Introduction
DNA-damaging drugs induce death in malignant tumor
cells. Although the majority of these drugs affect the primary
structure of DNA, their usefulness may be limited by the chromatin
structure that represents the physiological template of the DNA.
Since methylation of DNA or proteins occurs in the nucleus, the
anticancer effect of DNA-damaging drugs may be affected by cellular
methylation (1,2).
S-adenosyl methionine (SAM) is key as a methyl donor
in methylation reactions. SAM transfers the methyl group,
CH3, to cell components such as DNA, proteins and lipids
(3,4). It is made from adenosine triphosphate
and methionine by methionine adenosyltransferase. Removal of the
methyl group from SAM yields S-adenosyl homocysteine (SAH), which
acts as a methyltransferase inhibitor (5,6).
Metabolic SAM can be synthesized throughout the body, but most SAM
is consumed in the liver (7,8). DNA
methyltransferases (DNMTs) catalyze the transfer of a methyl group
from SAM to cytosine residues to form 5-methyl cytosine at CpG
sites in the genome, an important regulatory mechanism for
regulation of the gene expression (9). Histone methyltransferases (HMTases)
also utilize SAM to methylate lysine or the arginine residue of
histone proteins (10). As SAM is
a key metabolite of hepatocyte growth, death and differentiation,
its use in treatment may improve survival in liver disease
(11,12). Recently, multiple clinical trials
have also indicated that SAM is important in the treatment of
Alzheimer’s disease, depression and osteoarthritis (13,14).
However, therapeutic usages of SAM are not yet proven in
cancer.
5-Fluorouracil (5-FU) has been used in the treatment
of various types of cancer (15–17).
Since 5-FU acts as a thymidylate synthase inhibitor, it blocks the
synthesis of thymidine during DNA replication. However, the
correlation between thymidylate synthase levels and 5-FU
sensitivity remains controversial, although it is widely thought
that thymidylate synthase is the main molecular mechanism governing
5-FU sensitivity (18–20). Other investigators have suggested
that 5-FU resistance may also be induced by p53 gene mutation,
mismatch repair gene deficiency, deregulation of pyrimidine
metabolism-related enzymes and overexpression of anti-apoptotic
factors (19,21). Cisplatin is a platinum-based
chemotherapy drug that causes DNA crosslinking. Cisplatin is
frequently administered as part of a combination chemotherapy
regimen with other drugs as it sometimes acts in synergy
synergistic with other agents (22,23).
In the present study, we hypothesized that SAM has
an impact on the cytotoxic effect of DNA-damaging drugs. We
characterized and compared the effects of SAM and assessed whether
it affects the anticancer effects of 5-FU and cisplatin. Using
several cytotoxic assays, we showed that SAM specifically regulates
the anticancer effect of 5-FU but not that of cisplatin.
Materials and methods
Cell culture and reagents
TheA549 human lung cancer cell line was obtained
from ATCC (Baltimore, MD, USA). Cells were maintained in DMEM
medium supplemented with 10% (v/v) fetal bovine serum, and
penicillin-streptomycin (100 U/ml) at 37°C in a humidified
incubator containing 5% CO2. SAM, 5-FU and cisplatin
were purchased from Sigma (St. Louis, MO, USA) and dissolved in
dimethyl sulfoxide for further use. Combination treatment of SAM
and anticancer drugs was performed as indicated in figure
legends.
3-(4,5-dimethylthiazol-2-yl)-2,5-dephenyl
tetrazolium bromide (MTT) assay
Various concentrations of 5-FU (2–40 μM) or
cisplatin (2–32 μM) were treated. Cells (1,000 cells/well) were
seeded and treated with various concentrations of the indicated
drugs for 2 days. MTT (Sigma) was dissolved in phosphate-buffered
saline (PBS) and filtered through a 0.2 μm filter. The solution was
stored at 4°C for future use. To evaluate cell viability, A549
cells were seeded in 96-well plates at densities of 500-1000
cells/well. The following day, the cells were treated with the
indicated drugs. After washing with PBS, the cells were incubated
in MTT solution for 30 min. Absorbance was measured using a
microplate reader (Bio-Rad, Hercules, CA, USA) at a wavelength of
540 nm. After the experiment was performed, the mean and standard
deviation of the data were calculated. Statistical analysis was
performed using the Student’s t-test. P<0.05 was considered to
indicate a statistically significant difference.
FACS analysis
Cells were seeded in 1×106 cells in
100-mm dishes and treated with SAM, 5-FU and cisplatin. The cells
were harvested and fixed with 70% cold ethanol. The fixed cells
were then washed with PBS and stained with 50 μg/ml propidium
iodide containing 1 mg/ml RNase A for 15 min in a 37°C water bath.
Analyses of 10,000 events were obtained on a FACSCalibur flow
cytometer (Becton-Dickinson, Mountain View, CA, USA) and the cell
cycles were analyzed using ModFit DNA analysis software.
Western blot analysis
Cells were cultured in 100-mm dishes and treated
with the indicated drugs. The cells were collected by scraping and
lysed with RIPA lysis buffer (0.02 M Tris, 0.15 M NaCl, 0.1% sodium
dodecyl sulfate DS, 1% Triton X-100, 1% sodium deoxycholate, 0.02
mM phenylmethylsulfonyl fluoride, 0.1 mM NaF, 0.01 mg/ml leupeptin,
0.01 mg/ml pepstatin) and then centrifuged at 14,200 × g for 30 min
at 4°C. The amount of protein was determined with the Bradford
protein assay (Bio-Rad). The lysates were boiled for 5 min,
separated by SDS-polyacrylamide gel electrophoresis and transferred
to polyvinylidene difluoride membrane (Amersham Biosciences,
Piscataway, NJ, USA). The membranes were incubated for 1 h with
blocking buffer [5% non-fat milk and 0.1% Tween-20 in Tris-buffered
saline (TBS-T)] and then incubated with the specific antibodies.
The membranes were washed three times with TBS-T and incubated for
1 h with secondary antibodies (Santa Cruz Biotechnology Inc., Santa
Cruz, CA, USA). Proteins were detected with enhanced
chemiluminescence reagent (Amersham Biosciences).
RT-PCR
Cells were collected after treatment with the
indicated drugs. Total RNA was extracted using TRIzol-reagent
(Promega Co., Madison, WI, USA) according to the manufacturer’s
instructions. cDNA was synthesized using reverse-transcriptase
according to the manufacturer’s instructions and was used as the
template for PCR amplification. Primer sequences of DNMTs-specific
primer sets are as follows: DNMT1, sense: 5′-ATC TAC CAG TGT ACA
GAG TGT GA-3′, antisense: 5′-ATA CTG ACA GAA GTA ATC TCG AT-3′;
DNMT3A, sense: 5′-ATC TAC CAG TGT ACA GAG TGT GA-3′, antisense:
5′-ATA CTG ACA GAA GTA ATC TCG AT-3′; DNMT3B, sense: 5′-ATC TAC CAG
TGT ACA GAG TGT GA-3′, antisense: 5′-ATA CTG ACA GAA GTA ATC TCG
AT-3′; GAPDH, sense: 5′-ATG ACA ACT TTG GCA TTG TGG AA-3′, GAPDH
antisense: 5′-CTG TTG CTG TAG CCG TAT TCA TT-3′. GAPDH was used as
a loading control. Each sample was incubated at 95°C for 20 sec,
60°C for 25 sec and 72°C for 30 sec for 35 cycles. Reaction samples
were then incubated for an additional 7 min at 72°C and cooled to
4°C. PCR products were resolved on 1% agarose gel.
Results
SAM-modulates the anticancer effect of
5-FU but not cisplatin
DNA-damaging drugs activate apoptosis in cancer
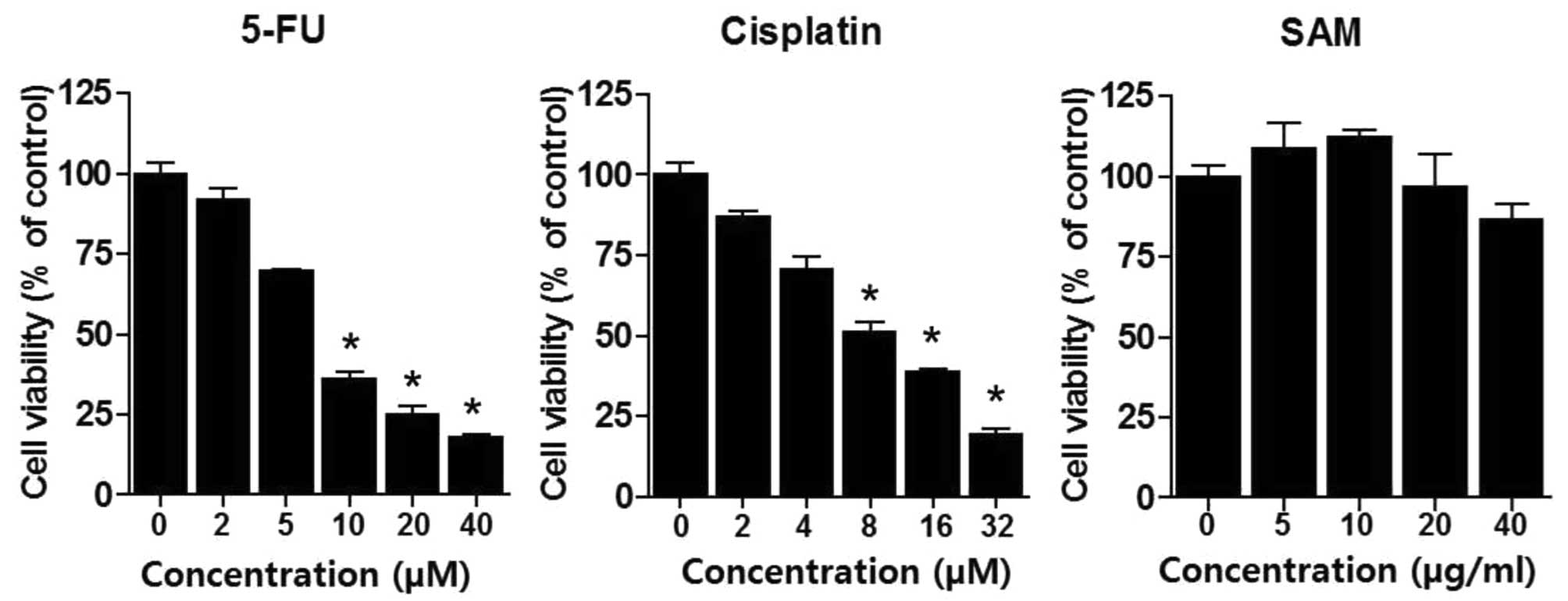
cells. We treated various concentrations of 5-FU (2–40 μM) or
cisplatin (2–32 μM) for 48 h in A549 lung cancer cells and
performed MTT assay to analyze drug sensitivity. As expected, 5-FU
or cisplatin resulted in cytotoxic effects in a dose-dependent
manner (Fig. 1). LD50
of 5-FU and cisplatin were ∼7 and 6 μM, respectively. We also
performed MTT assay by treatment using SAM (5–40 μg/ml). SAM
treatment produced no special morphological changes and had no
distinct cytotoxic effect in these ranges. However, since SAM
modulates cellular methylation of DNA or proteins, we hypothesized
that SAM affected DNA-damaging drugs. MTT assay was performed at
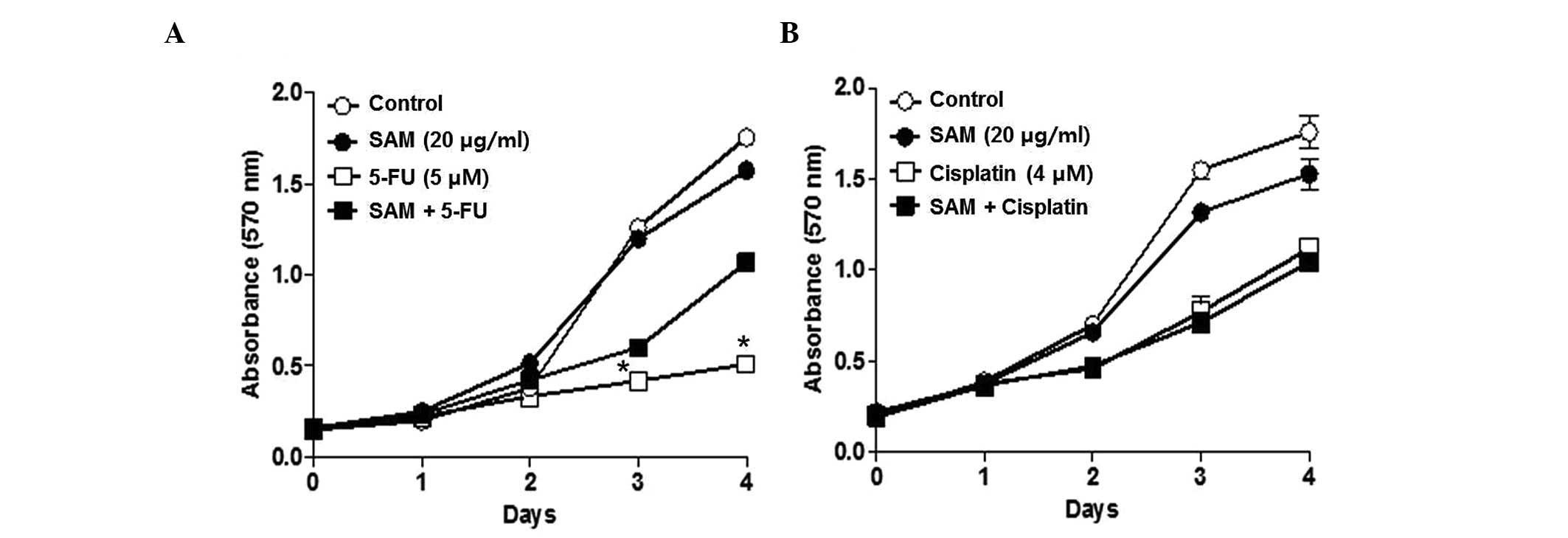
several concentrations of 5-FU and cisplatin in combination with
SAM of 20 μg/ml. Combination treatment of SAM and 5-FU somewhat
protected the anticancer effect of 5-FU. However, this phenomenon
was not observed in the combination treatment of SAM and cisplatin
(Fig. 2). The protection effect of
SAM on 5-FU was evident from 3 days after the combination treatment
of SAM and 5-FU. This result suggests that SAM specifically
modulates the anticancer effect of 5-FU but not for cisplatin.
Protective effect of SAM on the
anticancer effect of 5-FU does not require specific cell cycle
arrest
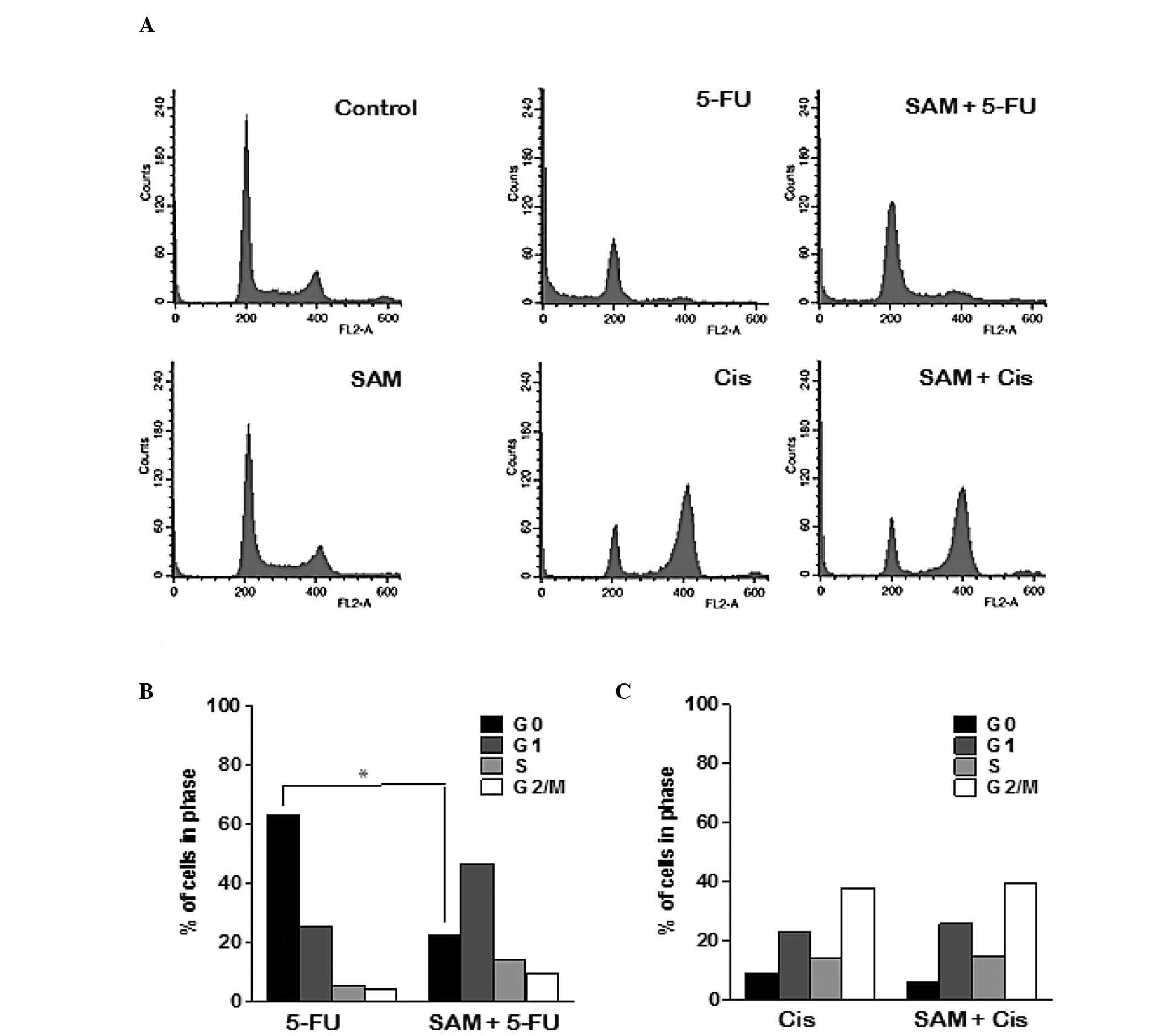
FACS analysis was performed to examine the manner in
which SAM affects cell cycle arrest or cell death by 5-FU or
cisplatin. Treatment with SAM only did not show any difference as
compared with the control. Treatment of 5-FU induced cell death,
but not specific cell cycle arrest, whereas cisplatin induced G2/M
arrest (Fig. 3A). However, the
combination treatment of SAM and 5-FU significantly decreased the
dead cell population, while the G1 cell population slightly
increased, compared with treatment with 5-FU alone. However,
combination treatment of SAM and cisplatin resulted in G2/M arrest,
similar to cisplatin alone. We quantified data obtained from FACS
analysis following treatment with a fixed concentration of SAM (20
μg) but with varying concentrations of 5-FU or cisplatin and the
results suggest that a moderate concentration of SAM has a specific
protective effect on 5-FU (Fig. 3B and
C). Therefore, we consider that the protective effect of SAM on
the anticancer effect of 5-FU does not require specific cell cycle
arrest.
5-FU decreases DNMTs expression but SAM
restores the reduction of DNMTs
To analyze which types of cellular methylation are
involved in SAM modulation of the anticancer effect of 5-FU, we
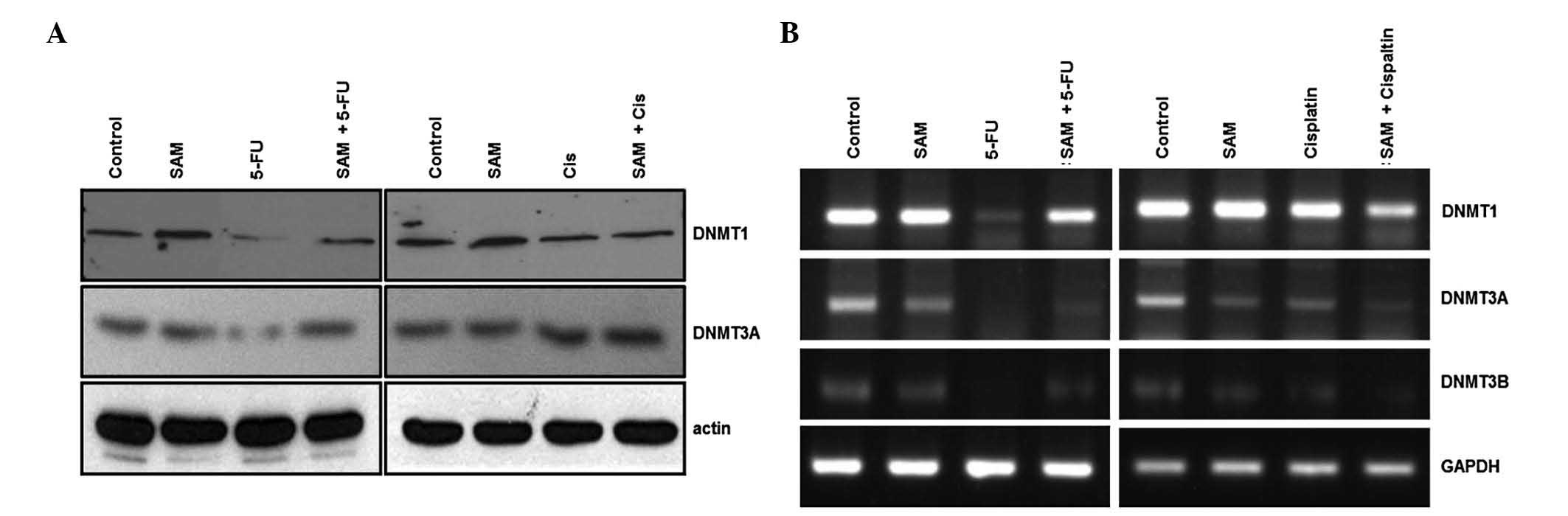
performed expression analyses on HMTases and DNMTs. The drugs were
treated for 3 days and western blot analysis was performed. SUV39H1
or G9a proteins were not detected during SAM treatment or
combination treatment with 5-FU, suggesting no induction by SAM or
5-FU (data not shown). However, the expression of DNMT1 and DNMT3A
were decreased in 5-FU but not in cisplatin treatment (Fig. 4A). Notably, the combination
treatment of SAM and 5-FU restored expression of DNMTs.
Expression of DNMTs was also analyzed using RT-PCR.
Consistent with the findings above, 5-FU treatment decreased DNMTs
expression in the RNA level but SAM restored the effect of 5-FU on
DNMTs expression (Fig. 4B). This
result means that protection of SAM on the anticancer effect of
5-FU is exhibited by regulating DNMT expression at the
transcriptional level.
Discussion
Resistance is one of the obstacles to the success of
DNA-damaging drug-based chemotherapy. Although the molecular
mechanisms of DNA damaging drugs remain controversial, a plausible
mechanism is that cellular metabolites regulate cytotoxicity of
anticancer drugs. SAM is a physiologic metabolite found in almost
every tissue and fluid in the body. Moreover, SAM concentration in
the body may also be determined by vitamin B12 and folate (vitamin
B6) obtained from dietary sources (24,25).
Since use of SAM in a therapeutic setting has not yet been proven
when SAM is combined with several anticancer drugs, we examined
whether the use of SAM is a regulatory mechanism for the direct
modulation of the anticancer effect of DNA-damaging drugs. It has
been reported that SAM has inhibitory effects on some carcinoma
cells, as well as the proliferation and migration of HUVEC cells at
high concentrations (10,26). In our analysis, however, SAM did
not exhibit distinct effects on cell growth in A549 lung cancer
cells at concentrations ranging up to 40 μg/ml. Instead, treatment
for a long period of time with SAM demonstrated a small inhibitory
effect on the cells. Therefore, the cytotoxic effect of SAM seems
to depend on the concentration and treatment time of SAM. Results
of the present study have shown that SAM has a protective effect
when combined with 5-FU. To achieve this, cytotoxic assays such as
MTT assay, cell counting and a viability test were performed. We
obtained similar results in that SAM specifically affected the
anticancer effect of 5-FU but not for cisplatin. Therefore, we
investigated differences between 5-FU and cisplatin to determine
this phenomenon. 5-FU treatment induced cell death without specific
cell cycle arrest, contrary to cisplatin treatment-induced G2/M
arrest. SAM itself had no specific cell cycle arrest or cytotoxic
effect. However, the G1 cell population was increased in the
combination of SAM and 5-FU, suggesting that protection of SAM is
likely involved in the G1 phase. As a pyrimidine analogue, 5-FU is
incorporated in DNA in the S phase of the cell cycle (27). The anticancer effect of 5-FU occurs
in an S phase-active manner, whereas its therapeutic effect is not
active when cells are in the G1 phase. As SAM methylates cellular
DNA, the anticancer effect of 5-FU is likely reduced during DNA
replication. In the case of cisplatin, SAM did not protect the
anticancer effect of cisplatin. The reason for this finding is that
cisplatin induced G2/M arrest, thus SAM was not able to affect the
anticancer effect of cisplatin because cells were already in the
G2/M phase.
5-FU treatment was found to regulate a group of cell
cycle-related genes such as cyclin and p53. SAM treatment may
regulate gene expression by reversing DNA hypomethylation on gene
promoter. As SAM treatment regulated SAM-utilizing genes, we
examined the expression of HMTases or DNMTs. 5-FU or SAM treatment
did not produce any induction of SUV39H1 or G9a. Instead, 5-FU
treatment decreased most DNMTs expression at the transcriptional
level, although SAM had no distinct effect on the expression of
DNMTs. However, the presence of SAM restored the effect of 5-FU on
DNMTs expression, suggesting that the protective effect of SAM was
mediated by the regulation of DNMTs expression. Although cell
levels of SAM might affect DNMTs expression, further studies are
needed to show the effect of SAM on the expression levels of
regulating genes by 5-FU.
The clinical applications of SAM remain
controversial in cancer due to low toxicity. Instead, a growing
number of trials have been conducted to find a new anticancer
effect of SAM metabolism-related analogues (28–30).
Sinefungin, which is a SAM analogue can compete for SAM binding and
inhibit the activity of the SAM-dependent methyltransferases.
Adenosine dialdehyde inhibits methylation reaction by hindering SAH
hydrolase activity.
SAM, as a methyl donor, plays a versatile regulatory
effect in the cells by methylating cell components. The effect of
SAM described in the present study may not be universal to the
anticancer drugs, but rather specific to 5-FU. Moreover, SAM causes
dysregulation of gene expression and cell death by 5-FU by
modulating aberrant DNA methylation. This opens new avenues of how
cellular metabolites regulate the anticancer effect of DNA-damaging
drugs.
Acknowledgements
This study was supported by grants
from the National Research Foundation of Korea (NRF 2011-0006047)
and the Ministry of Education Science and Technology (The Regional
Research Universities Program/Medical and Bio-Materials Research
Center), Republic of Korea.
References
|
1
|
Dunne AL, Price ME, Mothersill C, et al:
Relationship between clonogenic radiosensitivity, radiation-induced
apoptosis and DNA damage/repair in human colon cancer cells. Br J
Cancer. 89:2277–2283. 2003. View Article : Google Scholar
|
|
2
|
Mothersill C and Seymour C:
Radiation-induced bystander effects, carcinogenesis and models.
Oncogene. 22:7028–7033. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu SC and Mato JM: Role of methionine
adenosyltransferase and S-adenosylmethionine in alcohol-associated
liver cancer. Alcohol. 35:227–234. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Roje S: S-Adenosyl-L-methionine: beyond
the universal methyl group donor. Phytochemistry. 67:1686–1698.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim KC, Geng L and Huang S: Inactivation
of a histone methyltransferase by mutations in human cancers.
Cancer Res. 63:7619–7623. 2003.PubMed/NCBI
|
|
6
|
Fiskus W, Wang Y, Sreekumar A, et al:
Combined epigenetic therapy with the histone methyltransferase EZH2
inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor
panobinostat against human AML cells. Blood. 114:2733–2743. 2009.
View Article : Google Scholar
|
|
7
|
Mato JM, Alvarez L, Ortiz P and Pajares
MA: S-adenosylmethionine synthesis: molecular mechanisms and
clinical implications. Pharmacol Ther. 73:265–280. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ji L, Chen Y and Wang Z: Protection of
S-adenosyl methionine against the toxicity of clivorine on
hepatocytes. Environ Toxicol Pharmacol. 26:331–335. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wakefield L, Boukouvala S and Sim E:
Characterisation of CpG methylation in the upstream control region
of mouse Nat2: evidence for a gene-environment interaction in a
polymorphic gene implicated in folate metabolism. Gene. 452:16–21.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sahin M, Sahin E, Gumuslu S, et al:
Inhibition of angiogenesis by S-adenosylmethionine. Biochem Biophys
Res Commun. 408:145–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song Z, Zhou Z, Chen T, et al:
S-adenosylmethionine (SAMe) protects against acute alcohol induced
hepatotoxicity in mice small star, filled. J Nutr Biochem.
14:591–597. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang X and Cederbaum AI:
S-adenosyl-L-methionine attenuates hepatotoxicity induced by
agonistic Jo2 Fas antibody following CYP2E1 induction in mice. J
Pharmacol Exp Ther. 317:44–52. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Papakostas GI: Evidence for
S-adenosyl-L-methionine (SAM-e) for the treatment of major
depressive disorder. J Clin Psychiatry. 70:18–22. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Coppede F: One-carbon metabolism and
Alzheimer’s disease: focus on epigenetics. Curr Genomics.
11:246–260. 2010.
|
|
15
|
Iacopetta B, Kawakami K and Watanabe T:
Predicting clinical outcome of 5-fluorouracil-based chemotherapy
for colon cancer patients: is the CpG island methylator phenotype
the 5-fluorouracil-responsive subgroup? Int J Clin Oncol.
13:498–503. 2008. View Article : Google Scholar
|
|
16
|
Lombardi L, Gebbia V, Silvestris N, et al:
Adjuvant therapy in colon cancer. Oncology. 77:50–56. 2009.
View Article : Google Scholar
|
|
17
|
Patel PA: Evolution of
5-fluorouracil-based chemoradiation in the management of rectal
cancer. Anticancer Drugs. 22:311–316. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Giovannetti E, Backus HH, Wouters D, et
al: Changes in the status of p53 affect drug sensitivity to
thymidylate synthase (TS) inhibitors by altering TS levels. Br J
Cancer. 96:769–775. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kunz C, Focke F, Saito Y, et al: Base
excision by thymine DNA glycosylase mediates DNA-directed
cytotoxicity of 5-fluorouracil. PLoS Biol. 7:e912009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tominaga T, Iwahashi M, Takifuji K, et al:
Combination of p53 codon 72 polymorphism and inactive p53 mutation
predicts chemosensitivity to 5-fluorouracil in colorectal cancer.
Int J Cancer. 126:1691–1701. 2010.PubMed/NCBI
|
|
22
|
Bae-Jump VL, Zhou C, Boggess JF and Gehrig
PA: Synergistic effect of rapamycin and cisplatin in endometrial
cancer cells. Cancer. 115:3887–3896. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Taylor-Harding B, Orsulic S, Karlan BY and
Li AJ: Fluvastatin and cisplatin demonstrate synergistic
cytotoxicity in epithelial ovarian cancer cells. Gynecol Oncol.
119:549–556. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Taban-Shomal O, Kilter H, Wagner A, et al:
The cardiac effects of prolonged vitamin B12 and folate deficiency
in rats. Cardiovasc Toxicol. 9:95–102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shin W, Yan J, Abratte CM, et al: Choline
intake exceeding current dietary recommendations preserves markers
of cellular methylation in a genetic subgroup of folate-compromised
men. J Nutr. 140:975–980. 2010. View Article : Google Scholar
|
|
26
|
Luo J, Li YN, Wang F, et al:
S-adenosylmethionine inhibits the growth of cancer cells by
reversing the hypomethylation status of c-myc and H-ras in human
gastric cancer and colon cancer. Int J Biol Sci. 6:784–795. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Robinson HM, Jones R, Walker M, et al:
Chk1-dependent slowing of S-phase progression protects DT40
B-lymphoma cells against killing by the nucleoside analogue
5-fluorouracil. Oncogene. 25:5359–5369. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hong S, Heo J, Lee S, et al:
Methyltransferase-inhibition interferes with neuronal
differentiation of P19 embryonal carcinoma cells. Biochem Biophys
Res Commun. 377:935–940. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kuratani M, Hirano M, Goto-Ito S, et al:
Crystal structure of Methanocaldococcus jannaschii Trm4 complexed
with sine-fungin. J Mol Biol. 401:323–333. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Choudhury SR, Balasubramanian S, Chew YC,
et al: (−)-Epigallocatechin-3-gallate and DZNep reduce polycomb
protein level via a proteasome-dependent mechanism in skin cancer
cells. Carcinogenesis. 32:1525–1532. 2011.
|