Introduction
Chronic myeloproliferative neoplasms (MPNs) have
been considered diseases caused by aberrations in signaling, mainly
affecting the JAK-STAT pathway. However, mutations in other genes
that affect different cell processes have been described,
suggesting that they are diseases with more complex causes. Recent
findings suggest that MPNs are likely the result of combined
genetic dysregulation and can present mutations in epigenetic
associated-genes such as TET2, ASXL1, EZH2, IDH1/2 and
DNMT3A (1–3) described also in the related
myelodysplastic/myeloproliferative neoplasms (MDS/MPN).
SETBP1 encodes the SET binding protein
1, one component of the multi-functional SET complex in the
nucleus that plays a role in histone modifications. In 2013, whole
exome sequencing identified recurrent SETBP1 mutations in
approximately 25% patients with atypical chronic myeloid leukemia
(aCML) and in a small proportion of patients with chronic
myelomonocytic leukemia (CMML) and chronic neutrophilic leukemia
(CNL) (4). SETBP1 was the
first gene recurrently mutated in aCML, a disease previously
diagnosed by exclusion. All of the mutations described were located
in a small region of 14 amino acids (858–871) in the
highly-conserved SKI homologous region encoded by exon 4 of the
gene.
SETBP1 seems to interact with SET protein, leading
to the inhibition of the activity of PP2A and promoting the
proliferation of leukemic cells (5).
Overexpression of SETBP1 increases the expression of other
genes such as HOXA9 and HOXA10 leading to
immortalization of murine myeloid progenitors (6), and mutant SETBP1 forms could drive the
disease through similar mechanisms (7).
There are three SETBP1 paralogous genes
(NSD1, NSD2 and NSD3) that encode proteins with
similar functions, so that they could also be mutated in MPNs.
Indeed, NSD1 and NSD3 are involved in fusions with
NUP98 in myeloid malignancies. Nuclear receptor binding
SET domain protein 1 (NSD1) encodes a H3K36 and H4K20
methyltransferase that activates or represses transcription
depending on the cellular context. Some rare cases of acute myeloid
leukemia (AML) with poor prognosis have a t(5;11)(q35;p15) that
fuses NUP98 with NSD1, and show abnormally high
levels of H3K36 methylation and activation of transcription of
oncogenes such as HOXA9 (8–10). On
the other hand, nuclear receptor binding SET domain protein
3 (NSD3) encodes a H3K4 and H3K27 methyltransferase,
marks of activation or repression of transcription, respectively.
This gene has also been found fused with NUP98 in patients
with AML (11) and therapy-related
myelodysplastic syndrome (t-MDS) with poor prognosis (12). Finally, nuclear receptor binding
SET domain protein 2 (NSD2) encodes an H3K36 histone
methyltransferase that has also been implicated in gene fusions in
multiple myeloma (13) and is
mutated in pediatric acute lymphoblastic leukemia (ALL) (14). The p.E1099K mutation affects the SET
domain and increases H3K36 dimethylation, which may lead to the
transforming activity of NSD2 (14).
To date, little is known about the role of
individual histone modifications in MPNs, and current research is
focusing mainly on changes related to polycomb group proteins
(3). These proteins can remodel
chromatin; for example, polycomb repressive complex 2 (PRC2)
catalyzes di- and trimethylation of H3K27 leading to gene
silencing. Addition of sex combs like protein 1
(ASXL1) encodes an important mediator of PRC2 function, and
mutations in this gene have been described in MDS/AML (15) and in MPNs (16). Thus, we asked whether SETBP1
related genes could also be mutated in these diseases.
With this aim in mind, we analyzed the mutational
profile of NSD1, NSD2 and NSD3 in a selected group of
patients with MPNs.
Materials and methods
We performed denaturing high performance liquid
chromatography (dHPLC) to analyze exon 13 of NSD1, NSD2 and
NSD3, which are homologous to SETBP1 exon 4 in which
mutations had been previously described. This SETBP1 exon
was also included in the analysis. In addition, we also analyzed
exon 19 of NSD1 and NSD2 and exon 20 of NSD3,
homologous to the NSD2-mutated exon in ALL (p.E1099K)
(primers and conditions are shown in Table I).
 | Table I.Polymerase chain reaction primers used
for the mutational analysis of NSD1, NSD2, NSD3 and
SETBP1 by dHPLC. |
Table I.
Polymerase chain reaction primers used
for the mutational analysis of NSD1, NSD2, NSD3 and
SETBP1 by dHPLC.
| Primer | Tm (°C) | % GC | Sequence (5′→3′) | PCR product (bp) | dHPLC conditions
(°C) |
|---|
| NSD1-e13L | 61.0 | 29.17 |
AATTTATCTTCTTTTGGCTTCTCA | 285 | 57.3; 59.2; 61 |
| NSD1-e13R | 62.8 | 40.91 |
TCTGTTGCCAATTAAACTGAGG |
|
|
| NSD1-e13Lm | 63.7 | 33.33 |
AATTTATCTTCGTTTGGCTTCTCA |
|
|
| NSD1_e19L | 66.2 | 54.55 |
TGCTGCTGACAGTGGTAGGAGT | 350 | 53.1; 56.8;
55.8 |
| NSD1_e19R | 65.8 | 39.13 |
CAGTGAAAACAGCATTTCCCATT |
|
|
| NSD1_e19Lm | 70.3 | 59.10 |
TGCTGCTGACGGTGGTAGGAGT |
|
|
| NSD2_e13L | 65.4 | 54.55 |
ACCTCTCTCTCCACCCCTTCTT | 329 | 57.8; 58.7;
62.7 |
| NSD2_e13R | 70,1 | 54.55 |
ACAATCCCAACAGCCCACCTTC |
|
|
| NSD2_e13Rm | 73.7 | 59.10 |
ACAATCCCAACGGCCCACCTTC |
|
|
| NSD2_e19L | 69.6 | 54.55 |
TCATGATGGGGAGTCTTGAGCC | 285 | 61.2; 61.8 |
| NSD2_e19R | 68.5 | 54.55 |
CCACAGGGCAAAGTCCAGTTCT |
|
|
| NSD2_e19Rm | 64.3 | 50.00 |
CCACAGGTCAAAGTCCAGTTCT |
|
|
| NSD3_e13L | 64.6 | 47.83 |
GCTGTTTGATGTCTGTAGCTGCT | 308 | 57.3; 59.4;
60.6 |
| NSD3_e13R | 66,7 | 41.67 |
TCTTTGTCTCCTTCTTCAGCTGTT |
|
|
| NSD3_e13Lm | 64,3 | 52.17 |
GCTGTTTGGTGTCTGTAGCTGCT |
|
|
| NSD3_e20L | 61.9 | 37.50 |
ACCATTTCTTTCTAGGGTTGAAGT | 372 | 53.8; 56; 60 |
| NSD3_e20R | 62.8 | 41.67 |
TCTTTGTCTCCTTCTTCAGCTGTT |
|
|
| NSD3_e20Lm | 64,3 | 41.67 |
ACCATTTCTGTCTAGGGTTGAAGT |
|
|
| SETBP1-e4F9L | 65.8 | 40.91 |
CTCTTCCAACCAAAACCCAAAA | 394 | 60.7; 62 |
| SETBP1-e4F9R | 65.4 | 45.45 |
CTTTTCCGTTTCCTCTTGTGCT |
|
|
| SETBP1-e4F9Rm | 61.2 | 40.91 |
CTTTTCCGTTTACTCTTGTGCT |
|
|
We included samples from patients with different
types of MPNs (PV, ET, PMF) and MDS/MPNs with and without the
p.V617F mutation in Janus kinase 2 (JAK2). Ninety-two
samples (34 ET, 37 PMF and 21 MDS/MPNs) were from patients without
p.V617F JAK2 mutation, while 50 samples (8 ET, 8 PV, 4 PMF
and 30 with an unspecified MPN type) harbored the p.V617F mutation.
Finally, we also analyzed samples from 25 different cancer cell
lines (Table II), most of them from
hematological malignancies. As a control group, we included samples
from 13 patients with B-ALL, the disease in which the p.E1099K
NSD2 mutation had been described (14). Informed consent was obtained from all
patients and procedures were approved by the Ethical Committee on
Clinical Research of University of Navarra.
| Table II.Human cancer cell lines tested. |
Table II.
Human cancer cell lines tested.
| Cell line name | Repository
(number) | Origin |
|---|
| A-549 | DSMZ (ACC-107) | Lung carcinoma |
| BK-006 | ECACC | p.V617F JAK2
positive PV |
| BK-013 | ECACC
(98100924) | p.V617F JAK2
negative ET |
| BK015 | ECACC
(99092421) | p.V617F JAK2
negative ET |
| DAUDI | DSMZ (ACC-78) | Burkitt
lymphoma |
| EOL-1 | DSMZ (ACC-386) | Acute myeloid
eosinophilic leukemia |
| F-36P | DSMZ (ACC-543) | Acute myeloid
leukemia secondary to myelodysplastic syndrome (MDS) |
| HCC-1937 | DSMZ (ACC-513) | Breast
carcinoma |
| HEL | DSMZ ACC-11 |
Erythroleukemia |
| HL60 | DSMZ ACC-3 | Acute myeloid
leukemia (AML) |
| HU3 | Dr Morgan, USA | Acute
megakaryoblastic leukemia |
| K-562 | DSMZ ACC-10 | BCR-ABL1
positive chronic myeloid leukemia (CML) in blast crisis |
| KARPAS-299 | DSMZ ACC-31 | T cell
lymphoma |
| KARPAS-422 | DSMZ ACC-32 | B cell
lymphoma |
| M-07e | DSMZ ACC-104 | Acute
megakaryoblastic leukemia |
| MG-63 | ATCC: CRL-1427 | Osteosarcoma |
| MOLM-13 | DSMZ ACC-554 | Acute myeloid
leukemia (AML) |
| MOLT-16 | DSMZ ACC-29 | T cell acute
lymphoid leukemia (T-ALL) |
| MOLT-4 | DSMZ ACC-362 | Acute lymphoid
leukemia (ALL) |
| MV-411 | DSMZ ACC-102 | Acute
monoblastic/monocytic leukemia |
| RAJI | DSMZ ACC-319 | Burkitt
lymphoma |
| REH | DSMZ ACC-22 | B-ALL (t(12;21)
(p13;q22) (fusion ETV6-RUNX1)) |
| SET2 | DSMZ ACC-608 | p.V617F JAK2
positive ET |
| TF-1 | DSMZ ACC-334 |
Erythroleukemia |
| UKE-1 | Dr W. Fiedelr,
Hamburg, Germany | p.V617F JAK2
positive ET transformed to AML |
Results
Most of the nucleotide changes that we detected were
SNPs already described (data not shown). No mis-sense changes were
found in NSD1 and NSD3 so they do not seem to be
frequently mutated in these diseases. With regard to NSD2,
we did not detect any mis-sense mutations in samples from MPN and
MDS/MPN patients, except for the previously described p.E1099K
mutation that we observed in one patient with B-precursor ALL.
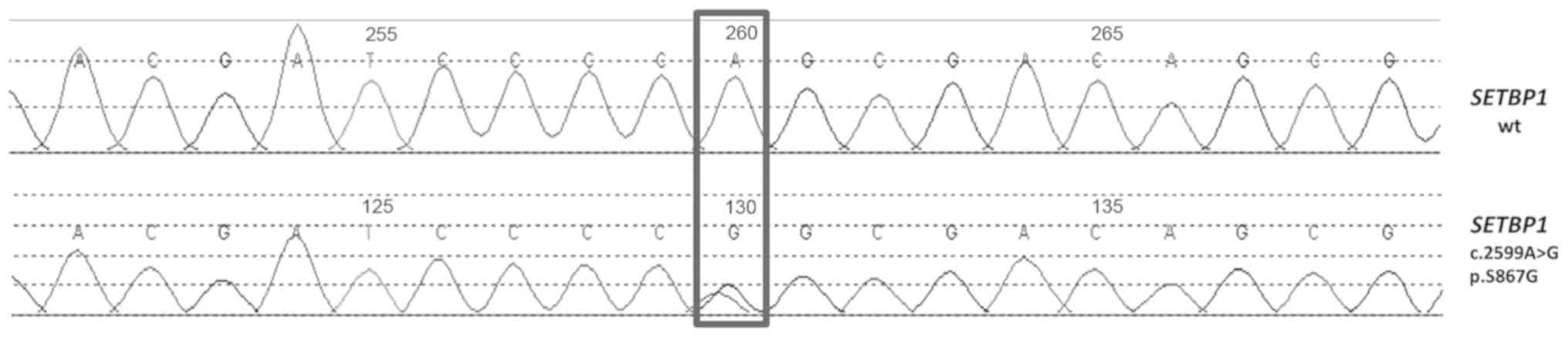
The analysis of SETBP1 found the mis-sense
mutation p.S867G (c.2599A>G according the first nucleotide of
the translation start codon of the CDS from NM_015559) (Fig. 1) in the sample from one patient with
a p.V617F JAK2 PMF This change is located in a residue that
is highly conserved between different species in the SKI homologous
region, the mutational hotspot described previously In
silico analysis by several bioinformatics tools like
PolyPhen-2, MutPred2, SIFT and FATHMM showed that it
is probably damaging (data not shown). No additional material was
available to determine the somatic status of this change. This
mutation has not been described as a polymorphism or a
cancer-related mutation in other databases. In fact, in COSMIC
(Catalogue of Somatic Mutations in Cancer, cancer.sanger.ac.uk/cosmic) there are other variants
affecting the same codon but not this specific nucleotide. These
mutations are p.S867N (c.2600G>A) a somatic change predicted as
pathogenic and found in an intestinal adenocarcinoma, p.S867R
(c.2601C>A) a non-confirmed somatic change predicted as neutral
and found in four samples from MDS and AML associated with MDS and
the silent p.S867S (c.2601C>T), a somatic change predicted also
as neutral and found in two samples from stomach and intestine with
adenocarcinoma but described as a SNP in dbSNP (rs376371660). This
is also the only change in ExAC database (Exome Aggregation
Consortium, http://exac.broadinstitute.org) affecting this codon,
found in 7 of 120746 alleles with a frequency of 0.00005797. This
is the only variant also described in 1000 Genomes Phase 3 database
(www.internationalgenome.org/category/phase-3)
with a frequency of 0.00039936 and in the ESP database (Exome
Sequencing Project, evs.gs.washington.edu) with a frequency of 0.00007688,
in both cases with only one allele T found. In gnomAD (Genome
Aggregation Database, gnomad.broadinstitute.org), that spans 125,748 exome
sequences and 15,708 whole-genome sequences from unrelated
individuals the mutation p.S867G is also not described.
Discussion
SETBP1 mutations can coexist with aberrations
in other genes frequently mutated in myeloid neoplasms such as
CBL, ASXL1, U2AF1 or RUNX1 (3,17,18) thus
cooperating in the progression of the disease. However, the
coexistence of mutations in SETBP1 and JAK2 in MPNs
has not been described to date. In the case of ASXL1 and
SETBP1 mutations, the former may initially inhibit cell
differentiation while mutations in SETBP1 would provide a
proliferative advantage (18).
Although a direct physical interaction between CBL and SETBP1 has
not been demonstrated, it seems plausible that both mutations could
cooperate indirectly by reducing cytokine dependence of leukemia
cells (17).
Furthermore, it is well established that mutations
in genes related to epigenetic regulation of gene expression can
coexist with mutations in JAK2 and could modulate the
progression of the disease (3,19). For
instance, TET2 encodes the Tet methylcytosine dioxygenase 2
which catalyzes the conversion of the 5-methylcytosine (5mC) into
5-hydroxymethylcytosine (5hmC) and plays a key role in active DNA
demethylation and it is frequently mutated in patients with MPNs
(20). TET2 mutations have
been found in 17% of patients with p.V617F JAK2 (20). Although both mutations are present in
different clones, data published to date show that TET2
somatic mutations might be associated with increased aggressiveness
and higher frequency of organomegaly in ET patients, but not in PV
or PMF patients (19). Although
SETBP1 mutations have not been associated with mutations in
JAK2 in MPNs to date, our finding suggests the possibility
of a similar interplay of mutational mechanisms.
Recently two cases have been reported with
Schinzel-Giedion syndrome (SGS, MIM 269150) and a milder phenotype
with the mutation at this residue p.S867R (21,22). SGS
is a rare dominant developmental disorder characterized by multiple
malformations including midface hypoplasia, cardiac defects,
hydronephrosis and skeletal abnormalities. It seems caused by
germline de novo mutations in SETBP1 in a hotspot of
12 bp coding for residues 868 to 871 of the protein, that overlaps
to the somatic mutations reported in myeloid malignancies and
support a gain-of-function effect. This dual role in cancer and
development is not new, there are other genes such as HRAS,
ASXL1, EZH2 and FGFR2 in which germline mutations cause
developmental disorders and the overlapping somatic mutations drive
cancer. The recurrently mutated region (868–871) in SETBP1 is
highly conserved and it has been identified as important to
initiate degradation by ubiquitination so the mutations could
increase protein stability (4,21,22). The
milder phenotype shown by both patients with SGS and the p.S867R
would point to a less activating gain-of-function of this mutant
protein.
The sample available in our case did not allow us to
determine whether SETBP1 and JAK2 mutations coexist
in the same clone. Lack of access to clinical data of this patient
prevents us from drawing definitive conclusions about the putative
effects of their presence in the progression of the disease.
SETBP1 mutations usually have poor prognosis and are
frequently acquired as secondary events in other myeloid neoplasms
(PMF can arise on its own or as a progression of PV or ET)
(18), so the p.S867G mutation in
SETBP1 could be relevant for the progression of the disease.
In addition, additional experiments would be required to
demonstrate the pathogenicity of the p.S867G mutation. These
experiments should be performed on cell lines that stably express
p.S867G SETBP1 and this mutation in combination with the
p.V617F JAK2 to test their possible cooperation for the
transformation of the disease.
In this study we show that NSD1, NSD2 and
NSD3 genes, that are paralogous to SETBP1 and encode
also epigenetic regulators, are not frequently mutated in MPNs in
the SETBP1 homologous region. However, we show that the
p.V617F JAK2 can coexist with mutations in SETBP1, a
gene mutated in some myeloid neoplasms. This fact adds support to
the notion that these diseases are genetically complex and shows
the need to identify additional oncogenic mechanisms that could
promote progression or phenotypic diversity, in order to design new
targeted therapies.
Acknowledgements
Not applicable.
Funding
The present study was funded by the PIUNA Program of
the University of Navarra. LE and DN have PhD Studentships granted
by the Ministry of Education, Culture and Sports of Spain (grant
nos. FPU14/03669 and FPU13/00424 respectively) within the
University Faculty Training (FPU) program.
Availability of data and materials
The datasets used and/or analyzed during this study
are available from the corresponding author on reasonable
request.
Authors' contributions
LEA, FJN and JLV designed the study. LEA and CH
performed the experiments. LEA, DNH, DC and JLV analyzed the data.
LEA, FJN and JLV wrote the manuscript. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all of
the patients and the procedures were approved by the Ethical
Committee on Clinical Research of University of Navarra (Pamplona,
Spain).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Murati A, Brecqueville M, Devillier R,
Mozziconacci MJ, Gelsi-Boyer V and Birnbaum D: Myeloid
malignancies: Mutations, models and management. BMC Cancer.
12:3042012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shih AH, Abdel-Wahab O, Patel JP and
Levine RL: The role of mutations in epigenetic regulators in
myeloid malignancies. Nat Rev Cancer. 12:599–612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McPherson S, McMullin MF and Mills K:
Epigenetics in myeloproliferative neoplasms. J Cell Mol Med.
21:1660–1667. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Piazza R, Valletta S, Winkelmann N,
Redaelli S, Spinelli R, Pirola A, Antolini L, Mologni L, Donadoni
C, Papaemmanuil E, et al: Recurrent SETBP1 mutations in atypical
chronic myeloid leukemia. Nat Genet. 45:18–24. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cristóbal I, Blanco FJ, Garcia-Orti L,
Marcotegui N, Vicente C, Rifon J, Novo FJ, Bandres E, Calasanz MJ,
Bernabeu C and Odero MD: SETBP1 overexpression is a novel
leukemogenic mechanism that predicts adverse outcome in elderly
patients with acute myeloid leukemia. Blood. 115:615–625. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oakley K, Han Y, Vishwakarma BA, Chu S,
Bhatia R, Gudmundsson KO, Keller J, Chen X, Vasko V, Jenkins NA, et
al: Setbp1 promotes the self-renewal of murine myeloid progenitors
via activation of Hoxa9 and Hoxa10. Blood. 119:6099–6108. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Trimarchi T, Ntziachristos P and Aifantis
I: A new player SETs in myeloid malignancy. Nat Genet. 45:846–847.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Akiki S, Dyer SA, Grimwade D, Ivey A,
Abou-Zeid N, Borrow J, Jeffries S, Caddick J, Newell H, Begum S, et
al: NUP98-NSD1 fusion in association with FLT3-ITD mutation
identifies a prognostically relevant subgroup of pediatric acute
myeloid leukemia patients suitable for monitoring by real time
quantitative PCR. Genes Chromosomes Cancer. 52:1053–1064. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hollink IH, van den Heuvel-Eibrink MM,
Arentsen-Peters ST, Pratcorona M, Abbas S, Kuipers JE, van Galen
JF, Beverloo HB, Sonneveld E, Kaspers GJ, et al: NUP98/NSD1
characterizes a novel poor prognostic group in acute myeloid
leukemia with a distinct HOX gene expression pattern. Blood.
118:3645–3656. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shiba N, Ichikawa H, Taki T, Park MJ, Jo
A, Mitani S, Kobayashi T, Shimada A, Sotomatsu M, Arakawa H, et al:
NUP98-NSD1 gene fusion and its related gene expression signature
are strongly associated with a poor prognosis in pediatric acute
myeloid leukemia. Genes Chromosomes Cancer. 52:683–693.
2013.PubMed/NCBI
|
|
11
|
Rosati R, La Starza R, Veronese A, Aventin
A, Schwienbacher C, Vallespi T, Negrini M, Martelli MF and Mecucci
C: NUP98 is fused to the NSD3 gene in acute myeloid leukemia
associated with t(8;11)(p11.2;p15). Blood. 99:3857–3860. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Taketani T, Taki T, Nakamura H, Taniwaki
M, Masuda J and Hayashi Y: NUP98-NSD3 fusion gene in
radiation-associated myelodysplastic syndrome with t(8;11)(p11;p15)
and expression pattern of NSD family genes. Cancer Genet Cytogenet.
190:108–112. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chesi M, Nardini E, Lim RS, Smith KD,
Kuehl WM and Bergsagel PL: The t(4;14) translocation in myeloma
dysregulates both FGFR3 and a novel gene, MMSET, resulting in
IgH/MMSET hybrid transcripts. Blood. 92:3025–3034. 1998.PubMed/NCBI
|
|
14
|
Jaffe JD, Wang Y, Chan HM, Zhang J,
Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP, et
al: Global chromatin profiling reveals NSD2 mutations in pediatric
acute lymphoblastic leukemia. Nat Genet. 45:1386–1391. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gelsi-Boyer V, Trouplin V, Adélaïde J,
Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M,
Sainty D, et al: Mutations of polycomb-associated gene ASXL1 in
myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br
J Haematol. 6:788–800. 2009. View Article : Google Scholar
|
|
16
|
Abdel-Wahab O, Tefferi A and Levine RL:
Role of TET2 and ASXL1 mutations in the pathogenesis of
myeloproliferative neoplasms. Hematol Oncol Clin North Am.
5:1053–1064. 2012. View Article : Google Scholar
|
|
17
|
Makishima H, Yoshida K, Nguyen N,
Przychodzen B, Sanada M, Okuno Y, Ng KP, Gudmundsson KO,
Vishwakarma BA, Jerez A, et al: Somatic SETBP1 mutations in myeloid
malignancies. Nat Genet. 45:942–946. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Makishima H: Somatic SETBP1 mutations in
myeloid neoplasms. Int J Hematol. 105:732–742. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saeidi K: Myeloproliferative neoplasms:
Current molecular biology and genetics. Crit Rev Oncol Hematol.
98:375–389. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tefferi A, Pardanani A, Lim KH,
Abdel-Wahab O, Lasho TL, Patel J, Gangat N, Finke CM, Schwager S,
Mullally A, et al: TET2 mutations and their clinical correlates in
polycythemia vera, essential thrombocythemia and myelofibrosis.
Leukemia. 23:905–911. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carvalho E, Honjo R, Magalhães M, Yamamoto
G, Rocha K, Naslavsky M, Zatz M, Passos-Bueno MR, Kim C and Bertola
D: Schinzel-Giedion syndrome in two Brazilian patients: Report of a
novel mutation in SETBP1 and literature review of the clinical
features. Am J Med Genet 167A. 1039–1046. 2015. View Article : Google Scholar
|
|
22
|
Acuna-Hidalgo R, Deriziotis P, Steehouwer
M, Gilissen C, Graham SA, van Dam S, Hoover-Fong J, Telegrafi AB,
Destree A, Smigiel R, et al: Overlapping SETBP1 gain-of-function
mutations in Schinzel-Giedion syndrome and hematologic
malignancies. PLoS Genet. 13:e10066832017. View Article : Google Scholar : PubMed/NCBI
|