponding NCs + pGEMHBV. Total RNA was extracted
after transfection for 24 h, and the expression of (A) miR-122 and
(B) APOBEC2 was determined via reverse transcription-quantitative
PCR analysis. Data are presented as the mean ± standard error of
the mean (n=3). *P<0.05, **P<0.01,
***P<0.001. APOBEC2, apoli-poprotein B mRNA-editing
enzyme catalytic subunit 2; AMO, anti-miR oligonucleotide; EGFP,
enhanced green fluorescent protein; HBV, hepatitis B virus; miR,
microRNA; NC, negative control. |
HBV infection and APOBEC2 induce chronic
inflammatory responses
Based on the fact that chronic inflammation is
closely associated with tumor development (35), in the present study, the levels of
IL-6, IKKε and TNF-α, three notable proteins involved in both
inflammatory and tumor-associated processes (36-39),
were measured in Huh7 cells following transfection with pGEMHBV or
pAPOBEC2. As presented in Fig. 8,
the expression of IL-6 was 2.6-fold higher, and the expression
levels of IKKε and TNF-α were 60 and 50% lower, respectively,
compared with the control group following HBV infection. APOBEC2
overexpression induced similar effects on the levels of these three
factors, with increased expression of IL-6, and downregulation of
IKKε and TNF-α.
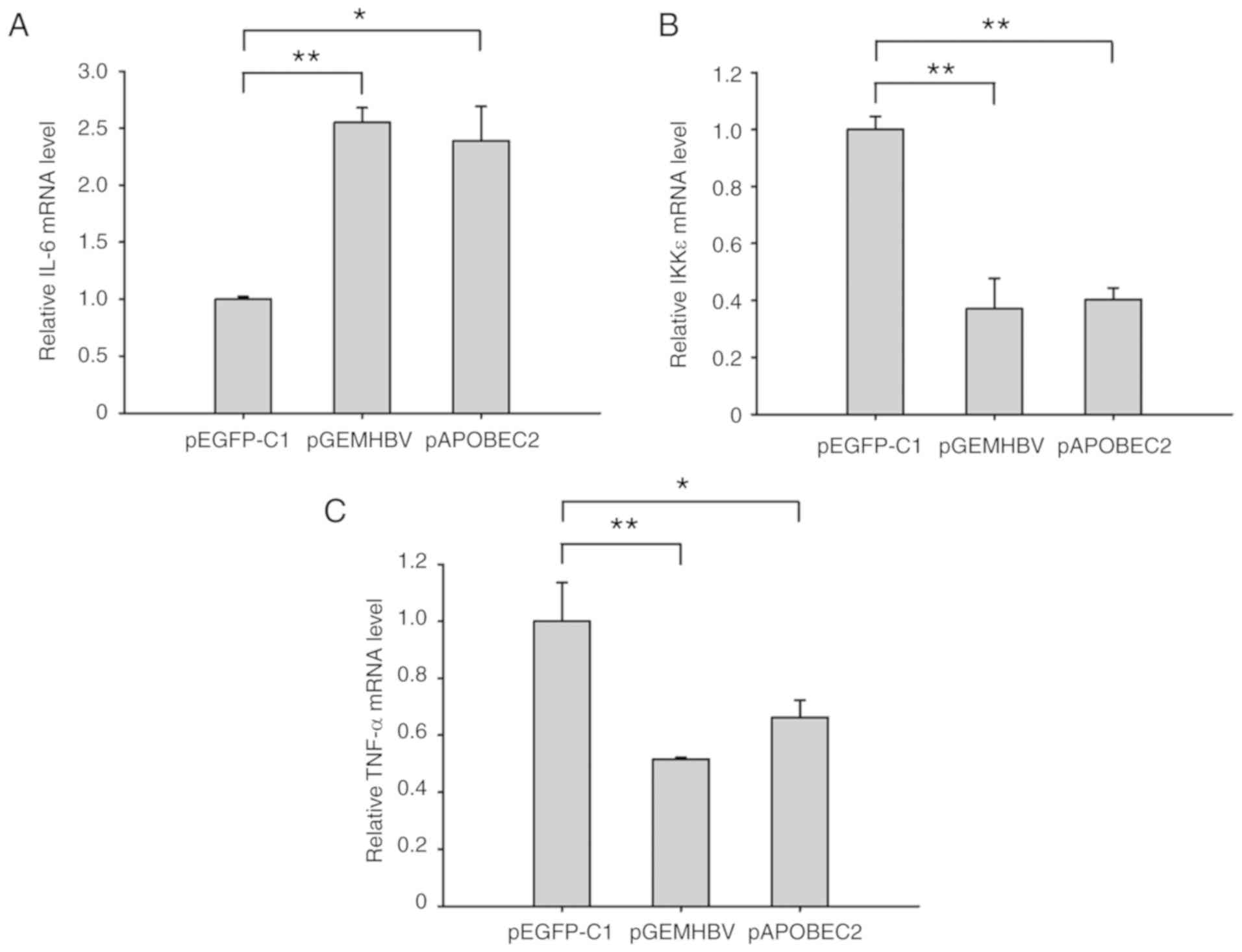 | Figure 8HBV and APOBEC2 modulate the
expression of IL-6, IKKε and TNF-α. Huh7 cells at 70% confluence
were transfected with pGEMHBV or pAPOBEC2 for 24 h. mRNA expression
of (A) IL-6, (B) IKKε and (C) TNF-α in Huh7 cells treated with
pGEMHBV or pAPOBEC2, as determined by reverse
transcription-quantitative polymerase chain reaction. pEGFP-C1
served as a negative control for pGEMHBV and pAPOBEC2. Data are
presented as the mean ± standard error of the mean (n=3).
*P<0.05, **P<0.01 and
***P<0.001. APOBEC2, apolipoprotein B mRNA-editing
enzyme catalytic subunit 2; EGFP, enhanced green fluorescent
protein; HBV, hepatitis B virus; IKK, IκB kinase; IL-6,
interleukin-6; TNF-α, tumor necrosis factor-α. |
Discussion
In the present study, the expression of APOBEC2 was
determined to be negatively associated with the liver-specific
miRNA, miR-122, in different liver cell lines. Furthermore, it was
demonstrated that miR-122 could specifically target the 3′UTR of
APOBEC2 mRNA and inhibit its expression. It has been
reported that HBV can inhibit miR-122 expression in hepatocytes
(40), which was consistent with
the present findings. In addition, this study demonstrated that the
transfection of HBV genes significantly induced APOBEC2 expression
via miR-122 inhibition. Thus, the results suggested that HBV
upregulates APOBEC2 expression and may promote cell
proliferation.
APOBEC2 is a member of the cytidine deaminase
family, APOBEC, with putative nucleotide editing activity (41). A total of 11 members comprise this
family, including APOBEC1, -2, -3A, -3B, -3C, -3DE, -3F, -3G, -3H
and -4, and AID in humans, which participate in various
physiological and pathological processes, such as lipid metabolism,
viral infection, spermato-genesis and immune gene diversity
(19,20,42).
At present, increasing evidence has indicated that the dysregulated
expression and abnormal activity of APOBEC members may be
associated with tumorigenesis via the nucleotide editing of
tumor-related genes. It has been reported that overexpression of
APOBEC1 induces dysplasia and the development of hepa-tocellular
carcinoma in transgenic mouse livers (43), whereas constitutive expression of
AID leads to frequent mutations of the T-cell receptor gene, Tp53
and c-myc gene, leading to the formation of malignant tumors in the
liver, lung, stomach and lymphatic system (44,45).
APOBEC2 was primarily reported as being exclusively expressed in
skeletal and cardiac muscle (28);
however, accumulating evidence has indicated that APOBEC2
transcripts are ubiquitous in various tissues in humans, including
the liver (29). Furthermore,
overexpres-sion of APOBEC2 in transgenic mice contributes to liver
tumorigenesis (31); consistent
with this, it was determined in the present study that
overexpression of APOBEC2 signifi-cantly promoted cell viability.
Additionally, the expression of cleaved-caspase-3 (a terminal shear
enzyme in apoptosis) was reduced following APOBEC2 overexpression,
indicating the suppression of cell apoptosis, whereas knockdown of
APOBEC2 induced opposing effects. These findings indicated that
APOBEC2 may increase the activity of liver cancer cells, suppress
apoptosis, and promote the proliferation and survival of liver
cells to contribute to HBV-induced liver cancer.
As a liver-specific miRNA, miR-122 constitutes 70%
of the total population of miRNAs in hepatocytes, which is
extensively involved in the physiological and pathological
processes of liver cells (46,47).
It has been reported that miR-122 can target a variety of genes to
inhibit liver cell growth and metastasis, such as cyclin G1, serum
response factor, insulin-like growth factor 1 receptor, N-myc
downstream-regulated gene 3, Bcl-w,
Glucose-6-phosphate-dehydrogenase, a disintegrin and
metalloprotease family (ADAM)10 and ADAM17 (48-53).
The serum levels of alanine transaminase and aspartate transaminase
were correlated with miR-122 expression, which suggested that the
severity of chronic hepatitis B is positively associated with the
expression of miR-122 (54).
Conversely, miR-122 expression was significantly decreased in human
liver cancer, and was associated with the prognosis of patients
(53). In the present study, the
expression of APOBEC2 was upregulated in HepG2 cells compared with
in Huh7 cells. Of note, the expression profile of miR-122 in these
cell lines opposed that of APOBEC2, which suggested a regulatory
mechanism underlying APOBEC2 expression mediated by miR-122.
Furthermore, it was reported that overexpression of miR-122
significantly suppressed APOBEC2 expression, whereas miR-122
knockdown resulted in opposing effects. These results indicated a
potential interaction between miR-122 and APOBEC2 mRNA.
After predicting potential target sites of miR-122 within the
APOBEC2 mRNA, it was hypothesized that miR-122 could bind to
the 3′UTR of APOBEC2 mRNA and inhibit its expression.
The mechanism underlying HBV-induced carcinogenesis
is complex. In the present study, it was investigated as to whether
the induction of APOBEC2 following HBV infection occurs via the
suppression of cellular miR-122 to facilitate the growth of liver
cancer cells. Thus, pGEMHBV was transfected into Huh7 cells; the
results revealed that the expression of miR-122 was decreased,
whereas that of APOBEC2 was increased. Furthermore, Huh7 cells were
co-transfected with pGEMHBV + miR-122 mimic or inhibitor. APOBEC2
exhibited the highest degree of upregulation in pGEMHBV and AMO-122
co-transfected cells; however, this was not observed in the pGEMHBV
and miR-122 co-transfection group. Thus, the present findings may
indicate a novel mechanism underlying the development of
HBV-induced liver cancer.
As chronic hepatitis has been associated with the
occurrence of liver cancer, the expression of several inflammatory
factors was also detected, including interleukin-6 (IL-6), IκB
kinase ε (IKKε), and tumor necrosis factor-α (TNF-α) after
transfecting cells with HBV genes or APOBEC2. The results revealed
that the expression of IL-6 was increased, whereas that of IKKε and
TNF-α was decreased following the expression of HBV genes. As a
multifunctional and pleiotropic inflammatory cytokine, the
expression of IL-6 is upregulated in response to viral infection
and the presence of certain tumors (55,56).
It has been reported that the serum levels of IL-6 are increased in
HBV-infected patients; this increase was proposed to serve a
crucial role in the induction of immune tolerance against HBV, and
could be applied to determine the outcomes of HBV nfections
(57). Members of the NF-κB family
serve crucial roles in various biological process, including
inflammation, immune responses, carcinogenesis and apoptosis. It
was previously reported that HBV could disrupt the interaction
between IKKε and DEAD-box RNA helicase, subsequently inhibiting the
induction of interferon-β, which suggests a novel strategy of
immune evasion in HBV infections (58). A previous study demonstrated that
TNF can inhibit HBV replication and stimulate HBV-specific T-cell
responses, which are involved in clearing HBV from infected
hepatocytes (59). Chronic
inflammation of the liver has been proposed to contribute to the
development of liver cancer (60);
activation of NF-κB promoted APOBEC2 expression in hepatocytes
(30). Similar to the effects of
HBV gene expression, including the signifi-cant upregulation of
APOBEC2, overexpression of APOBEC2 induced similar effects on the
aforementioned inflammatory factors. This suggested that APOBEC2
may act as a factor linking inflammation and the oncogenicity of
HBV; however, further investigation is required.
In conclusion, the findings of the present study may
provide novel insight the molecular mechanisms underlying
HBV-associated carcinogenesis, comprising downregulation of
miR-122, which targets APOBEC2, inducing its expression to promote
the growth of liver cancer cells. Due to the interactions between
miRNAs and their targets in host cells, these findings may serve as
a basis for future investigations into the development of liver
cancer under conditions of HBV infection, potentially improving
understanding regarding how HBV modulates host cell signaling
pathways via a variety of mechanisms.
Supplementary Data
Funding
This work was supported by grants from the National
Natural Science Foundation of China (grant nos. 81402267, 81501737,
81202296, 81601428, 81773267 and 81301703), General Financial Grant
from the China Postdoctoral Science Foundation (grant nos.
2014M551271 and 2014M561372), Heilongjiang Postdoctoral Fund (grant
no. LBH-Z14164), The Fundamental Research Funds for the Provincial
Universities (grant no. 2017JCZX27), The Young Innovative Talent
Training Program for Heilongjiang Province Undergraduate Colleges
and Universities (grant no. UNPYSCT-2018052), and the Harbin
Special Fund for the Scientific and Technological Innovation
Scholars (grant no. RC2016QN004073).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Author's contributions
AL, ZZ and FZ were involved in the conception of the
study. AL, JW, XW and WK performed the experiments. AL, AZ and JQ
analyzed the data. AL drafted the primary manuscript. AL, AZ, QZ,
MAQ, YF, YL, WS, FZ and ZZ analyzed and interpreted data, produced
the figures, further edited the manuscript and revised it
critically. FZ and ZZ supervised the whole study and approved the
final version of the manuscript to be published. All authors
reviewed the results and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Professor Zhaohua
Zhong (Harbin Medical University) for technical assistance.
References
|
1
|
Ferlay J, Soerjomataram I, Ervik M,
Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and
Bray F: GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and
Prevalence Worldwide in 2012 v1.0. IARC CancerBase No. 11. IARC;
Lyon: 2012, https://publications.iarc.fr/Databases/Iarc-Cancerbases/GLOBOCAN-2012-Estimated-Cancer-Incidence-Mortality-And-Prevalence-Worldwide-In-2012-V1.0-2012.
|
|
2
|
Motola-Kuba D, Zamora-Valdes D, Uribe M
and Mendez-Sanchez N: Hepatocellular carcinoma. An overview Ann
Hepatol. 5:16–24. 2006. View Article : Google Scholar
|
|
3
|
Fattovich G, Stroffolini T, Zagni I and
Donato F: Hepatocellular carcinoma in cirrhosis: Incidence and risk
factors. Gastroenterology. 127(5 Suppl 1): S35–S50. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Venook AP, Papandreou C, Furuse J and de
Guevara LL: The incidence and epidemiology of hepatocellular
carcinoma: A global and regional perspective. Oncologist. 15(Suppl
4): S5–S13. 2010. View Article : Google Scholar
|
|
5
|
Wei Y, Neuveut C, Tiollais P and Buendia
MA: Molecular biology of the hepatitis B virus and role of the X
gene. Pathol Biol (Paris). 58:267–272. 2010. View Article : Google Scholar
|
|
6
|
Luo N, Cai Y, Zhang J, Tang W, Slagle BL,
Wu X and He S: The C-terminal region of the hepatitis B virus X
protein is required for its stimulation of HBV replication in
primary mouse hepato-cytes. Virus Res. 165:170–178. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Geng X, Huang C, Qin Y, McCombs JE, Yuan
Q, Harry BL, Palmer AE, Xia NS and Xue D: Hepatitis B virus X
protein targets Bcl-2 proteins to increase intracellular calcium,
required for virus replication and cell death induction. Proc Natl
Acad Sci USA. 109:18471–18476. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sze KM, Chu GK, Lee JM and Ng IO:
C-terminal truncated hepatitis B virus x protein is associated with
metastasis and enhances invasiveness by C-Jun/matrix
metalloproteinase protein 10 activation in hepatocellular
carcinoma. Hepatology. 57:131–139. 2013. View Article : Google Scholar
|
|
9
|
Yip WK, Cheng AS, Zhu R, Lung RW, Tsang
DP, Lau SS, Chen Y, Sung JG, Lai PB, Ng EK, et al:
Carboxyl-terminal truncated HBx regulates a distinct microRNA
transcription program in hepatocellular carcinoma development. PLoS
One. 6:e228882011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xiao CX, Yang XN, Huang QW, Zhang YQ, Lin
BY, Liu JJ, Liu YP, Jazag A, Guleng B and Ren JL: ECHS1 acts as a
novel HBsAg-binding protein enhancing apoptosis through the
mitochondrial pathway in HepG2 cells. Cancer Lett. 330:67–73. 2013.
View Article : Google Scholar
|
|
11
|
Liu YP, Yang XN, Jazag A, Pan JS, Hu TH,
Liu JJ, Guleng B and Ren JL: HBsAg inhibits the translocation of
JTB into mitochondria in HepG2 cells and potentially plays a role
in HCC progression. PLoS One. 7:e369142012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pan JS, Zhou F, Xie CX, Cai JY, Chen JM,
Zhang ZP, Dong J, Xu HZ, Shi HX and Ren JL: Aldolase A-HBsAg
interaction and its effect on ultraviolet radiation induced
apoptosis in 293FT cells. J Gastroenterol Hepatol. 25:1702–1709.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sung WK, Zheng H, Li S, Chen R, Liu X, Li
Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, et al: Genome-wide
survey of recurrent HBV integration in hepatocellular carcinoma.
Nat Genet. 44:765–769. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shafritz DA, Shouval D, Sherman HI,
Hadziyannis SJ and Kew MC: Integration of hepatitis B virus DNA
into the genome of liver cells in chronic liver disease and
hepatocellular carcinoma. Studies in percutaneous liver biopsies
and post-mortem tissue specimens. N Engl J Med. 305:1067–1073.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park YM, Jang JW, Yoo SH, Kim SH, Oh IM,
Park SJ, Jang YS and Lee SJ: Combinations of eight key mutations in
the X/preC region and genomic activity of hepatitis B virus are
associated with hepatocellular carcinoma. J Viral Hepat.
21:171–177. 2014. View Article : Google Scholar
|
|
16
|
Xu HZ, Liu YP, Guleng B and Ren JL:
Hepatitis B virus-related hepatocellular carcinoma: Pathogenic
mechanisms and novel therapeutic interventions. Gastrointest
Tumors. 1:135–145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sell S and Leffert HL: Liver cancer stem
cells. J Clin Oncol. 26:2800–2805. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Das A and Maini MK: Innate and adaptive
immune responses in hepatitis B virus infection. Dig Dis.
28:126–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rogozin IB, Basu MK, Jordan IK, Pavlov YI
and Koonin EV: APOBEC4, a new member of the AID/APOBEC family of
polynucleotide (deoxy)cytidine deaminases predicted by
computational analysis. Cell Cycle. 4:1281–1285. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Prochnow C, Bransteitter R and Chen XS:
APOBEC deami-nases-mutases with defensive roles for immunity. Sci
China C Life Sci. 52:893–902. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Teng B, Burant CF and Davidson NO:
Molecular cloning of an apolipoprotein B messenger RNA editing
protein. Science. 260:1816–1819. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Navaratnam N, Morrison JR, Bhattacharya S,
Patel D, Funahashi T, Giannoni F, Teng BB, Davidson NO and Scott J:
The p27 catalytic subunit of the apolipoprotein B mRNA editing
enzyme is a cyti-dine deaminase. J Biol Chem. 268:20709–20712.
1993.PubMed/NCBI
|
|
23
|
Mangeat B, Turelli P, Caron G, Friedli M,
Perrin L and Trono D: Broad antiretroviral defence by human
APOBEC3G through lethal editing of nascent reverse transcripts.
Nature. 424:99–103. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Har ris RS, Bishop KN, Sheehy AM, Craig
HM, Petersen-Mahrt SK, Watt IN, Neuberger MS and Malim MH: DNA
deamination mediates innate immunity to retroviral infection. Cell.
113:803–809. 2003. View Article : Google Scholar
|
|
25
|
Suspene R, Guetard D, Henry M, Sommer P,
Wain-Hobson S and Vartanian JP: Extensive editing of both hepatitis
B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in
vivo. Proc Natl Acad Sci USA. 102:8321–8326. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Muramatsu M, Sankaranand VS, Anant S,
Sugai M, Kinoshita K, Davidson NO and Honjo T: Specific expression
of activation-induced cytidine deaminase (AID), a novel member of
the RNA-editing deaminase family in germinal center B cells. J Biol
Chem. 274:18470–18476. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Neuberger MS, Harris RS, Di Noia J and
Petersen-Mahrt SK: Immunity through DNA deamination. Trends Biochem
Sci. 28:305–312. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liao W, Hong SH, Chan BH, Rudolph FB,
Clark SC and Chan L: APOBEC-2, a cardiac- and skeletal
muscle-specific member of the cytidine deaminase supergene family.
Biochem Biophys Res Commun. 260:398–404. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Anant S, Mukhopadhyay D, Sankaranand V,
Kennedy S, Henderson JO and Davidson NO: ARCD-1, an
apobec-1-related cytidine deaminase, exerts a dominant negative
effect on C to U RNA editing. Am J Physiol Cell Physiol.
281:C1904–C1916. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matsumoto T, Marusawa H, Endo Y, Ueda Y,
Matsumoto Y and Chiba T: Expression of APOBEC2 is transcriptionally
regulated by NF-kappaB in human hepatocytes. FEBS Lett.
580:731–735. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Okuyama S, Marusawa H, Matsumoto T, Ueda
Y, Matsumoto Y, Endo Y, Takai A and Chiba T: Excessive activity of
apolipoprotein B mRNA editing enzyme catalytic polypeptide 2
(APOBEC2) contributes to liver and lung tumorigenesis. Int J
Cancer. 130:1294–1301. 2012. View Article : Google Scholar
|
|
32
|
Bandopadhyay M, Sarkar N, Datta S, Das D,
Pal A, Panigrahi R, Banerjee A, Panda CK, Das C, Chakrabarti S and
Chakravarty R: Hepatitis B virus X protein mediated suppression of
miRNA-122 expression enhances hepatoblastoma cell proliferation
through cyclin G1-p53 axis. Infect Agent Cancer. 11:402016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Scaglioni PP, Melegari M and Wands JR:
Posttranscriptional regulation of hepatitis B virus replication by
the precore protein. J Virol. 71:345–353. 1997.PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
35
|
Vallee A, Lecarpentier Y and Vallee JN:
Targeting the canonical WNT/β-catenin pathway in cancer treatment
using non-steroidal anti-inflammatory drugs. Cells. 8:E7262019.
View Article : Google Scholar
|
|
36
|
Kumari N, Dwarakanath BS, Das A and Bhatt
AN: Role of interleukin-6 in cancer progression and therapeutic
resistance. Tumour Biol. 37:11553–11572. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Göktuna SI, Diamanti MA and Chau TL: IKKs
and tumor cell plasticity. FEBS J. 285:2161–2181. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zubair H, Azim S, Srivastava SK, Ahmad A,
Bhardwaj A, Khan MA, Patel GK, Arora S, Carter JE, Singh S and
Singh AP: Glucose metabolism reprogrammed by overexpression of
IKKepsilon promotes pancreatic tumor growth. Cancer Res.
76:7254–7264. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Balkwill F: TNF-alpha in promotion and
progression of cancer. Cancer Metastasis Rev. 25:409–416. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li C, Wang Y, Wang S, Wu B, Hao J, Fan H,
Ju Y, Ding Y, Chen L, Chu X, et al: Hepatitis B virus mRNA-mediated
miR-122 inhibition upregulates PTTG1-binding protein, which
promotes hepatocellular carcinoma tumor growth and cell invasion. J
Virol. 87:2193–2205. 2013. View Article : Google Scholar :
|
|
41
|
Cascalho M: Advantages and disadvantages
of cytidine deamination. J Immunol. 172:6513–6518. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen SH, Habib G, Yang CY, Gu ZW, Lee BR,
Weng SA, Silberman SR, Cai SJ, Deslypere JP and Rosseneu M:
Apolipoprotein B-48 is the product of a messenger RNA with an
organ-specific in-frame stop codon. Science. 238:363–366. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yamanaka S, Balestra ME, Ferrell LD, Fan
J, Arnold KS, Taylor S, Taylor JM and Innerarity TL: Apolipoprotein
B mRNA-editing protein induces hepatocellular carcinoma and
dysplasia in transgenic animals. Proc Natl Acad Sci USA.
92:8483–8487. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Okazaki IM, Hiai H, Kakazu N, Yamada S,
Muramatsu M, Kinoshita K and Honjo T: Constitutive expression of
AID leads to tumorigenesis. J Exp Med. 197:1173–1181. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Morisawa T, Marusawa H, Ueda Y, Iwai A,
Okazaki IM, Honjo T and Chiba T: Organ-specific profiles of genetic
changes in cancers caused by activation-induced cytidine deaminase
expression. Int J Cancer. 123:2735–2740. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Girard M, Jacquemin E, Munnich A, Lyonnet
S and Henrion-Caude A: miR-122, a paradigm for the role of
microRNAs in the liver. J Hepatol. 48:648–656. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lewis AP and Jopling CL: Regulation and
biological function of the liver-specific miR-122. Biochem Soc
Trans. 38:1553–1557. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gramantieri L, Ferracin M, Fornari F,
Veronese A, Sabbioni S, Liu CG, Calin GA, Giovannini C, Ferrazzi E,
Grazi GL, et al: Cyclin G1 is a target of miR-122a, a microRNA
frequently down-regulated in human hepatocellular carcinoma. Cancer
Res. 67:6092–6099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fan CG, Wang CM, Tian C, Wang Y, Li L, Sun
WS, Li RF and Liu YG: miR-122 inhibits viral replication and cell
proliferation in hepatitis B virus-related hepatocellular carcinoma
and targets NDRG3. Oncol Rep. 26:1281–1286. 2011.PubMed/NCBI
|
|
50
|
Lin CJ, Gong HY, Tseng HC, Wang WL and Wu
JL: miR-122 targets an anti-apoptotic gene, Bcl-w, in human
hepatocellular carcinoma cell lines. Biochem Biophys Res Commun.
375:315–320. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Barajas JM, Reyes R, Guerrero MJ, Jacob
ST, Motiwala T and Ghoshal K: The role of miR-122 in the
dysregulation of glucose-6-phosphate dehydrogenase (G6PD)
expression in hepatocellular cancer. Sci Rep. 8:91052018.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bai S, Nasser MW, Wang B, Hsu SH, Datta J,
Kutay H, Yadav A, Nuovo G, Kumar P and Ghoshal K: MicroRNA-122
inhibits tumorigenic properties of hepatocellular carcinoma cells
and sensitizes these cells to sorafenib. J Biol Chem.
284:32015–32027. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tsai WC, Hsu PW, Lai TC, Chau GY, Lin CW,
Chen CM, Lin CD, Liao YL, Wang JL, Chau YP, et al: MicroRNA-122, a
tumor suppressor microRNA that regulates intrahepatic metastasis of
hepatocellular carcinoma. Hepatology. 49:1571–1582. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Waidmann O, Bihrer V, Pleli T, Farnik H,
Berger A, Zeuzem S, Kronenberger B and Piiper A: Serum microRNA-122
levels in different groups of patients with chronic hepatitis B
virus infection. J Viral Hepat. 19:e58–e65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Song le H, Binh VQ, Duy DN, Kun JF, Bock
TC, Kremsner PG and Luty AJ: Serum cytokine profiles associated
with clinical presentation in vietnamese infected with hepatitis B
virus. J Clin Virol. 28:93–103. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Porta C, De Amici M, Quaglini S, Paglino
C, Tagliani F, Boncimino A, Moratti R and Corazza GR: Circulating
interleukin-6 as a tumor marker for hepatocellular carcinoma. Ann
Oncol. 19:353–358. 2008. View Article : Google Scholar
|
|
57
|
Lan T, Chang L, Wu L and Yuan YF: IL-6
plays a crucial role in HBV infection. J Clin Transl Hepatol.
3:271–276. 2015. View Article : Google Scholar
|
|
58
|
Wang H and Ryu WS: Hepatitis B virus
polymerase blocks pattern recognition receptor signaling via
interaction with DDX3: Implications for immune evasion. PLoS
Pathog. 6:e10009862010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Marinos G, Naoumov NV, Rossol S, Torre F,
Wong PY, Gallati H, Portmann B and Williams R: Tumor necrosis
factor receptors in patients with chronic hepatitis B virus
infection. Gastroenterology. 108:1453–1463. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Thorgeirsson SS and Grisham JW: Molecular
pathogenesis of human hepatocellular carcinoma. Nat Genet.
31:339–346. 2002. View Article : Google Scholar : PubMed/NCBI
|