Introduction
Colorectal cancer (CRC) is the third most common
cancer in Western countries in terms of incidence and mortality.
With a trend of increasing incidence, it constitutes 8% of all new
diagnoses of cancer in adulthood in Europe, placing itself in third
place for men and second place for women (1). The disease is diagnosed in 80% of cases
at an early stage, susceptible to curative surgery and possible
adjuvant chemotherapy (CT), with a 5-year recurrence rate that
exceeds 35%. In the remaining 20% of cases, the disease is
diagnosed at an advanced stage [metastatic CRC (mCRC)], with a
trend in overall survival (OS) that is closely dependent on the
molecular characterization and the resection of metastatic sites
(1).
Over the last 15 years, mCRC has been associated
with an increase in survival rates, reaching an average of 31
months courtesy of the introduction of doublets/triplets of CT in
combination with monoclonal antibodies (mAbs) directed against the
vascular endothelial growth factor (VEGF) and the epidermal growth
factor receptor (EGFR). Although biomarkers to select patients
responding to anti-VEGF mAbs are not yet available, mutations in
exons 2, 3 and 4 of the Kirsten rat sarcoma 2 viral oncogene
homolog (KRAS) and neuroblastoma RAS viral oncogene homolog (NRAS)
genes are predictive of resistance to anti-EGFR mAbs, whereas the
Val600èGln (V600E) mutation of the B-Raf proto-oncogene (BRAF) has
a strong prognostic role, predicting resistance to standard CT
(2–6).
Therefore, mutations in the RAS and BRAF genes may
identify subgroups of tumors with specific biological, pathological
and clinical features, a phenomenon referred to as ‘inter-tumor
heterogeneity’. However, CRC is a complex disease, characterized by
a number of genetic alterations that may also co-exist in the same
tumor. In particular, it has been suggested that CRC is likely to
be formed by different clones of cancer cells with distinct
genotypic profiles. This intratumor heterogeneity is likely to
markedly affect the progression of the disease, as well as the
sensitivity and resistance to therapies. Notably, recent studies
have also suggested that treatment of CRC patients with anti-EGFR
agents produces an increase in intratumor heterogeneity, leading to
the emergence of clones with different genetic alterations
(7).
An additional important consequence of intratumor
heterogeneity is the possible difference in genetic alterations
between primary tumors (PTs) and metastases (8). Although a mutational concordance of
KRAS, NRAS and BRAF between the PT and metastases in excess of 95%
has been highlighted in different cases, this agreement begins to
falter with the introduction of high-throughput genotyping
techniques, such as targeted resequencing applications of
next-generation sequencing (NGS) (9–11).
Focusing on the key role of the heterogeneity on the
development and spread of the tumor, the authors of the present
study speculated on whether the PT tissue of CRC should continue to
offer a trustworthy overview of metastatic tissue, how the de
novo resistance may impact on treatments with anti-EGFR mAbs,
and the nature of the role of the secondary resistance on the
progress of the disease.
Recent data have suggested that acquired resistance
to anti-EGFR mAbs is driven by a number of molecular alterations,
including mutations in KRAS, NRAS, BRAF, and other driver genes
(12,13). Furthermore, the higher sensitivity of
NGS may permit the identification of mutations in RAS not
identified by the standard Sanger sequencing technique, as
highlighted by the analysis conducted in a subpopulation of the
CAPRI-GOIM multicenter study (13,14). In
this regard, it was also suggested that low levels of KRAS
mutations could justify an intrinsic resistance mechanism to
anti-EGFR mAbs (15,16).
In the present study, the genetic profile of the
colorectal PT prior to and after CT, and of resected liver
metastases removed post-CT in association with cetuximab, was
assessed in order to investigate the genetic heterogeneity and the
intrinsic and acquired resistance mechanisms in patients with
mCRC.
Patients and methods
Study design and patient
population
The working hypothesis adopted for the present study
was to investigate the impact of intra- and inter-tumoral molecular
heterogeneity between colorectal PTs and metastatic sites prior to
and after treatment with cetuximab, in combination with doublet
(folinic acid, fluorouracil and irinotecan, or FOLFIRI) or triplet
(folinic acid, 5-fluorouracil, oxaliplatin and irinotecan, or
FOLFOXIRI) CT in KRAS exon 2 wild-type chemo-naive patients with
synchronous potentially resectable liver metastases.
Seven cases of patients with wild-type KRAS exon 2
mCRC were evaluated at the Oncology Medical Unit of St.
Orsola-Malpighi Hospital, Bologna, Italy, between June 2010 and
February 2014 for a total of 54 analyzed lesions. The selected time
period justifies the population selected only for the absence of
mutations in KRAS exon 2.
All the patients provided their informed consent for
the treatment of their genetic material for research purposes, and
the present study was approved by the Ethical Committee of the S.
Orsola-Malpighi Hospital.
Two patients (patient nos. 2 and 5) had undergone
two-stage hepatectomy surgery interspersed with CT in combination
with cetuximab, whereas others underwent surgery only after
conversion treatment. The average number of treatment cycles
carried out prior to the surgical resection was agreed for each
individual case during the multidisciplinary meeting of the study,
and for the treatment of liver metastases prior to
clinical-instrumental re-evaluation, and was set equal to seven
(range, 6–8 cycles).
Gene mutation analysis by NGS
NGS analysis was performed using genomic DNA
extracted either from PT tissue obtained prior to systemic
treatment or on liver metastases following either liver resection
or biopsy. Formalin-fixed, paraffin-embedded (FFPE), circled
tumor-rich (>70%) areas (10-µm thick) were scraped off the
slides using a sterile scalpel by manual microdissection,
deparaffinized in xylene, and DNA was isolated using the GeneRead
DNA FFPE kit (Qiagen GmbH, Hilden, Germany), according to the
manufacturer's protocol. DNA quantification was performed using a
Quantifiler® Human DNA Quantification kit (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). NGS was performed using the
Ion System Personal Genome Machine (PGM) system (Life Technologies;
ThermoFisher Scientific, Inc.), with all reagents supplied by
Thermo Fisher Scientific, Inc. An amplicon library was produced
from 10 ng DNA from each sample using the Ion AmpliSeq™ Colon and
Lung Research panel v2 (Thermo Fisher Scientific, Inc.), which
generates 92 amplicons for analyses of ‘hotspot’ and targeted
regions of 22 genes implicated in colon and lung cancers [namely,
KRAS, EGFR, BRAF, p110α catalytic subunit of phosphoinositide
3-kinase (PIK3CA), serine/threonine kinase 1 (AKT1), erb-b2
receptor tyrosine kinase (ERBB)2, phosphatase and tensin homolog
(PTEN), NRAS, serine/threonine kinase 11 (STK11), mitogen-activated
protein kinase kinase 1 (MAP2K1), anaplastic lymphoma kinase (ALK),
discoidin domain-containing receptor 2 (DDR2), catenin β1 (CTNNB1),
MET proto-oncogene, tumor protein p53 (TP53), SMAD4, F-box/WD
repeat-containing protein 7 (FBXW7), fibroblast growth factor
receptor (FGFR)3, NOTCH1, ERBB4, FGFR1 and FGFR2].
The manufacturer's protocols were followed without
modification. Briefly, after amplification of the target sequences,
primer digestion was performed, and barcode adapters (Ion Xpress
Barcode Adapters; Thermo Fisher Scientific, Inc.) were ligated to
the amplicons. Amplicons were purified using Agencourt AMPure XP
(Beckman Coulter, Brea, CA, USA). Library quantification was
subsequently performed using the Ion Library TaqMan™ Quantitation
kit (Thermo Fisher Scientific, Inc.). The library was diluted in
nuclease-free water to obtain a final concentration of 8 pM.
Emulsion polymerase chain reaction (PCR) was performed using an Ion
PGM™ Template OT2 200 kit on the Ion OneTouch™ 2 instrument (Thermo
Fisher Scientific, Inc.). The quality of the DNA following PCR was
measured using the Qubit Ion Sphere™ Quality Control kit (Thermo
Fisher Scientific, Inc.). Selective ion spheres with DNA were
isolated (Ion PGM™ Enrichment Beads on an Ion OneTouch™ ES
instrument), and sequenced on an Ion 316™ Chip kit v2 (5
samples/chip) or an Ion 318™ Chip kit v2 (10 samples/chip) using
the Ion PGM™ Sequencing 200 kit v2 (Thermo Fisher Scientific,
Inc.). Successful sequencing of a sample required at least 500,000
reads with a quality score ≥ Q20.
As tumor specimens were admixed with normal tissue,
a minimum coverage of 500X with at least 10% frequency was used as
cutoff for a variant to be considered true.
Sequence alignment and base calling was performed
using Torrent Suite software v.4.4.3 (Thermo Fisher Scientific,
Inc.), with Human Genome Build 19 (hg19) as the reference. Variant
calling was performed with the Variant Caller v.4.4.3.3 plug-in
using default ‘Somatic-Low Stringency’ settings. Variants were
further filtered using Ion Reporter software v.4.4 (Thermo Fisher
Scientific, Inc.) to meet the following criteria: Non-synonymous
coding, an allele frequency ≥10%, a total amplicon coverage of
≥500, each variant coverage >20, a Phred-based quality score of
≥30, and P<0.001. This software also included the ClinVar, dpSNP
(National Center for Biotechnology Information, Bethesda, MD, USA)
and COSMIC (Wellcome Trust Sanger Institute, Cambridge, UK)
databases, and, for missense mutations of unknown significance,
in silico prediction tools, including SIFT (Sorting
Intolerant From Tolerant), PolyPhen (Polymorphism Phenotyping),
PhyloP and Grantham.
The Integrative Genomics Viewer (IGV; Broad
Institute, Cambridge, MA, USA) was used to visualize variants.
Additionally, several of the detected missense mutations were
confirmed using Sanger's sequencing.
Results
After the initial treatment, an objective partial
response was achieved in all 7 patients (according to RECIST
Criteria v.1.1), which allowed the previously planned surgical
resection to be performed for all our patients.
Following the resection of the liver metastases,
patients had a liver recurrence rate of 71%; two patients (28%;
patient nos. 3 and 6) were subjected to further liver surgery, with
an expected benefit in terms of progression free survival (PFS; 25
and 9 months, respectively) and OS (40 and 37 months,
respectively). Three out of seven patients (42%) were subjected to
a second line of treatment, containing an anti-VEGF antibody; two
out of seven (28%) patients were subjected to a third line of
treatment, and 14% (one patient) was subjected to successive lines
of treatment.
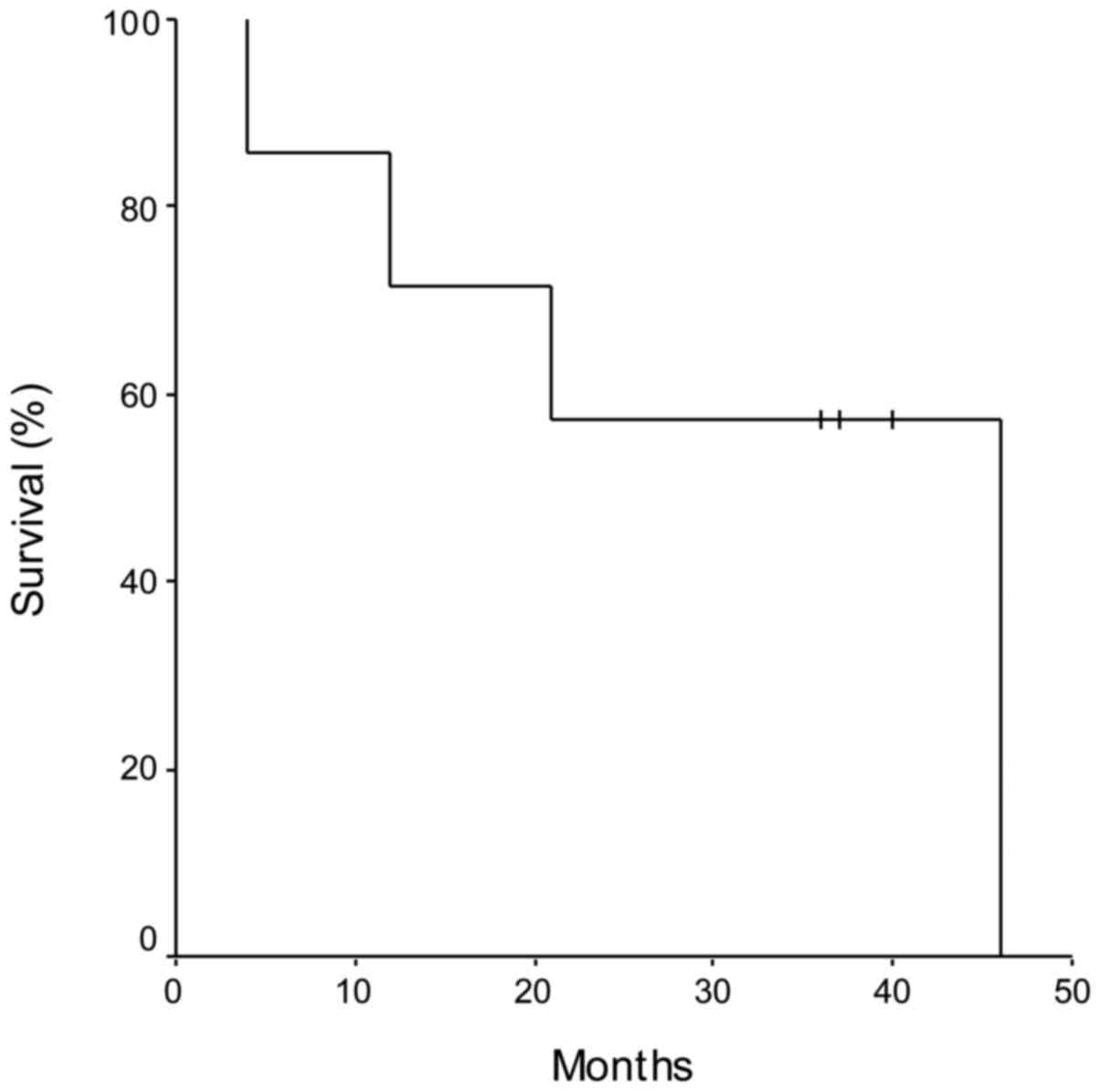
Using Kaplan-Meier analysis, the PFS, calculated as
the time from liver resection and the disease progression, was 11
months (range, 3–21 months), whereas the OS was 31 months (range,
4–46 months) (Fig. 1). Patients who
demonstrated a significant advantage in OS were those subjected to
further liver surgery (patient nos. 3 and 6, equal to 40 and 37
months, respectively), and to subsequent lines of CT, confirming
the importance of a multidisciplinary approach to the treatment of
mCRC (Table I).
 | Table I.Disease progression and mutation
percentages, comparing PTs with srLm. |
Table I.
Disease progression and mutation
percentages, comparing PTs with srLm.
| Patient no. | PFS (months) | OS (months) | Number of CT lines
subsequent to the first | Number of liver
resections after the first | PT (%) mutated genes
prior to CT | PT (%) mutated genes
after CT | srLm (%) mutated
genes after CT |
|---|
| 1 | 14 | 46 | 4 | 1 | 9 | 14 | 9 |
| 2 | 3 | 4 | 0 | 1 | 9 | – | 9 |
| 3 | 9 | 40 | 1 | 1 | 0 | 0 | 0 |
| 4 | 14 | 36 | 2 | 0 | 4 | – | 4 |
| 5 | 9 | 21 | 2 | 1 | 4 | – | 9 |
| 6 | 21 | 37 | 2 | 1 | 4 | – | 4 |
| 7 | 8 | 12 | 2 | 0 | 14 | 14 | 14 |
The NGS analysis of pre- and post-treatment
available tissues revealed 50 mutations in 8 genes: KRAS (22%),
NRAS (8%), PIK3CA (8%), MET (4%), FBXW7 (6%), PTEN (2%), SMAD4 (8%)
and TP53 (42%) (Table II).
| Table II.Genomic analysis performed on 54
lesions revealed 50 mutations in 8 genes. |
Table II.
Genomic analysis performed on 54
lesions revealed 50 mutations in 8 genes.
| Patient no. | PT (biopsy or
specimen) mutations detected prior to CT | % mutations detected
by NGS | PT specimen mutations
detected following CT | % mutations detected
by NGS | srLm mutations
detected following CT | % mutations detected
by NGS |
|---|
| 1 | FBXW7 (ex.8):
p.R465C/c.1393C>T | 25.1 | FBXW7 (ex.8):
p.R465C/c.1393C>T | 25.9 | 1a. KRAS (ex.2):
p.G12D/c.35G>A | 14.2 |
|
| KRAS (ex.2):
p.K5N/c.15A>T | 17.1 | KRAS (ex.2):
p.K5N/c.15A>T | 13.8 | 2a. PIK3CA (ex.10):
p.D538G/c.1613A>G | 17.0 |
|
|
|
| PTEN (ex.7):
p.K267R/c.795delA | 23.1 | 3a. KRAS (ex.2):
p.G12D/c.35G>A | 28.4 |
|
|
|
|
|
| 4a. KRAS (ex.2):
p.G12D/c.35G>A | 20.0 |
|
|
|
|
|
| 5a. KRAS (ex.2):
p.G12D/c.35G>A | 30.5 |
|
|
|
|
|
| 6a. KRAS (ex.2):
p.G12D/c.35G>A | 27.5 |
|
|
|
|
|
| 7a. KRAS (ex.2):
p.G12D/c.35G>A | 16.6 |
|
|
|
|
|
| PIK3CA (ex.20):
p.H1047R/c.3140A>G | 7.6 |
|
|
|
|
|
| 8a. KRAS (ex.2):
p.G12D/c.35G>A | 27.5 |
|
|
|
|
|
| 9a. KRAS (ex.2):
p.G12D/c.35G>A | 4.9 |
|
|
|
|
|
| PIK3CA (ex.20):
p.H1047R/c.3140A>G | 6.4 |
| 2 | TP53 (ex.7):
p.R248W/c.742C>T | 52.2 | Not available |
| 1a. TP53 (ex.7):
p.R248W/c.742C>T | 87.7 |
|
| SMAD4 (ex.5): p.D351V
c.1052A>T | 46.4 |
|
| SMAD4 (ex.5): p.D351V
c.1052A>T | 82.6 |
|
|
|
|
|
| 2a. TP53 (ex.7):
p.R248W/c.742C>T | 82.1 |
|
|
|
|
|
| SMAD4 (ex.5):
p.D351V c.1052A>T | 80.2 |
| 3 | Any mutations |
| Any mutations |
| 1a. any mutations |
|
|
|
|
|
|
| 2a. any mutations |
|
|
|
|
|
|
| 3a. any mutations |
|
|
|
|
|
|
| 4a. any mutations |
|
|
|
|
|
|
| 5a. any mutations |
|
|
|
|
|
|
| 6a. any mutations |
|
|
|
|
|
|
| 7a. any mutations |
|
|
|
|
|
|
| 8a. any
mutations |
|
|
|
|
|
|
| 9a. any mutations |
|
| 4 | TP53 (ex.8):
p.R306a/c.916C>T | 64.3 | Not available |
| 1a. TP53 (ex.8): p.R306a/c.916C>T | 45.1 |
|
|
|
|
|
| 2a. TP53 (ex.8): p.R306a/c.916C>T | 35.2 |
|
|
|
|
|
| 3a. TP53 (ex.8): p.R306a/c.916C>T | 71.6 |
|
|
|
|
|
| 4a. TP53 (ex.8): p.R306a/c.916C>T | 48.4 |
|
|
|
|
|
| 5a. TP53 (ex.8): p.R306a/c.916C>T | 17.9 |
|
|
|
|
|
| 6a. TP53 (ex.8): p.R306a/c.916C>T | 11.1 |
|
|
|
|
|
| 7a. TP53 (ex.8): p.R306a/c.916C>T | 59.6 |
|
|
|
|
|
| 8a. TP53 (ex.8): p.R306a/c.916C>T | 37.1 |
| 5 | NRAS (ex.2):
p.G12V/c.35G>T | 30.5 | Not available |
| 1a. NRAS (ex.2):
p.G12V/c.35G>T | 38.1 |
|
|
|
|
|
| 2a. any mutations |
|
|
|
|
|
|
| 3a. PIK3CA (ex.20):
p.H1047R/c.3140A>G | 39.4 |
|
|
|
|
|
| 4a.any mutations |
|
|
|
|
|
|
| 5a. any mutations |
|
| 6 | TP53 (ex.6):
p.S215I/c.644G>T | 46.9 | Not available |
| 1a. TP53
(ex.6):p.S215I/c.644G>T | 55.4 |
|
|
|
|
|
| 2a.TP53 (ex.6):
p.S215I/c.644G>T | 63.7 |
|
|
|
|
|
| 3a. TP53 (ex.6):
p.S215I/c.644G>T | 15.0 |
|
|
|
|
|
| 4a. TP53 (ex.6):
p.S215I/c.644G>T | 30.2 |
|
|
|
|
|
| 5a. TP53 (ex.6):
p.S215I/c.644G>T | 58.7 |
| 7 | NRAS (ex.3):
p.Q61L/c.182A>T | 37 | NRAS (ex.3):
p.Q61L/c.182A>T | 10.9 | 1a. any mutations |
|
|
| MET (ex.2):
p.E168D/c.504G>T | 38.8 | MET (ex.2):
p.E168D/c.504G>T | 49.5 | 2a. NRAS (ex.3):
p.Q61L/c.182A>T | 65.0 |
|
|
|
|
|
| MET (ex.2):
p.E168D/c.504G>T | 28.7 |
|
| TP53 (ex.8):
p.V274F/c.820G>T | 26.4 | TP53 (ex.8):
p.V274F/c.820G>T | 4.1 | TP53 (ex.8):
p.V274F/c.820G>T | 65.3 |
|
| TP53 (ex.5):
p.R175H/c.524G>A | 7.7 |
|
|
|
|
Five of the 7 treated patients were of the RAS
wild-type. Two had NRAS mutations that were retrospectively
assessed, since the label of the drug allowed treatment of patients
with KRAS exon 2 wild-type tumor at the time of the treatment.
Mutations in TP53, SMAD4, FBXW7 and MET were also present in the PT
from four of the seven patients prior to treatment.
As shown in Table
III, the treatment produced marked changes in the mutational
profile in 3 of the 7 patients, namely patient nos. 1, 5, and
7.
| Table III.KRAS (K), NRAS (N), and PIK3CA (P)
discrepancies between the PT and srLm. |
Table III.
KRAS (K), NRAS (N), and PIK3CA (P)
discrepancies between the PT and srLm.
|
|
| Patient 1 |
|
| Patient 2 |
|
| Patient 3 |
|
| Patient 4 |
|
| Patient 5 |
|
| Patient 6 |
|
| Patient 7 |
|
|---|
| CT regimen |
| Folfiri/cet |
|
| Folfiri/cet |
|
| Folfiri/cet |
|
| Folfoxiri/cet |
|
| Folfoxiri/cet |
|
| Folfoxiri/cet |
|
| Folfiri/cet |
|
| PFS (months) |
| 14 |
|
| 3 |
|
| 9 |
|
| 14 |
|
| 9 |
|
| 21 |
|
| 8 |
|
| OS (months) |
| 46 |
|
| 4 |
|
| 40 |
|
| 36 |
|
| 20 |
|
| 37 |
|
| 12 |
|
| NGS profile | K | N | P | K | N | P | K | N | P | K | N | P | K | N | P | K | N | P | K | N | P |
| PT pre-CT | − | − | − | − | − | − | − | − | − | − | − | − | − | + | − | − | − | − | − | + | − |
| PT post-CT | + | − | − | n.a. | n.a. | n.a | − | − | − | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | − | + | − |
| srLmCRC no. 1 | + | − | − | − | − | − | − | − | − | − | − | − | − | + | − | − | − | − | − | − | − |
| srLmCRC no. 2 | − | − | + | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | + | − |
| srLmCRC no. 3 | + | − | − |
|
|
| − | − | − | − | − | − | − | − | + | − | − | − |
|
|
|
| srLmCRC no. 4 | + | − | − |
|
|
| − | − | − | − | − | − | − | − | − | − | − | − |
|
|
|
| srLmCRC no. 5 | + | − | − |
|
|
| − | − | − | − | − | − | − | − | − | − | − | − |
|
|
|
| srLmCRC no. 6 | + | − | − |
|
|
| − | − | − | − | − | − |
|
|
|
|
|
|
|
|
|
| srLmCRC no. 7 | + | − | + |
|
|
| − | − | − | − | − | − |
|
|
|
|
|
|
|
|
|
| srLmCRC no. 8 | + | − | − |
|
|
| − | − | − | − | − | − |
|
|
|
|
|
|
|
|
|
| srLmCRC no. 9 | + | − | + |
|
|
| − | − | − |
|
|
|
|
|
|
|
|
|
|
|
|
Patient no.1 demonstrated genetic heterogeneity of
the PT prior to and after CT, with the appearance following CT plus
cetuximab of exon 2 KRAS (p.K5N/c.15A>T) and exon 7 PTEN
(p.K267R/c.795delA) mutations, and a marked increase in a mucinous
pattern. In addition, the exon 2 KRAS mutation was identified in 7
of 9 resected liver metastases. In 3 of 9 metastases, different
PIK3CA mutations were also detected (exon 10: p.D538
G/c.1613A>G; exon 20: p.H1047R/c.3140A>G). Notably, the exon
8 FBXW7 (p.R465C/c.1393C>T) mutation present in the PT prior to
and after CT was not detected in the liver metastases.
Patient no. 5 had a mutation in NRAS exon 2 (p.G12
V/c.35G>T) in the PT prior to and after treatment and in two of
the five resected metastases, whereas the exon 20 PIK3CA variant
(p.H1047R/c.3140A>G) was observed in one single metastasis.
Patient no. 7 had mutations in NRAS exon 3
(p.Q61L/c.182A>T), TP53 exon 8 (p.V274F/c.820G>T) and MET
exon 2 (p.E168D/c.504G>T) in the PT prior to and after
treatment, whereas the identical mutations were present in only one
of the two resected liver metastases. An additional TP53 exon 5
(p.R175H/c.524G>A) mutation was observed only in the PT prior to
CT.
Discussion
NGS is a powerful technique that allows the study of
multiple biomarkers in a single analysis. By applying this
technique to tissue specimens obtained prior and after CT, it has
been possible to follow the molecular evolution of the disease in a
small cohort of patients.
In agreement with previous studies, significant
changes in KRAS mutation status were identified in one patient
(patient no. 1). Indeed, several reports have revealed that RAS
wild-type patients might develop RAS mutations following exposure
to anti-EGFR mAbs (15). However, in
the present study, loss of the FBXW7 mutation and the gain of a
PIK3CA mutation in liver metastases were also identified. Changes
in PIK3CA mutation status during treatment with anti-EGFR drugs
have previously been described, although a clear correlation with
resistance to anti-EGFR mAbs was not identified (17). Mutations in FBXW7 have been recently
reported as potential markers of resistance to anti-EGFR mAbs
(18). However, the disappearance of
the FBXW7 mutation from cells following response to cetuximab-based
therapy argues against this hypothesis. Such differences could also
be due to a relatively lower ability of cells with the FBXW7
mutation to establish distant metastases in the liver. Another
noteworthy observation is the switch to a mucinous pattern that was
not observed prior to therapy. Such a histological change may
represent a novel pathway of resistance to anti-EGFR mAbs.
In two distinct patients, NRAS mutations that were
evident prior to treatment were not detected in all, or in
selected, liver metastases. Again, the lack of liver tissue prior
to treatment in the present study prevents the conclusion from
being drawn that changes in the mutational pattern have occurred
after therapy, rather than representing a different ability of
tumor clones to establish liver metastases. Nevertheless, these
findings highlight how the dynamics of clonal evolution between the
PT and distant localizations of the disease are highly complex.
No mutations of the extracellular domain of the EGFR
were identified in this small cohort of patients after exposure to
cetuximab. Different studies have suggested that development of
these mutations is one of the main mechanisms of acquired, but not
intrinsic, resistance to cetuximab (19–21). It
must be emphasized that the majority of the available data have
been obtained with liquid biopsy, which still requires further
validation (22).
In conclusion, the present study has highlighted
marked differences between pre- and post-treatment biopsies from
patients with CRC. These changes are likely to be due to the
selection of sub-clones by therapy, thus suggesting a high level of
intratumor heterogeneity that could markedly affect the response to
therapy.
References
|
1
|
Arnold M, Sierra MS, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global patterns and trends in
colorectal cancer incidence and mortality. Gut. 66:683–691. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prenen H, Tejpar S and Van Cutsem E:
Impact of molecular markers on treatment selection in advanced
colorectal cancer. Eur J Cancer. 45 suppl 1:S70–S78. 2009.
View Article : Google Scholar
|
|
3
|
Bokemeyer C, van Cutsem E, Rougier P,
Ciardiello F, Heeger S, Schlichting M, Celik I and Köhne CH:
Addition of cetuximab to chemotherapy as first-line treatment for
KRAS wild-type metastatic colorectal cancer: Pooled analysis of the
CRYSTAL and OPUS randomised clinical trials. Eur J Cancer.
48:1466–1475. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Douillard JY, Oliner KS, Siena S,
Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham
D, Jassem J, et al: Panitumumab-FOLFOX4 treatment and RAS mutations
in colorectal cancer. N Engl J Med. 369:1023–1034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stintzing S, Jung A, Rosssius L and
Heinemann V: Analysis of KRAS/NRAS and BRAF mutations in FIRE-3: A
randomized phase III study of Folfiri plus cetuximab or bevacizumab
as first-line treatment for wild-type (WT) KRAS (exon 2) metastatic
colorectal cancer (mCRC) patients. Eur J Cancer. 49 suppl
3:Abstract 17. 2013.
|
|
6
|
Schwartzberg LS, Rivera F, Karthaus M,
Fasola G, Canon JL, Yu H, Oliner KS and Go WY: Analysis of
KRAS/NRAS mutations in PEAK: A randomized phase II study of FOLFOX6
plus panitumumab (pmab) or bevacizumab (bev) as first-line
treatment (tx) for wild-type (WT) KRAS (exon 2) metastatic
colorectal cancer (mCRC). J Clin Oncol. 13 suppl 3:abstr 3631.
2013.
|
|
7
|
de Roock W, De Vriendt V, Normanno N,
Ciardiello F and Tejpar S: KRAS, BRAF, PIK3CA, and PTEN mutations:
Implications for targeted therapies in metastatic colorectal
cancer. Lancet Oncol. 12:594–603. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gerlinger M, Rowan AJ, Horswell S, Larkin
J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A,
Tarpey P, et al: Intratumor heterogeneity and branched evolution
revealed by multiregion sequencing. N Engl J Med. 366:883–892.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kopetz S, Overman MJ, Chen K,
Lucio-Eterovic AK, Kee BK, Fogelman DR, Dasari A, Raghav KPS,
Sanchez EV, Phillips J, et al: Mutation and copy number discordance
in primary versus metastatic colorectal cancer (mCRC). J Clin
Oncol. 32 Suppl: abstr 3509:5S2014.
|
|
10
|
Adua D, Altimari A, Gruppioni E, Ercolani
G, Llimpe FLR, Fabio FD, Fiorentino M, Pinna AD, Pinto C; Medical
Oncology Unit, ; S. Orsola-Malpighi Hospital, Bologna, Italy, ; et
al: Molecular evaluation of primary tumor (PT) and synchronous
liver metastasis in colorectal cancer (srLmCRC) patients after
cetuximab-based chemotherapy. J Clin Oncol. 32 Suppl:abstr 3624.
5S2014.
|
|
11
|
Mao C, Wu XY, Yang ZY, Threapleton DE,
Yuan JQ, Yu YY and Tang JL: Concordant analysis of KRAS BRAF,
PIK3CA mutations, and PTEN expression between primary colorectal
cancer and matched metastases. Sci Rep. 5:80652015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sartore-Bianchi A, Di Nicolantonio F,
Nichelatti M, Molinari F, De Dosso S, Saletti P, Martini M, Cipani
T, Marrapese G, Mazzucchelli L, et al: Multi-determinants analysis
of molecular alterations for predicting clinical benefit to
EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One.
4:e72872009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Normanno N, Rachiglio AM, Lambiase M,
Martinelli E, Fenizia F, Esposito C, Roma C, Troiani T, Rizzi D,
Tatangelo F, et al: Heterogeneity of KRAS NRAS, BRAF and PIK3CA
mutations in metastatic colorectal cancer and potential effects on
therapy in the CAPRI GOIM trial. Ann Oncol. 26:1710–174. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ciardiello F, Normanno N, Maiello E,
Martinelli E, Troiani T, Pisconti S, Giuliani F, Barone C, Cartenì
G, Rachiglio AM, et al: Clinical activity of FOLFIRI plus cetuximab
according to extended gene mutation status by next-generation
sequencing: Findings from the CAPRI-GOIM trial. Ann Oncol.
25:1756–1761. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ciardiello F and Normanno N: HER2
signaling and resistance to the anti-EGFR monoclonal antibody
cetuximab: A further step toward personalized medicine for patients
with colorectal cancer. Cancer Discov. 1:472–474. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Misale S, Di Nicolantonio F,
Sartore-Bianchi A, Siena S and Bardelli A: Resistance to anti-EGFR
therapy in colorectal cancer: From heterogeneity to convergent
evolution. Cancer Discov. 4:1269–1280. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang ZY, Wu XY, Huang YF, Di MY, Zheng DY,
Chen JZ, Ding H, Mao C and Tang JL: Promising biomarkers for
predicting the outcomes of patients with KRAS wild-type metastatic
colorectal cancer treated with anti-epidermal growth factor
receptor monoclonal antibodies: A systematic review with
meta-analysis. Int J Cancer. 133:1914–1925. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guinney J, Ferté C, Dry J, McEwen R,
Manceau G, Kao KJ, Chang KM, Bendtsen C, Hudson K, Huang E, et al:
Modeling RAS phenotype in colorectal cancer uncovers novel
molecular traits of RAS dependency and improves prediction of
response to targeted agents in patients. Clin Cancer Res.
20:265–272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Montagut C, Dalmases A, Bellosillo B,
Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S,
Tsai SP, et al: Identification of a mutation in the extracellular
domain of the epidermal growth factor receptor conferring cetuximab
resistance in colorectal cancer. Nat Med. 18:221–223. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Arena S, Bellosillo B, Siravegna G,
Martínez A, Cañadas I, Lazzari L, Ferruz N, Russo M, Misale S,
González I, et al: Emergence of multiple EGFR extracellular
mutations during cetuximab treatment in colorectal cancer. Clin
Cancer Res. 21:2157–2166. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
van Emburgh BO, Arena S, Siravegna G,
Lazzari L, Crisafulli G, Corti G, Mussolin B, Baldi F, Buscarino M,
Bartolini A, et al: Acquired RAS or EGFR mutations and duration of
response to EGFR blockade in colorectal cancer. Nat Commun.
7:136652016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Montagut C, Siravegna G and Bardelli A:
Liquid biopsies to evaluate early therapeutic response in
colorectal cancer. Ann Oncol. 26:1525–1527. 2015. View Article : Google Scholar : PubMed/NCBI
|