Introduction
Rosai-Dorfman disease (RDD), or sinus histiocytosis
with massive lymphadenopathy, is a relatively rare self-limited
benign disease (1,2). This atypical cellular disorder was
described by Pierre-Paul Louis Lucien Destombes for the first time
in 1965 and was labeled as a distinct pathological entity by Rosai
and Dorfman in 1969 (2). Although
RDD predominantly affects children <10 years of age (66% of
cases) and young adults <20 years old (80% of cases) with male
predilection, cutaneous disease is also encountered in women in
their fourth decade (3). The
systemic form of RDD is commonly observed in African-American
individuals, whereas the purely cutaneous form is more common in
Asian and Caucasian ethnicities (4).
The etiology of RDD is unknown; possible causes may be viral
infections (including Epstein-Barr virus, parvovirus B19 and human
herpes virus), immunodeficiency, autoimmune disease and neoplastic
processes (5,6). The clinical course of RDD is often
benign, though lethal outcomes are also possible when multiple
organs are involved (7). The
treatment strategies may be different according to the severity or
vital organ involvement (8). In
patients with RDD requiring systemic treatment, steroids are a
first-line therapeutic option that produce responses in nodal and
extranodal disease. Radiation may be used as a palliative option
for symptomatic disease. Surgery is an appropriate option for
disease that may be excised, such as primary central nervous system
involvement. In cases of disseminated RDD or those refractory to
surgery, radiotherapy or steroids, chemotherapy has been used with
varying degrees of success (9).
Clinical outcome depends on the affected organs, as well as the
number of extranodal sites involved (10).
The present report described a rare case of
18F-fluoro-2-deoxyglucose positron emission-computed
tomography (18FDG PET-CT) performed in a young adult
male with a rare purely cutaneous form of RDD.
Case report
A 33-year-old male soldier, previously healthy,
presented to dermatology clinics of the Central Military Hospital
(Beirut, Lebanon) in June 2014 for evaluation of two new onset
subcutaneous enlarged and asymptomatic masses located along the
left cheek and the right upper gluteal region. On physical
examination, masses were firm with overlying indurated skin,
non-tender and painless. Extensive biological test results,
including complete blood count, platelet count, erythrocyte
sedimentation rate, c-reactive protein, complement component 3,
complement component 4, rheumatoid factor and lactate
dehydrogenase, were unremarkable. According to their tumor feature,
and as cutaneous lymphoma was considered as a differential
diagnosis, staging with 18FDG PET-CT was ordered prior
to excisional biopsies of the aforedescribed masses, and pathology
reported RDD.
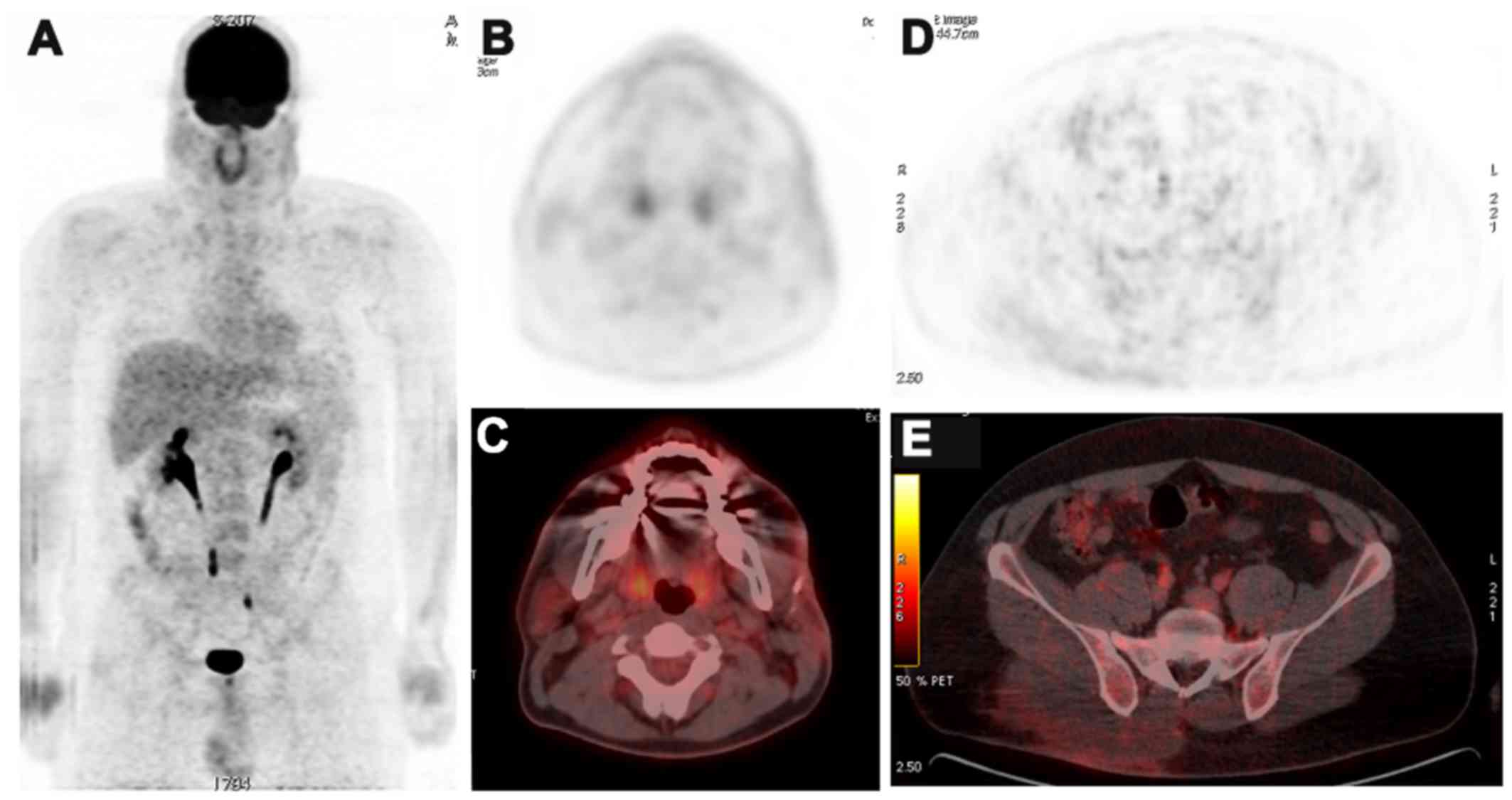
A total body 18FDG PET-CT scan was
obtained for staging purposes and to guide therapy initiation.
Cheek and gluteal masses were of irregular contours with equivalent
metabolic level of radioglucose (Fig.
1). They measured 55×24 mm and 70×32 mm in size, respectively,
with a standardized uptake value average of 4 and 4.2,
respectively, and a standardized uptake value maximum
(SUVmax) of 8.9 and 9, respectively. Initially, no
evidence of active lymph node involvement was noted on both sides
of the diaphragm nor other cutaneous or extranodal active
sites.
Microscopy of excisional biopsy demonstrated dermal
inflammatory infiltrate, essentially macrophages with large
cytoplasm, vesicular nuclei and evident nucleoli. On
immunohistochemistry, the large pale macrophages demonstrated
strong, diffuse cytoplasmic and nuclear staining for S100 protein,
with focal and less intense staining for cluster of differentiation
(CD) 68 (histiocytic marker). The cells were negative for CD1a, A1K
protein, vimentin and smooth muscle actin. Of note was the
recognition of non-staining of lymphocytes in proximity to the
macrophages, suggesting intracytoplasmic phagocytosis.
A conservative approach (clinical observation) was
chosen following surgical excision, and an 8-month 18FDG
PET-CT follow-up demonstrated no evidence of local recurrence or
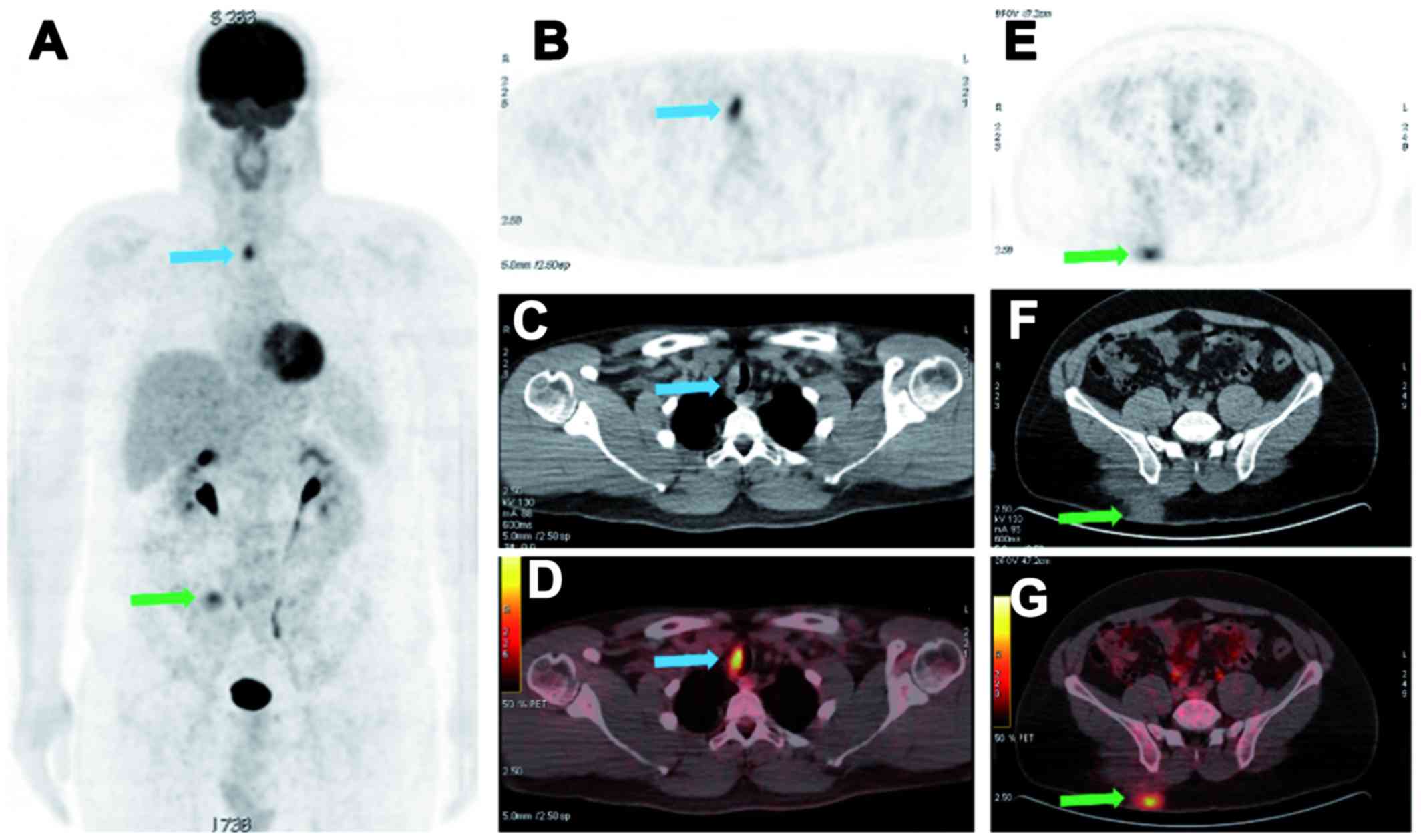
new abnormal focal uptake in the rest of the body (Fig. 2). However, 18 months later, a local
recurrence of the major RDD mass of the gluteal region was
suspected on dermatological examination, with the interval
appearance of one paratracheal active lymph node (diameter, 17 mm;
SUVmax, 4.5) on follow-up 18FDG PET-CT
imaging. Again, the RDD subcutaneous mass of the right gluteal
region was excised surgically, with the initiation of steroid
therapy, a first-line therapeutic option (9). Following the completion of steroid
therapy after 24 months, the 18FDG PET-CT follow-up
performed at 24 months demonstrated a second recurrence of the
right gluteal mass with an increase in size and metabolic activity
of the single right paratracheal lymph node (diameter, 22 mm;
SUVmax, 9.3). The surgical bed of the left cheek RDD
mass remained completely inactive (Fig.
3). The paratracheal lymph node was considered as the nodal
part of RDD according to clinical feature progression, and its
management did not warrant histological documentation by means of
an invasive act of thoracoscopy. Written informed consent was
obtained from the patient for publication of the present case
report.
Discussion
The most common symptoms on presentation of RDD are
painless cervical lymphadenopathy associated with fever, night
sweats and weight loss; other symptoms are related to sites of
involvement (11). Lymphatic disease
manifests predominantly with bilateral massive cervical adenopathy
(12). Neurological disease reveals
as intracranial masses and, less frequently, intraspinal
dural-based masses (13). RDD may
affect the breasts, lungs, gastrointestinal tract and, less often,
the bones (12). Involvement of the
eyes, nose and trachea have also been described (8). Patients with RDD may present to
rheumatologists due to bone or joint pain (14).
Mixed nodal and extranodal involvement is the most
common presentation of RDD and the skin is the most common
extranodal site affected; however, it is rarely isolated (13). The cutaneous form was first described
in 1978 by Thawerani et al (15). Cutaneous RDD is distinct from the
systemic form and is confined to the skin without lymphadenopathy
and with different dermographic features (16). Skin lesions are often papules or
nodules that are firm, indurated and ranging in size from 1–10 cm.
Pustular, psoriasiform and acneiform presentations have also been
documented (17). Cutaneous RDD has
a benign course usually with spontaneous regression in the majority
of cases (9). Therapy may be
required for relapsed cases and for cosmetic reasons only.
The present report detailed a rare case of cutaneous
RDD, isolated initially and presenting as bifocal
cutaneous/subcutaneous masses involving the left cheek and right
gluteal region. The dermatologic pattern of the subcutaneous firm
masses with overlying skin indurated violaceous plaque has
previously been described in the literature (18). The two masses in the present case of
RDD measured >5 cm in diameter and were likely present for
several years.
RDD was previously defined by the accumulation of
histiocytes that do not meet the phenotypic criteria for the
diagnosis of Langerhans cells (19,20). A
recent classification of histiocytic disorders and neoplasms of the
macrophage-dendritic cell lineage has been proposed with RDD
forming its own subtype (R group) due to its unique characteristics
(21). In this more recent
classification, histiocyte proliferation is presumably reactive and
polyclonal (22). The diagnosis
requires the presence of large histiocytic cells displaying
hypochromatic nuclei and pale cytoplasm, often with abundant
emperipolesis with positive immunohistological staining for S100,
fascin, CD68, CD14, human leukocyte antigen-antigen D related and
CD163. Typically, negative staining for CD1a and CD207
distinguishes it from Langerhans histiocytosis (22).
In lymph nodes, the large S100-positive histiocytes
are predominantly localized within the sinuses, and the cortex
often contains numerous plasma cells and activated B cells
(21). The cutaneous lesions are
characterized by proliferation of polygonal S100-positive
histiocytes and mixed inflammatory infiltrates (23) that are composed predominantly of
epithelioid histiocytes with a pale to eosinophilic cytoplasm as
Russel bodies (2). The pathognomonic
RDD cells demonstrate abundant granular and palely eosinophilic
cytoplasm with feathery borders and medium nuclei (24). However, the most important feature is
phagocytosis of intact lymphocytes, plasma cells and
polymorphonuclear leukocytes within the cytoplasm, a process termed
as emperipolesis or lymphophagocytosis (24).
In the present case, the histological diagnosis of
RDD evoked on soft tissue, skin and deep margin excisions, was
based on the strong histiocyte expression of S100 marker.
Furthermore, the recognition of non-staining lymphocytes in
proximity to the macrophages suggested emperipolesis. Langerhans's
cell histiocytosis, Erdheim-Chester disease and inflammatory
myofibroblastic tumor were considered in the differential diagnoses
list; however, the immunostaining profile was not supportive of any
of these considerations.
RDD usually affects multiple nodal and extranodal
sites with variable radiological aspect on imaging studies,
commonly affecting the head and neck areas. On ultrasound, nodal
disease appears as multiple large, round, well-defined hypoechoic
nodes, with loss of echogenicity in the hilum (12). On contrast-enhanced CT scan, it
appears as homogenously enhancing lymph nodes and, in some cases,
as hypodense owing to cystic changes (12). On gadolinium-enhanced magnetic
resonance imaging (MRI), lymph nodes are hypointense on T1- and
T2-weighted images, and homogeneously enhancing following
gadolinium administration (12). In
such cases, the main radiological differential diagnoses would be
non-Hodgkin lymphoma, reactive lymph nodes, tuberculous
adenopathies, Langerhans histiocytosis or Castleman disease
(25).
Skin RDD appears on ultrasounds as ill-defined,
hypoechoic cutaneous and subcutaneous nodules (26). On CT scans, it appears as ill-defined
enhancing lesions of the skin, while on gadolinium-enhanced MRI
scans, lesions are hypointense on T1-weighted MRI, hypointense to
hyperintense on T2-weighted MRI relative to underlying muscle, and
homogeneously enhancing following gadolinium administration
(12).
Similar to various benign or malignant
lymphoproliferative disorders, nodal and extranodal RDD lesions
have been demonstrated to be 18FDG-avid (27–30).
This avidity is attributed to the high level of the radioglucose
metabolism of the proliferating histiocytes as well as the
infiltrating inflammatory cells (27–30).
Various studies have reported the utility of 18FDG
PET-CT for monitoring steroid therapy, particularly in the visceral
form of the disease (29,31). 18FDG PET-CT is also the
method of choice for staging and follow-up on treatment response in
extranodal disease (29). Multiple
and various case reports that diagnosed or managed diseases
according to 18FDG PET-CT follow-up scans are available
in literature. Notably, a case report of RDD mimicking a lymphoma
on 18FDG PET-CT in a pediatric patient demonstrated that
the exact diagnosis was only made following histological sampling
(32). Additionally, a case of a
hypothalamic lesion with 18FDG-avidity was diagnosed as
RDD of the central nervous system (CNS) (33). Follow-up on treatment responses using
18FDG PET-CT have also been described for a
gastrointestinal case presenting as Crohn's disease of ileum, for a
case of local and isolated CNS relapse following neurosurgery of a
primary RDD of the brain (34), and
a case of bone involvement by RDD (28). In a rare case report, the
18FDG PET-CT demonstrated abnormal hyperactivities
limited to the spleen and the liver, without active
lymphadenopathy, and was used to indicate the splenectomy alone, as
both a diagnostic and therapeutic approach (30).
In the present case, the first 18FDG
PET-CT examination demonstrated initially the only cutaneous form
of RDD, which is a rare finding previously described in a single
case report by Huang et al (35). It demonstrated high and equivalent
metabolic activity of radioglucose in the cheek and gluteal
cutaneous masses, which shared similar histology in the present
case. 18FDG PET-CT results were a prerequisite based on
which the therapeutic strategy was planned. As no nodal or systemic
disease was associated initially with the two sites of cutaneous
lesions, they were excised surgically and no medical treatment was
offered. The first recurrence was local in the surgical bed of the
major lesion affecting the right gluteal region, which was
re-excised, with the interval appearance of hypermetabolic
paratracheal lymph node. Therefore, findings prompted steroid
initiation. The right gluteal mass relapsed again, and was
suspected on dermatological examination and confirmed by
18FDG PET-CT criteria. The elevated metabolic activity
recorded in the present case of cutaneous lesions
(SUVmax, 9) may have represented an aggressive nature,
which would explain the local relapses following surgical
excisions. The cutaneous mass of the left cheek was less voluminous
than the right gluteus one, and did not locally relapse, indicating
that surgery was successful at fully removing the mass. Therefore,
the present case highlighted the importance of 18FDG
PET-CT scanning in staging and decision making or therapeutic
recommendations.
Prognosis of RDD is variable with a large spectrum,
starting with spontaneous healing within weeks to months, and
finishing at the other end with persistence or recurrence following
surgical excision or medical treatments (36). Often, RDD regresses spontaneously and
aggressive therapeutic measures are not recommended (36). When the disease progression affects
the kidneys, the lower respiratory tract, the pancreas or the
liver, during its long-term clinical course with remissions and
exacerbations, end organ damage may occur and may have a fatal
outcome (2).
Localized RDD of skin may be treated with complete
surgical resection, with a risk of local recurrence (37). It is important to note that the
cosmetic appearance of skin represents a therapeutic challenge for
surgery. Recently, a study by Li et al (38) reported the possible mechanisms of
action of in situ photoimmunotherapy for RDD, which
demonstrated no obvious side effects (38).
Surgical excision is also considered when vital
organs or vascular/neurological compromise is imminent, but also
for cosmetic reasons when the skin is involved (39). Otherwise, conservative approaches and
regular follow-up is preferred. Radiotherapy, chemotherapy or
steroids may lead to a transient response; however, they do not
provide long-term benefits (2).
Surgery and steroid therapy are associated with adverse effects
(40).
In the present case, the disease was initially
limited to the skin, and the initial treatment was a surgical
excision. Follow-up 18FDG PET-CT was performed three
times over a 2-year period, and it documented a complete response
following the initial surgical excision of the cutaneous lesions,
local recurrences in the skin treated surgically, and an interval
appearance of a paratracheal lymph adenopathy, which required
systemic therapy.
In conclusion, to the best of our knowledge, there
is only one prior case of a cutaneous-only form of RDD diagnosed by
18FDG PET-CT scan in English literature (35). Therefore, the present report
described the second case of isolated cutaneous RDD evaluated by
18FDG PET-CT scan, which displayed high radioglucose
metabolism. The present case demonstrated the importance of
18FDG PET-CT imaging in the screening of extranodal RDD,
even with cutaneous involvement. In the latter case,
18FDG-avidity is usually attributed to the infiltrative
and inflammatory changes caused by the disease process. Ultimately,
this imaging modality may aid with making therapeutic
recommendations.
References
|
1
|
Rosai J and Dorfman RF: Sinus
histiocytosis with massive lymphadenopathy. A newly recognized
benign clinicopathological entity. Arch Pathol. 87:63–70.
1969.PubMed/NCBI
|
|
2
|
Foucar E, Rosai J and Dorfman R: Sinus
histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease):
Review of the entity. Semin Diagn Pathol. 7:19–73. 1990.PubMed/NCBI
|
|
3
|
Chappell JA, Burkemper NM, Frater JL and
Hurley MY: Cutaneous rosai-dorfman disease and morphea: Coincidence
or association? Am J Dermatopathol. 31:487–489. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brenn T, Calonje E, Granter SR, Leonard N,
Grayson W, Fletcher CD and McKee PH: Cutaneous rosai-dorfman
disease is a distinct clinical entity. Am J Dermatopathol.
24:385–391. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pulsoni A, Anghel G, Falcucci P, Matera R,
Pescarmona E, Ribersani M, Villivà N and Mandelli F: Treatment of
sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman
disease): Report of a case and literature review. Am J Hematol.
69:67–71. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Elbuluk N, Egbers R, Taube JM and Wang TS:
Cutaneous Rosai-Dorfman Disease in a Patient with Human
Immunodeficiency Virus. Dermatol Online J.
22:13030/qt2162h3fj2016.PubMed/NCBI
|
|
7
|
Chen J, Tang H, Li B and Xiu Q:
Rosai-Dorfman disease of multiple organs, including the epicardium:
An unusual case with poor prognosis. Heart Lung. 40:168–171. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ottaviano G, Doro D, Marioni G, Mirabelli
P, Marchese-Ragona R, Tognon S, Marino F and Staffieri A:
Extranodal Rosai-Dorfman disease: Involvement of eye, nose and
trachea. Acta Otolaryngol. 126:657–660. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dalia S, Sagatys E, Sokol L and Kubal T:
Rosai-Dorfman Disease: Tumor Biology, Clinical Features, Pathology,
and Treatment Cancer Control. October;2014.21(4)
|
|
10
|
McClain KL, Natkunam Y and Swerdlow SH:
Atypical cellular disorders. Hematology (Am Soc Hematol Educ
Program). 2004:283–296. 2004.
|
|
11
|
La Barge DV III, Salzman KL, Harnsberger
HR, Ginsberg LE, Hamilton BE, Wiggins RH III and Hudgins PA: Sinus
histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease):
Imaging manifestations in the head and neck. AJR Am J Roentgenol.
191:W299–306. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zaveri J, La Q, Yarmish G and Neuman J:
More than just Langerhans cell histiocytosis: A radiologic review
of histiocytic disorders. Radiographics. 34:2008–2024. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu JQ, Zhuang H, Xiu Y, Talati E and Alavi
A: Demonstration of increased FDG activity in Rosai-Dorfman disease
on positron emission tomography. Clin Nucl Med. 29:209–210. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mosheimer BA, Oppl B, Zandieh S, Fillitz
M, Keil F, Klaushofer K, Weiss G and Zwerina J: Bone Involvement in
Rosai-Dorfman Disease (RDD): A Case Report and Systematic
Literature Review. Curr Rheumatol Rep. 19:292017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thawerani H, Sanchez RL, Rosai J and
Dorfman RF: The cutaneous manifestations of sinus histiocytosis
with massive lymphadenopathy. Arch Dermatol. 114:191–197. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang KH, Chen WY, Liu HN, Huang CC, Lee WR
and Hu CH: Cutaneous Rosai-Dorfman disease: Clinicopathological
profiles, spectrum and evolution of 21 lesions in six patients. Br
J Dermatol. 154:277–286. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cole S and Finlay J:
2-Chlorodeoxyadenosine for adults with multi-system Langerhans cell
histiocytosis. Med Pediatr Oncol. 33:5121999.
|
|
18
|
Rubenstein MA, Farnsworth NN, Pielop JA,
Orengo IF, Curry JL, Drucker CR and Hsu S: Cutaneous Rosai-Dorfman
disease. Dermatol Online J. 12:82006.PubMed/NCBI
|
|
19
|
Hervier B, Haroche J, Arnaud L, Charlotte
F, Donadieu J, Néel A, Lifermann F, Villabona C, Graffin B, Hermine
O, et al French Histiocytoses Study Group, : Association of both
Langerhans cell histiocytosis and Erdheim-Chester disease linked to
the BRAFV600E mutation. Blood. 124:1119–1126. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weitzman S and Jaffe R: Uncommon
histiocytic disorders: The non-Langerhans cell histiocytoses.
Pediatr Blood Cancer. 45:256–264. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Emile JF, Abla O, Fraitag S, Horne A,
Haroche J, Donadieu J, Requena-Caballero L, Jordan MB, Abdel-Wahab
O, Allen CE, et al Histiocyte Society, : Revised classification of
histiocytoses and neoplasms of the macrophage-dendritic cell
lineages. Blood. 127:2672–2681. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
O'Malley DP, Duong A, Barry TS, Chen S,
Hibbard MK, Ferry JA, Hasserjian RP, Thompson MA, Richardson MS,
Jaffe R, et al: Co-occurrence of Langerhans cell histiocytosis and
Rosai-Dorfman disease: Possible relationship of two histiocytic
disorders in rare cases. Mod Pathol. 23:1616–1623. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Juskevicius R and Finley JL: Rosai-Dorfman
disease of the parotid gland: Cytologic and histopathologic
findings with immunohistochemical correlation. Arch Pathol Lab Med.
125:1348–1350. 2001.PubMed/NCBI
|
|
24
|
Kong YY, Kong JC, Shi DR, Lu HF, Zhu XZ,
Wang J and Chen ZW: Cutaneous rosai-dorfman disease: A clinical and
histopathologic study of 25 cases in China. Am J Surg Pathol.
31:341–350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bonetti F, Chilosi M, Menestrina F, Scarpa
A, Pelicci PG, Amorosi E, Fiore-Donati L and Knowles DM II:
Immunohistological analysis of Rosai-Dorfman histiocytosis. A
disease of S-100 + CD1-histiocytes. Virchows Arch A Pathol Anat
Histopathol. 411:129–135. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ying M, Ahuja AT and Yuen HY: Grey-scale
and power Doppler sonography of unusual cervical lymphadenopathy.
Ultrasound Med Biol. 30:449–454. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karunanithi S, Singh H, Sharma P, Naswa N
and Kumar R: 18F-FDG PET/CT imaging features of Rosai Dorfman
disease: A rare cause of massive generalized lymphadenopathy. Clin
Nucl Med. 39:268–269. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsang JS, Anthony MP, Wong MP and Wong CS:
The use of FDG-PET/CT in extranodal Rosai-Dorfman disease of bone.
Skeletal Radiol. 41:715–717. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Albano D, Bosio G and Bertagna F: 18F-FDG
PET/CT follow-up of Rosai-Dorfman disease. Clin Nucl Med.
40:e420–e422. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ha H, Kim KH, Ahn YJ, Kim JH, Kim JE and
Yoon S-S: A rare case of Rosai-Dorfman disease without
lymphadenopathy. Korean J Intern Med. 31:802–804. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shaikh F, Awan O, Mohiuddin S, Farooqui S,
Khan SA and McCartney W: 18F-FDG PET/CT Imaging of Extranodal
Rosai-Dorfman Disease with Hepatopancreatic Involvement - A
Pictorial and Literature Review. Cureus. 7:e3922015.PubMed/NCBI
|
|
32
|
Liu B, Lee NJ, Otero HJ, Servaes S and
Zhuang H: Rosai-Dorfman disease mimics lymphoma on FDG PET/CT in a
pediatric patient. Clin Nucl Med. 39:206–208. 2014.PubMed/NCBI
|
|
33
|
Deshayes E, Le Berre JP, Jouanneau E,
Vasiljevic A, Raverot G and Seve P: 18F-FDG PET/CT findings in a
patient with isolated intracranial Rosai-Dorfman disease. Clin Nucl
Med. 38:e50–e52. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Le Guenno G, Galicier L, Uro-Coste E,
Petitcolin V, Rieu V and Ruivard M: Successful treatment with
azathioprine of relapsing Rosai-Dorfman disease of the central
nervous system. J Neurosurg. 117:486–489. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang JY, Lu CC, Hsiao CH and Tzen KY: FDG
PET/CT findings in purely cutaneous Rosai-Dorfman disease. Clin
Nucl Med. 36:e13–e15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Madhunapantula SV, Gowda R, Inamdar GS and
Robertson GP: In situ photoimmunotherapy: A new hope for cutaneous
melanoma patients. Cancer Biol Ther. 10:1088–1090. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Adeleye AO, Amir G, Fraifeld S, Shoshan Y,
Umansky F and Spektor S: Diagnosis and management of Rosai-Dorfman
disease involving the central nervous system. Neurol Res.
32:572–578. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li M, Shi L, Luo M, Chen J, Wang B, Zhang
F, Keyal U, Bhatta AK, Chen WR and Wang X: Successful treatment of
Rosai-Dorfman disease using in situ photoimmunotherapy. Indian J
Dermatol Venereol Leprol. 83:332–336. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lu LY, Ju WT, Cai M and Lu XF: Cutaneous
Rosai-Dorfman disease recurrence in infraorbital region. J
Craniofac Surg. 23:e509–e510. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dalia S, Sagatys E, Sokol L and Kubal T:
Rosai-Dorfman disease: Tumor biology, clinical features, pathology,
and treatment. Cancer Control. 21:322–327. 2014. View Article : Google Scholar : PubMed/NCBI
|