Introduction
Lung cancer is one of the most common malignant
diseases and a major threat to public health on a global scale. The
main types of lung cancer are small-cell lung cancer (SCLC) and
non-small-cell lung cancer (NSCLC). NSCLC has a higher degree of
malignancy and a lower 5-year survival rate compared with SCLC, and
may be divided into two major histopathological subtypes, namely
adenocarcinoma (ADC) and squamous cell carcinoma (SCC). As NSCLC is
a complex disease, the identification of disease genes has been
among the main goals of biologists and clinicians. Although our
understanding of lung cancer, particularly NSCLC, has improved
significantly in recent years, several questions require further
elucidation.
With the completion of the Human Genome Project,
scientific research has entered the post-genome era. Furthermore,
with the advances in biological research and the development of
high throughput biotechnologies, the quantities of candidate genes
and numerous genomic loci have been reported to play a vital role
in the pathogenesis of NSCLC. However, the mechanism underlying
this disease has not yet been fully elucidated. As is well-known,
different analytical methods may result in different consequences
due to the ‘small N large P’ problem in microarray data, i.e., the
gene expression data usually come with only dozens of tissue
samples, but with up to tens of thousands of gene features. This
extreme sparseness is considered to significantly compromise the
performance of a classifier (1). As
a result, the ability to extract a subset of informative genes
while removing irrelevant or redundant genes is crucial for
accurate classification (2). Hence,
the methodology research is of great importance and the reuse of
microarray data is feasible and extremely valuable. It is
meaningful to identify disease-specific genes to help provide a
novel therapeutic target for tumor treatment and elucidate the
mechanism of the disease.
The aim of the present study was to identify the
NSCLC subtype-related genes based on the classification method and
the shortest path analysis based on the protein-protein interaction
(PPI) network, and to investigate their underlying functions by
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway enrichment analysis with the Database for
Annotation, Visualization and Integrated Discovery (DAVID). This
workflow may be of value for identifying disease genes and may
provide insight into the pathogenesis of malignant tumors. To the
best of our knowledge, this investigation is the first of its type
in lung cancer studies, and it may be useful for further lung
cancer candidate gene verification, thus enabling a better
understanding of the molecular mechanisms of NSCLC and the
development more effective diagnostic and intervention methods.
Materials and methods
Data source
The gene expression dataset (GDS3627, accession no.
GSE10245) was downloaded from the National Center for Biotechnology
Information Gene Expression Omnibus (GEO) from a research conducted
by Kuner et al (3) on lung
cancer. The data were processed with the Affymatrix GPL570 platform
and generated from 58 tissue samples (40 ADC and 18 SCC). All the
tissue samples underwent careful analysis of the histopathology,
estimation of tumor and stromal content and subsequent
macro-dissection of the tumor regions prior to enrollment in the
microarray experiment. No ethics committee approval is required to
obtain these data, since they are publicly available. In addition,
only expression data are presented, and no personal patient
information is revealed.
Preprocessing of microarray data
Data preprocessing, including quantile normalization
and log2 transformation was completed by the GEOquery package
(4). In order to minimize false
positives, the GeneSelector package in R programme was used to rank
gene lists. Briefly, selection of differential expressed genes may
be highly affected by the statistical methods used, and the overlap
of genes selected using the same significance value may be quite
low. GeneSelector ranks genes on the basis of how well they perform
in a selected number of statistical tests and minimize the number
of false positives (5). Ranked lists
based on ordinary Student's t-test, significance analysis of
microarrays, and fold-change were first generated for each of the
criteria. The Markov chain model within the GeneSelector package
was used to aggregate the ranked lists of all criteria into a list
of genes that are significant, independently of the method used. To
reduce noise and due to the fact that prediction methods such as
Naïve Bayes Classifier prefer categorical data, the gene expression
data were discretized using the respective µ (mean) and σ (standard
deviation): Any data larger than µ + σ/2 were transformed to state
1; any data between µ - σ/2 and µ + σ/2 were transformed to state
0; and any data smaller than µ - σ/2 were transformed to state −1.
These three states correspond to the overexpression, baseline, and
underexpression of genes, respectively. According to the previous
studies, discretization of gene expression data generally leads to
better prediction accuracy.
Maximum relevance minimum redundancy
(mRMR) feature selection method
The mRMR feature selection method was proposed by
Ding et al (6). For discrete
variables, the mutual information I of two variables
x and y is defined based on their joint probabilistic
distribution p(x,y) and the respective marginal
probabilities p(x) and p(y).1)
I(x,y)=∑i,jp(xi,yj)logp(xi,yj)p(xi)p(yj)
Let S denote the subset of features we are
seeking. The maximum relevance condition is
maxD(S,c),D=1|S|∑xi∈SI(xi;c)
The minimum redundancy condition is
minR(S),R=1|S|2∑xi,xi∈SI(xi;xj)
where |S| is used to represent the number of
features in S, and c represents the targeted
classes.
The mutual information quotient criterion may be
described as follows.
maxФ(D,R),Ф=D/R
This optimization may be computed efficiently by
heuristic algorithm
max∇MIQ,∇MIQ=max{I(xj:c)/1m–1∑xi∈Sm–1I(xj;xi)}
where xj ∈ XF
-Sm-1, XF represents the
original feature set.
Naïve Bayes Classifier
The Naïve Bayes Classifier is a simple probabilistic
classifier based on applying Bayes' theorem with strong
independence assumptions between features. For a sample s
with m gene expression levels {g1,
g2,….., gm} for the m features, the
posterior probability that s belongs to class
ck is
p(ck|s)∝∏i∈sp(gi|Ck)
where
p(gt|ck) are conditional
tables estimated from training examples.
The Naïve Bayes Classifier was used after all the
features ranked by the mRMR. A series of classifiers were then
constructed when the features were added into the classifier one by
one. For example, the first classifier was constructed by the first
feature, the second classifier by the first two features, and the
rest in the same manner.
Using incremental feature selection (IFS), the
number N may be determined. Its idea is to compare
prediction accuracy defined in the following selection among
different Ns, and select the one with the highest
accuracy.
The prediction accuracy was formulated by
Accuracy=TP+TNTP+TN+FP+FN
where TP represents the number of true positives, TN
represents the number of true negatives, FP represents the number
of false positives, and FN represents the number of false
negatives.
Shortest path analysis
The PPI data have been widely used for gene function
prediction with the assumption that interacting proteins share the
same or have similar functions and, hence, may be involved in the
same pathway (7–9). The initial weighted graph G =
(V,E), where V represented all human proteins
occurring in PPI retrieved from STRING (version 10.0) (http://string-db.org/) (10), and E contained all pairs of
nodes such that the corresponding proteins comprised a PPI in
STRING, was constructed according to the information retrieved from
STRING. In a network, shortest path measures the least nodes
required to pass through from one node to another (11). The Dijkstra's algorithm in the igraph
package was applied to identify the shortest path connecting each
pair of genes selected by the mRMR in the PPI network. The genes
occurring in any shortest path were considered as candidate
genes.
Function and pathway enrichment
analysis
GO and KEGG pathway enrichment analysis was
performed with the functional annotation tool DAVID (12). All the protein-coding genes in the
human genome were taken as background during the enrichment
analysis.
Results
Genes selected by Naïve Bayes
Classifier
A total of 4,015 genes remained for the following
analysis after preprocessing. Naïve Bayes Classifier with mRMR was
used to select genes associated with the discrimination of ADC and
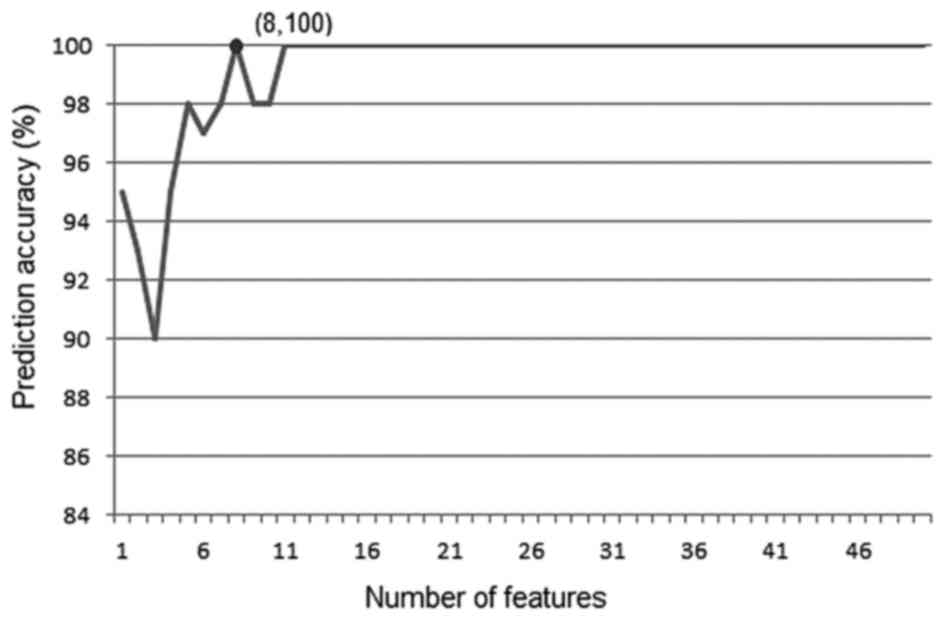
SCC. The IFS result is provided in Fig.
1. In the IFS curve, the x-axis is the number of genes used for
classification and the y-axis is the prediction accuracy. As shown,
the accuracy reached the maximum (100%) when 8 features were
included. Those genes are listed in Table I, along with their results of
fold-change and t-test. LOC100508342 was not included for
the shortest path analysis and bioinformatics analysis since it is
a hypothetical gene, which means its biological function remains
unknown to date.
 | Table I.Differentially expressed genes
selected based on the gene expression profiles. |
Table I.
Differentially expressed genes
selected based on the gene expression profiles.
| Gene symbol | Ensembl gene
ID | Description | Fold
changea | t | P-value |
|---|
| TP63 |
ENSG00000073282 | Tumor protein
p63 | 0.158 | 2.63 | 8.77E-03 |
| SPC24 |
ENSG00000161888 | SPC24, NDC80
kinetochore complex component | 0.343 | 5.60 | 1.11E-06 |
| RGS12 |
ENSG00000159788 | Regulator of
G-protein signaling 12 | 0.034 | 0.94 | 1.79E-01 |
| CTDSPL2 |
ENSG00000137770 | CTD
(carboxy-terminal domain, RNA polymerase II, polypeptide A) small
phosphatase-like 2 | 0.400 | 5.30 | 2.40E-06 |
| TMEM62 |
ENSG00000137842 | Transmembrane
protein 62 | 0.347 | 8.20 | 8.62E-10 |
| SDCCAG8 |
ENSG00000054282 | Serologically
defined colon cancer antigen 8 | 0.127 | 3.88 | 2.88E-04 |
|
LOC100508342 | – | – | 0.509 | 6.63 | 2.09E-07 |
| EFS |
ENSG00000100842 | Embryonal
Fyn-associated substrate | 0.405 | 7.11 | 1.30E-09 |
Shortest path analysis
Dijkstra's algorithm was applied to select genes in
the shortest path between any two genes described in Table I. Finally, 21 genes were found in 21
shortest paths, where AURKA and SLC7A2 were selected
three and two times, respectively. Those genes, along with their
Ensembl peptide ID, Ensembl gene ID and descriptions, are listed in
Table II.
| Table II.Genes selected by the shortest path
analysis. |
Table II.
Genes selected by the shortest path
analysis.
| Ensembl peptide
ID | Ensembl gene
ID | Gene symbol | Description |
|---|
|
ENSP00000004531 |
ENSG00000003989 | SLC7A2 | Solute carrier
family 7 (cationic amino acid transporter, y+ system),
member 2 |
|
ENSP00000005340 |
ENSG00000004975 | DVL2 | Dishevelled segment
polarity protein 2 |
|
ENSP00000052754 |
ENSG00000011465 | DCN | Decorin |
|
ENSP00000160827 |
ENSG00000079616 | KIF22 | Kinesin family
member 22 |
|
ENSP00000171887 |
ENSG00000079308 | TNS1 | Tensin 1 |
|
ENSP00000215904 |
ENSG00000241360 | PDXP | Pyridoxal
(pyridoxine, vitamin B6) phosphatase |
|
ENSP00000216911 |
ENSG00000087586 | AURKA | Aurora kinase
A |
|
ENSP00000222248 |
ENSG00000105641 | SLC5A5 | Solute carrier
family 5 (sodium/iodide cotransporter), member 5 |
|
ENSP00000228928 |
ENSG00000111331 | OAS3 |
2′-5′-oligoadenylate synthetase 3, 100
kDa |
|
ENSP00000251337 |
ENSG00000134183 | GNAT2 | Guanine nucleotide
binding protein (G protein), α transducing activity polypeptide
2 |
|
ENSP00000261884 |
ENSG00000103671 | TRIP4 | Thyroid hormone
receptor interactor 4 |
|
ENSP00000264126 |
ENSG00000121957 | GPSM2 | G-protein signaling
modulator 2 |
|
ENSP00000264977 |
ENSG00000073711 | PPP2R3A | Protein phosphatase
2, regulatory subunit B', α |
|
ENSP00000266970 |
ENSG00000123374 | CDK2 | Cyclin-dependent
kinase 2 |
|
ENSP00000288266 |
ENSG00000157500 | APPL1 | Adaptor protein,
phosphotyrosine interaction, PH domain and leucine zipper
containing 1 |
|
ENSP00000306330 |
ENSG00000170027 | YWHAG | Tyrosine
3-monooxygenase/tryptophan 5-monooxygenase activation protein,
γ |
|
ENSP00000308339 |
ENSG00000120341 | SEC16B | SEC16 homolog B
(S. cerevisiae) |
|
ENSP00000308450 |
ENSG00000117399 | CDC20 | Cell division cycle
20 |
|
ENSP00000322775 |
ENSG00000129667 | RHBDF2 | Rhomboid 5 homolog
2 (Drosophila) |
|
ENSP00000332816 |
ENSG00000120899 | PTK2B | Protein tyrosine
kinase 2 β |
|
ENSP00000362556 |
ENSG00000169155 | ZBTB43 | Zinc finger and BTB
domain containing 43 |
Function and pathway enrichment
analysis
Using the functional annotation tool provided by
DAVID, GO and KEGG pathway enrichment analysis was performed on the
genes selected by Naïve Bayes Classifier and the shortest path
analysis. The results demonstrated that several genes are
significantly enriched in several biological processes such as
mitosis, nuclear division, M phase of the mitotic cell cycle and
two cellular components, intracellular non-membrane-bound organelle
and non-membrane-bound organelle. There was no significant
enrichment in molecular function (Table III).
| Table III.Gene ontology analysis based on the
genes associated with lung adenocarcinoma and squamous cell
carcinoma. |
Table III.
Gene ontology analysis based on the
genes associated with lung adenocarcinoma and squamous cell
carcinoma.
| Term | Pathway name | Number of
genes |
P-valuea |
|---|
| Biological
process |
GO:0007067~mitosis | 5 | 3.96E-04 |
|
| GO:0000280~nuclear
division | 5 | 3.96E-04 |
|
| GO:0000087~M phase
of mitotic cell cycle | 5 | 4.23E-04 |
|
|
GO:0048285~organelle fission | 5 | 4.60E-04 |
|
| GO:0000279~M
phase | 5 | 1.78E-03 |
|
| GO:0000278~mitotic
cell cycle | 5 | 2.73E-03 |
|
| GO:0022403~cell
cycle phase | 5 | 4.08E-03 |
| Cellular
component |
GO:0043232~intracellular
non-membrane-bounded organelle | 12 | 2.82E-03 |
|
|
GO:0043228~non-membrane-bounded
organelle | 12 | 2.82E-03 |
The KEGG pathway enrichment analysis revealed that
these genes are significantly enriched in oocyte meiosis, cell
cycle, and pathways in cancer (Table
IV).
| Table IV.Signaling pathways associated with
the genes selected from lung adenocarcinoma and squamous cell
carcinoma. |
Table IV.
Signaling pathways associated with
the genes selected from lung adenocarcinoma and squamous cell
carcinoma.
| Pathway | Gene symbols |
P-valuea |
|---|
| hsa04114:oocyte
meiosis | YWHAG,
CDC20, CDK2, AURKA | 3.24E-04 |
| hsa04110:cell
cycle | YWHAG,
CDC20, CDK2 | 1.16E-02 |
| hsa05200:pathways
in cancer | APPL1,
DVL2, CDK2 | 7.02E-02 |
Discussion
Lung cancer is one of the leading causes of
cancer-related mortality worldwide. The Naïve Bayes Classifier
approach with mRMR was applied followed by IFS to a microarray data
generated from 40 ADC and 18 SCC tissue samples. With this
approach, 8 genes that could optimally discriminate between ADC and
SCC were identified. Those genes not only included known
NSCLC-related genes, such as RGS12 (13), TP63 (14) and EFS (15), but also included genes that were not
found to be closely associated with NSCLC. Fortunately, several
genes, such as TP63, were found to be differentially
expressed between ADC and SCC (16).
Previously reported experimental evidence is provided for the
expression and functions of these inferred genes in the
discrimination of NSCLC, indicating that our method is effective
for the discovery of new candidate genes.
Liu et al (17) compared the protein profiles of LC5
cell lines with MASPIN overexpression and knockdown using
comparative two-dimensional gel electrophoresis, and found that
SDCCAG8 was unique in MASPIN-expressing cell lines, but absent in
knockout cell lines. Therefore, the authors hypothesized that
SDCCAG8 may play a significant role in the invasion of
cancer cells. Jadoon et al (18) used the iterative threading refinement
programme I-TASSER to match the TMEM62 sequence with known protein
structures. The top 10 structural analogues and five enzyme
homologues for the TMEM62 model in the Protein Data Bank were all
lipoxygenases (LOXs). According to the previous studies, LOXs form
a heterogeneous class of lipid peroxidizing enzymes and were found
to be implicated in the pathogenesis of various cancers (19,20).
Thus, TMEM62 may play a vital role in the diagnosis and
treatment of NSCLC. Unlike the two genes discussed above,
CTDSPL2 was reported in only two studies. Ma et al
(21) preliminarily confirmed that
the CTDSPL2 gene obviously improved the expression of ε- and
γ-globin genes, which provides a new candidate target for effective
treatment of sickle cell disease and β-thalassemia. Zhao et
al (22) demonstrated that
CTDSPL2, as a Smad phosphatase, plays a critical role in bone
morphogenetic protein (BMP)-induced signaling and cellular
functions. Experimental evidence demonstrated that the mRNA level
of BMP5 was significantly higher in ADC tissues compared with that
in SCC tissues, and the immunohistochemistry analysis confirmed
this result at the protein level (23). However, a correlation between the
CTDSPL2 expression level and the occurrence of NSCLC has not
been reported to date; it may be a novel biomarker of NSCLC
development and progression.
Ndc80, which is composed of two heterodimers,
CDCA1-KNTC2 and SPC24-SPC25, has been shown to play an important
role in stable microtubule-kinetochore attachment, chromosome
alignment and spindle checkpoint activation mitosis. The study
conducted by Hayama et al (24) reported that CDCA1 and KNTC2 appear to
belong in the category of cancer-testis antigens, and that their
simultaneous upregulation is a frequent and important
characteristic of cell growth/survival in lung cancer, and
selective suppression of CDCA1 or KNTC2 activity and/or inhibition
of the CDCA1-KNTC2 complex formation may be a promising therapeutic
target for the treatment of lung cancers. Kaneno et al
(25) reported the mRNA
overexpression of CDCA1, KNTC2, SPC24 and
SPC25 was observed in colorectal and gastric cancers.
Therefore, SPC24 may be associated with the development and
discrimination of NSCLC based on the abovementioned experimental
evidence.
Rather than the traditional approach to studying
individual genes or loci, a systematic investigation of cancer
proteins in the human PPI network may provide important biological
information for uncovering the molecular mechanisms of cancer.
Since the discrimination genes must have some common
characteristics related to NSCLC, it is reasonable to identify
novel genes based on the PPI network and may provide insights into
the comprehensive biological systems. Therefore, molecular
interaction networks were constructed based on the PPI data from
the STRING database, and 21 genes were identified in the shortest
paths among the genes identified with the Naïve Bayes Classifier
with mRMR. The AURKA (fold-change = 0.168; t=2.59, P=0.006)
and SLC7A2 (fold-change = 0.007; t=1.28, P=0.104) genes were
selected three and two times, respectively, in the shortest path
analysis.
In humans, the aurora kinase family consists of
highly conserved serine-threonine kinases, which play a critical
role in the regulation of mitotic events such as spindle assembly,
function of centrosomes and cytoskeleton and cytokinesis (26,27). Lo
Iacono et al (28) confirmed
that AURKA expression was significantly upregulated in tumor
samples compared with matched lung tissue [P<0.01, mean
log2 (FC) = 1.5]. Moreover, AURKA was principally
upregulated in moderately and poorly differentiated lung cancers
(P<0.01), as well as in SCC and ADC compared with the
non-invasive bronchioloalveolar histotype (P=0.029). Only a limited
number of studies have reported the association of SLC7A2 with the
development of NSCLC and future experimental studies are required
to investigate this association.
Additionally, KEGG pathway enrichment analysis
revealed that all those genes participate in a number important
pathways, such as oocyte meiosis, cell cycle and pathways in
cancer, among which CDK2 was found to appear in three pathways
simultaneously. In fact, a large number of previous studies have
demonstrated the importance of the role of CDK family members, such
as CDK2, in the cell cycle control of tumor cells (29,30).
These results are helpful for cancer candidate gene
verification, biomarker discovery and understanding the etiology of
cancer at the biological level. However, further experiments are
required to confirm these findings.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81373103) and the
Science Committee Basic and Cutting-edge Research Program of
Chongqing (no. cstc2013jcyjA10009).
References
|
1
|
Golub TR, Slonim DK, Tamayo P, Huard C,
Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caliqiuri
MA, et al: Molecular classification of cancer: Class discovery and
class prediction by gene expression monitoring. Science.
286:531–537. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tang Y, Zhang YQ and Huang Z: Development
of two-stage SVM-RFE gene selection strategy for microarray
expression data analysis. IEEE/ACM Trans Comput Biol Bioinform.
4:365–381. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuner R, Muley T, Meister M, Ruschhaupt M,
Buness A, Xu EC, Schnabel P, Warth A, Poustka A, Sültmann H and
Hoffmann H: Global gene expression analysis reveals specific
patterns of cell junctions in non-small cell lung cancer subtypes.
Lung Cancer. 63:32–38. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sean D and Meltzer P: GEOquery: a bridge
between the gene expression omnibus (GEO) and BioConductor.
Bioinformatics. 23:18462007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Konsak BSD, Haring V, Geier M, Hughes R,
Howarth G, Crowley T and Moore R: Identification of differential
duodenal gene expression levels and microbiota abundance correlated
with differences in energy utilization in chickens. Anim Prod Sci.
53:72013.
|
|
6
|
Ding C and Peng H: Minimum redundancy
feature selection from microarray gene expression data. J Bioinform
Comput Biol. 3:185–205. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao P, Wang QP, Chen L and Huang T:
Prediction of human genes' regulatory functions based on
proteinprotein interaction network. Protein Pept Lett. 19:910–916.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao YF, Chen L, Cai YD, Feng KY, Huang T
and Jiang Y: Predicting metabolic pathways of small molecules and
enzymes based on interaction information of chemicals and proteins.
PloS One. 7:e459442012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang J, Jiang M, Yuan F, Feng KY, Cai YD,
Xu X and Chen L: Identification of age-related macular degeneration
related genes by applying shortest path algorithm in
protein-protein interaction network. Biomed Res Int.
2013:5234152013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Szklarczyk D, Franceschini A, Kuhn M,
Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork
P, et al: The STRING database in 2011: Functional interaction
networks of proteins, globally integrated and scored. Nucleic Acids
Res. 39(Database Issue): D561–D568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Du ZP, Wu BL, Wang SH, Shen JH, Lin XH,
Zheng CP, Wu ZY, Qiu XY, Zhan XF, Xu LY and Li EM: Shortest path
analyses in the protein-protein interaction network of NGAL
(neutrophil gelatinase-associated lipocalin) overexpression in
esophageal squamous cell carcinoma. Asian Pac J Cancer Prev.
15:6899–6904. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dai J, Gu J, Lu C, Lin J, Stewart D, Chang
D, Roth JA and Wu X: Genetic variations in the regulator of
G-protein signaling genes are associated with survival in
late-stage non-small cell lung cancer. PloS One. 6:e211202011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jin YX, Jiang GN, Zheng H, Duan L and Ding
JA: Common genetic variants on 3q28 contribute to non-small cell
lung cancer susceptibility: Evidence from 10 case-control studies.
Mol Genet Genomics. 290:573–584. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y, McKay JD, Rafnar T, Wang Z,
Timofeeva MN, Broderick P, Zong X, Laplana M, Wei Y, Han Y, et al:
Rare variants of large effect in BRCA2 and CHEK2 affect risk of
lung cancer. Nat Genet. 46:736–741. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Choy B, Findeis-Hosey JJ, Li F, McMahon
LA, Yang Q and Xu H: High frequency of coexpression of maspin with
p63 and p53 in squamous cell carcinoma but not in adenocarcinoma of
the lung. Int J Clin Exp Pathol. 6:2542–2547. 2013.PubMed/NCBI
|
|
17
|
Liu Y, Geng Y, Li K, Wang F, Zhou H, Wang
W, Hou J and Liu W: Comparative proteomic analysis of the function
and network mechanisms of MASPIN in human lung cells. Exp Ther Med.
3:470–474. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jadoon A, Cunningham P and McDermott LC:
Arachidonic acid metabolism in the human placenta: Identification
of a putative lipoxygenase. Placenta. 35:422–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Catalano A and Procopio A: New aspects on
the role of lipoxygenases in cancer progression. Histol
Histopathol. 20:969–975. 2005.PubMed/NCBI
|
|
20
|
Nie D: Cyclooxygenases and lipoxygenases
in prostate and breast cancers. Front Biosci. 12:1574–1585. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma YN, Zhang X, Yu HC and Zhang JW: CTD
small phosphatase like 2 (CTDSPL2) can increase ε- and γ-globin
gene expression in K562 cells and CD34+ cells derived from
umbilical cord blood. BMC Cell Biol. 11:752010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao Y, Xiao M, Sun B, Zhang Z, Shen T,
Duan X, Yu PB, Feng XH and Lin X: C-terminal domain (CTD) small
phosphatase-like 2 modulates the canonical bone morphogenetic
protein (BMP) signaling and mesenchymal differentiation via Smad
dephosphorylation. J Biol Chem. 289:26441–26450. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Deng T, Lin D, Zhang M, Zhao Q, Li W,
Zhong B, Deng Y and Fu X: Differential expression of bone
morphogenetic protein 5 in human lung squamous cell carcinoma and
adenocarcinoma. Acta Biochim Biophys Sin (Shanghai). 47:557–563.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hayama S, Daigo Y, Kato T, Ishikawa N,
Yamabuki T, Miyamoto M, Ito T, Tsuchiya E, Kondo S and Nakamura Y:
Activation of CDCA1-KNTC2, members of centromere protein complex,
involved in pulmonary carcinogenesis. Cancer Res. 66:10339–10348.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaneko N, Miura K, Gu Z, Karasawa H,
Ohnuma S, Sasaki H, Tsukamoto N, Yokoyama S, Yamamura A, Nagase H,
et al: siRNA-mediated knockdown against CDCA1 and KNTC2, both
frequently overexpressed in colorectal and gastric cancers,
suppresses cell proliferation and induces apoptosis. Biochem
Biophys Res Commun. 390:1235–1240. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brown JR, Koretke KK, Birkeland ML,
Sanseau P and Patrick DR: Evolutionary relationships of Aurora
kinases: Implications for model organism studies and the
development of anti-cancer drugs. BMC Evol Biol. 4:392004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carmena M and Earnshaw WC: The cellular
geography of aurora kinases. Nat Rev Mol Cell Biol. 4:842–854.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lo Iacono M, Monica V, Saviozzi S, Ceppi
P, Bracco E, Papotti M and Scaqliotti GV: Aurora kinase a
expression is associated with lung cancer histological-subtypes and
with tumor de-differentiation. J Transl Med. 9:1002011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu L, Wang C, Wen Z, Yao X, Liu Z, Li Q,
Wu Z, Xu Z, Liang Y and Ren T: Selective up-regulation of CDK2 is
critical for TLR9 signaling stimulated proliferation of human lung
cancer cell. Immunol Lett. 127:93–99. 2010. View Article : Google Scholar : PubMed/NCBI
|