Introduction
The μ-opioid receptor, a member of the G
protein-coupled receptor (GPCR) family characterized by a
seven-transmembrane structure, has been extensively investigated
(1). Activation of μ-opioid
receptors leads to the inhibition of adenylyl cyclase (AC) and thus
the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA)
pathway is suppressed (2). The
prolonged opioid exposure results in a comprehensive adaptation of
opioid receptor trafficking and signaling, including receptor
downregulation and desensitization (3). Furthermore, naloxone
precipitation-induced cAMP overshoot following chronic opioid
treatment has become an approved marker of cellular opioid
dependence (4). In addition to
inhibiting the cAMP/PKA pathway through the Gαi/o subunit, the
μ-opioid receptor was also revealed to crosstalk with the
extracellular signal-regulated kinase (ERK) cascade (Raf-MEK-ERK).
The stimulation of opioid receptors in cells may induce a notable
enhancement of phosphorylated ERK (pERK), which results in the
activation of the ERK cascade (5,6).
Furthermore, ERK activation was observed in the neurons in several
reward-associated brain regions and the administration of the MEK
inhibitor eliminated long-term potentiation in the hippocampus,
which is considered a classic example of synaptic plasticity
(7).
The crosstalk between the μ-opioid receptor pathway
and the ERK cascade involves multiple important molecules,
including PKA (8), β-arrestins
(9) and protein kinase C (PKC)
(10), although the exact
mechanism remains to be elucidated. PKC is a potent activator of
the ERK cascade. Early studies suggested that PKC directly
phosphorylated and activated Raf-1 (11,12),
however, subsequent studies indicated that PKC activated the ERK
cascade through a Ras-dependent pathway rather than direct
phosphorylation of Raf-1 (13,14).
Phosphatidylethanolamine-binding protein (PEBP),
also termed Raf kinase inhibitor protein, was identified to be an
endogenous negative regulator of the ERK cascade by binding to
Raf-1 kinase but not the B-Raf isoform (15,16).
The phosphorylation of PEBP at Ser153 induced by PKC resulted in
the release of PEBP from Raf-1 kinase and thus the PEBP-induced
inhibition of the ERK cascade was rescued (17). Previously, our research group found
that hippocampal PEBP was involved in morphine dependence in rats
and downregulation of hippocampal PEBP levels induced by antisense
oligodeoxynucleotides resulted in aggravated morphine dependence
(18). Due to the crucial role of
ERK in drug dependence, it is possible that PEBP phosphorylation
serves as a mechanism involved in μ-opioid receptor-mediated ERK
activation.
Materials and methods
Cell lines
The present study was approved by the ethics
committee of Beijing Institute of Pharmacology and Toxicology
(Beijing, China). SH-SY5Y cells were obtained from the Cell Culture
Center, Chinese Academy of Medical Science (Beijing, China) and
maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium
(Gibco-BRL, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (Gibco-BRL), 100 U/ml penicillin and 100 U/ml streptomycin
(Sigma-Aldrich, St. Louis, MO, USA) at 37°C in a humidified
atmosphere containing 5% CO2. Chinese hamster ovary
(CHO) cells, which stably express the rat μ-opioid receptor (CHO/μ
cells) were setup previously (19)
and were maintained in the same conditions as that for SH-SY5Y
cells with the addition of 200 μg/ml geneticin. Human embryonic
kidney (HEK)293 cells that stably express the human D1 dopamine
receptor or α1A adrenergic receptor were setup as previously
described (20,21) and cultured in DMEM medium,
supplemented with the same as that for CHO/μ cells.
Drug administration
All the drugs used in the present study, including
morphine, D-Ala2, MePhe4, Gly5-ol
enkephalin (DAMGO), naloxone, Gö6983, dopamine and phenylephrine
(PE) were purchased from Sigma-Aldrich. For acute drug treatment,
cells were seeded onto six-well plates for 24 h at densities of
5×105 cells/well (CHO/μ cells and HEK 293 cells) or
1×106 cells/well (SH-SY5Y cells), and were serum-starved
overnight upon reaching 80% confluence prior to stimulation by
different concentration of drugs or vehicles. For morphine chronic
treatment, cells were seeded onto six-well plates for 12 h prior to
treatment, at densities of 2×105 cells/well (CHO/μ
cells) or 5×105 cells/well (SH-SY5Y cells), and then
treated with morphine or vehicle for 36 h.
Immunoblotting analysis
Immunoblot was performed following drug treatment as
described previously (22).
Briefly, following stimulation, cells were washed twice with
chilled phosphate-buffered saline (Sigma-Aldrich). The cells were
placed on ice and 60 μl chilled lysis buffer [50 mM Tris, 150 mM
NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1 mg/l aprotinin, 1 mg/l
pepstatin, 1 mg/l leupeptin, 1 mM phenylmethylsulfonyl fluoride, 1
mM dithiothreitol, 2 mM NaF and 1 mM sodium vanadate (pH 7.4;
Sigma-Aldrich)] per well was added. Cells were scraped from plates
and transferred to a 1.5-ml Eppendorf tube. Following incubation on
ice for 30 min, the lysis was centrifuged at 14,000 × g for 20 min
at 4°C. Supernatant protein concentrations were determined using a
Bicinchoninic Acid Protein Assay kit (Pierce Biotechnology, Inc.,
Rockford, IL, USA). Aliquots of sample were boiled for 5 min in the
presence of 1X loading buffer (Pierce Biotechnology, Inc.). 40 μg
of protein was resolved using SDS-PAGE on 15% tricine gels and then
was transferred onto a polyvinylidene difluoride membrane
(Millipore, Billerica, MA, USA) for immunoblotting. Rabbit
monoclonal antibody against PEBP (1:1,000) and rabbit monoclonal
antibody against phospho-PEBP (S153) (1:200) were obtained from
Abcam (Cambridge, UK). Mouse monoclonal antibody against
phospho-ERK1/2 (1:2,000) and rabbit polyclonal antibody against
ERK1/2 (1:5,000) were obtained from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Subsequently, the phosphorylated proteins
were visualized and the phospho-antibodies were stripped from the
blots by incubating in stripping buffer for 1 h at 37°C. Blots were
subsequently reblocked and probed with antibodies against ERK or
PEBP. For ERK or PEBP activity, the quantity of pERK (combined
pERK1 and 2) or pPEBP protein was normalized to total ERK (combined
ERK1 and 2) or PEBP.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism® version 5.02 (GraphPad Software Inc., La Jolla,
CA, USA). All data are expressed as the mean ± standard error of
the mean and optical density values were determined using a gel
imaging system (Alpha Innotech, San Leandro, CA, USA). Student’s
t-test was used to compare the differences between two groups and
statistics between groups were assessed using analysis of variance
followed by Dunnett’s-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Acute opioid treatment induces transient
ERK, but not PEBP phosphorylation in CHO/μ cells and SH-SY5Y
cells
Initially, a CHO cell line was selected that
expressed exogenous rat μ-opioid receptors (2.9 pmol/mg membrane
protein) to examine the effect of acute opioid exposure on the
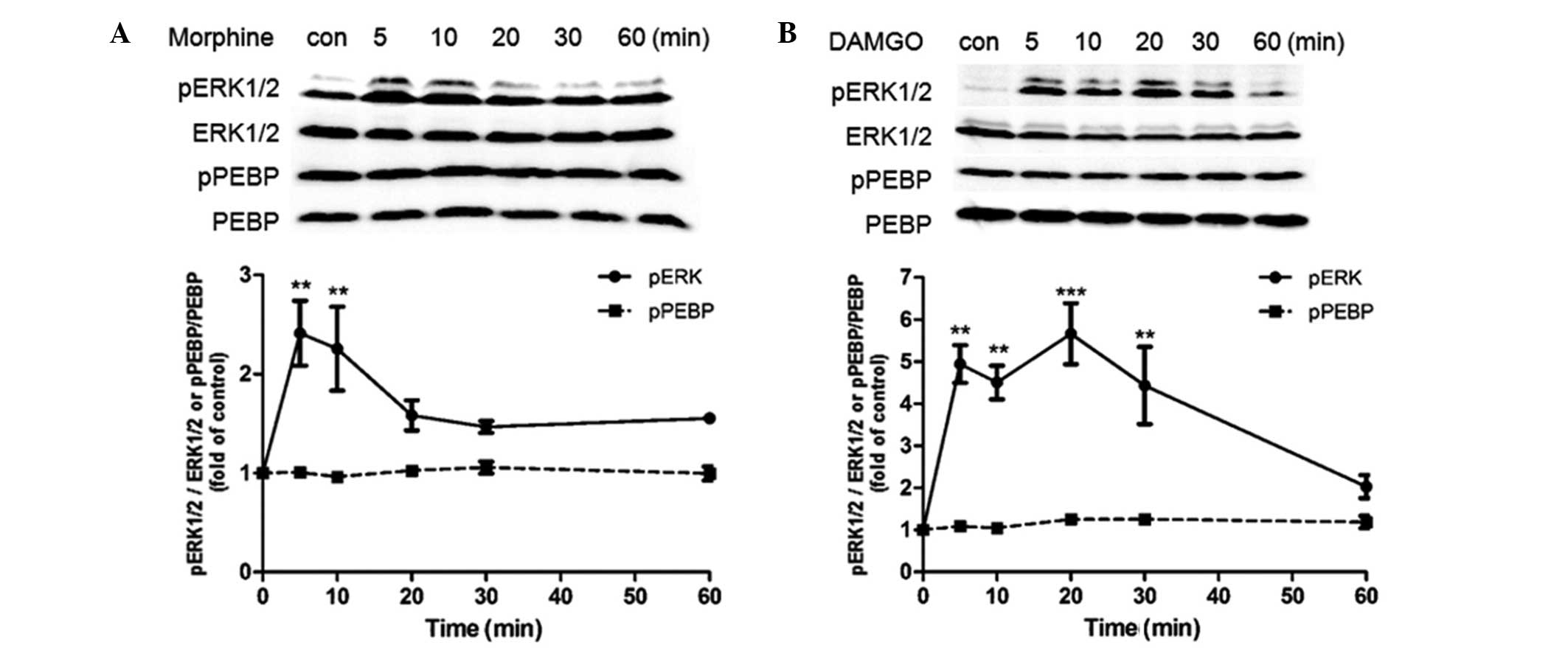
phosphorylation of ERK and PEBP. It was observed that 1 μM morphine
caused transient activation of ERK at 5 min (~2-fold compared with
the control treatment), which then decreased to the basal level.
However, no significant difference in expression of pPEBP was
observed during 1 h morphine treatment (Fig. 1A). Similarly to morphine, 1 μM
DAMGO also induced rapid ERK activation, which was sustained for a
longer time period than that induced by morphine (>30 min);
however, no significant difference in pPEBP was observed during 1 h
of DAMGO treatment (Fig. 1B). To
confirm that the activation of ERK is mediated by the μ-opioid
receptor, the μ-opioid receptor antagonist Naloxone (5 μM) was
selected to preincubate cells for 30 min prior to DAMGO treatment
and the result revealed that naloxone completely eradicated
DAMGO-induced ERK activation (data not shown), indicating that the
activation of ERK was mediated by μ-opioid receptors.
A similar investigation was also conducted in
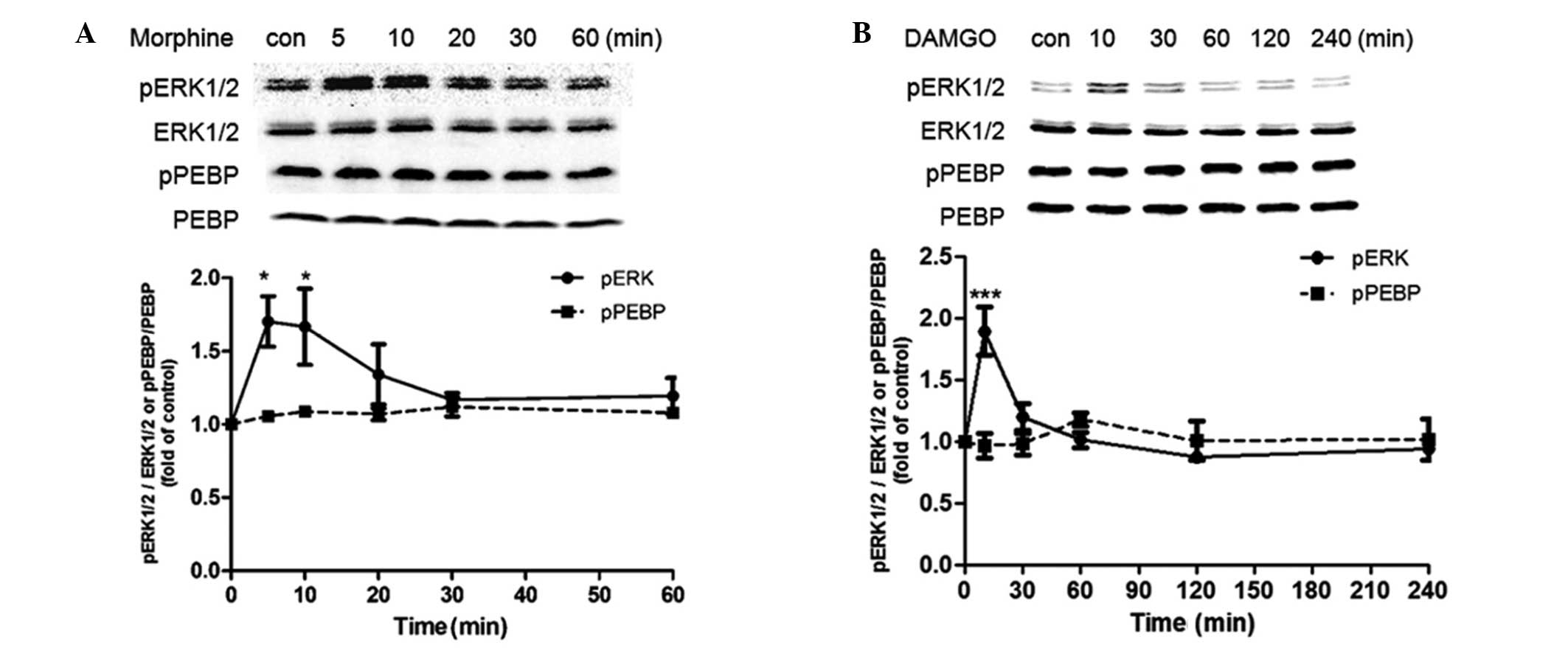
SH-SY5Y cells. Acute treatment with 10 μM morphine for 1 h caused
transient activation of ERK at 5 min (~1.7-fold compared with the
control treatment), which returned to basal levels after 30 min.
During this time course, however, the level of pPEBP exhibited
little change compared with the control treatment (Fig. 2A). For DAMGO treatment, a 4 h
treatment duration was investigated. It was found that DAMGO
induced a transient activation of ERK at 10 min (~1.9-fold compared
with the control treatment), which then returned to basal levels
after 30 min, but the level of pPEBP did not alter significantly
during 4 h of DAMGO treatment (Fig.
2B).
Chronic morphine treatment has no effect
on PEBP expression and phosphorylation
Since short-term opioid treatment did not induce any
significant change in pPEBP, whether chronic treatment with
morphine may affect the level of PEBP or pPEBP in these two cell
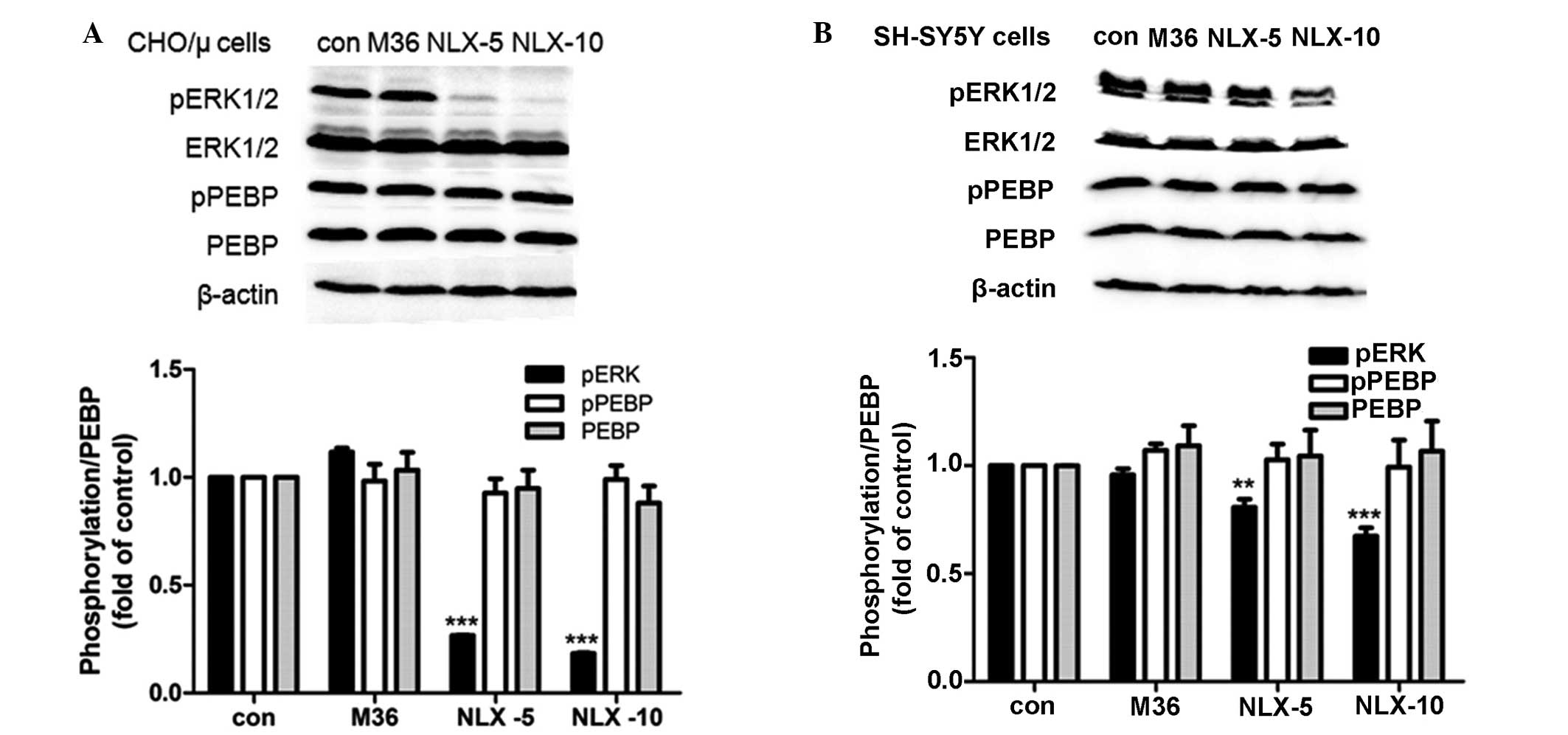
lines was investigated. It was found that chronic treatment with 10
μM morphine for 36 h did not affect PEBP expression level in CHO/μ
cells and SH-SY5Y cells, as well as the level of pPEBP. When the
cells were precipitated with 5 μM naloxone following chronic
morphine treatment, significantly decreased phosphorylation of ERK
was observed after 10 or 20 min of naloxone precipitation and
furthermore, the decreased phosphorylation of ERK in CHO/μ cells
(Fig. 3A) was greater than that in
SH-SY5Y cells (Fig. 3B). However,
the level of pPEBP did not alter in the two cell lines during the
course of naloxone precipitation (Fig.
3A and B).
Opioid-induced ERK phosphorylation is
inhibited by Gö6983
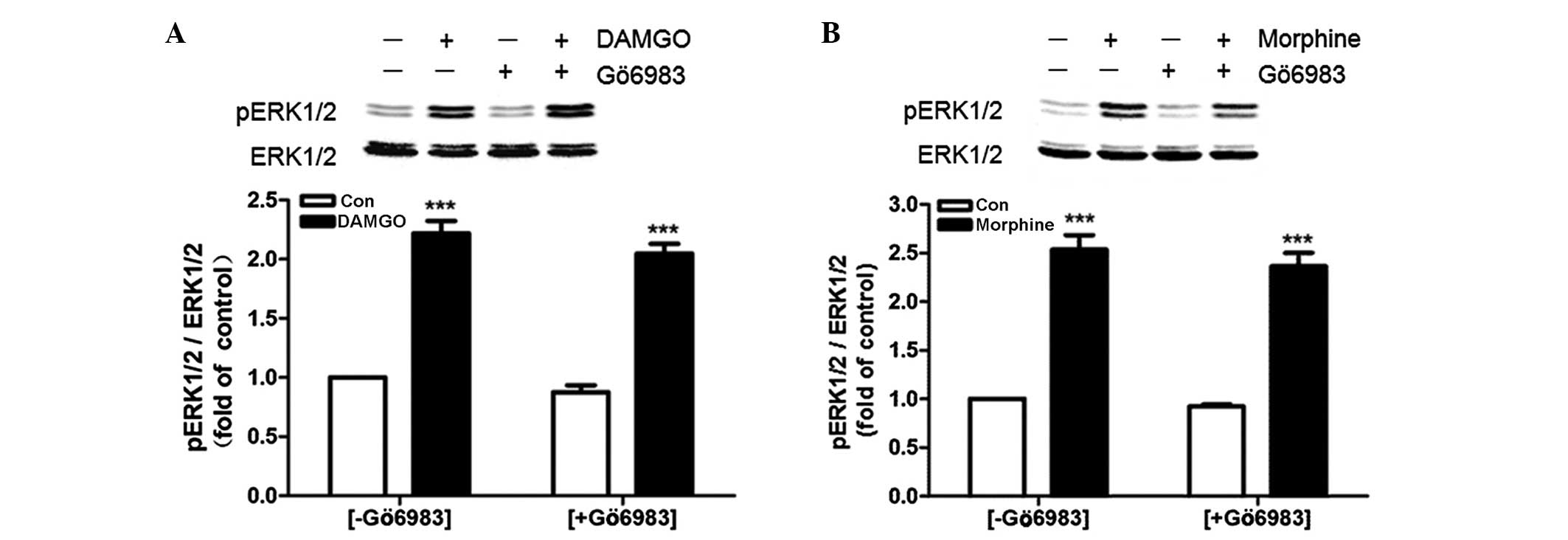
Pretreatment with Gö6983 (1μM) for 30 min did not
eradicate the rapid activation of ERK induced by 1 μM DAMGO
(Fig. 4A) or 1 μM morphine
(Fig. 4B).
Activation of the adrenergic α1A receptor
but not the dopamine D1 receptor induces phosphorylation of
PEBP
The present study continued to investigate whether
the activation of two other types of GPCR may affect the
phosphorylation of PEBP. A total of two HEK293 cell lines that
stably express dopamine D1 receptors (HEK293/D1 cells, 2.9 pmol/mg
membrane protein) and adrenergic α1A receptors (HEK293/α1A cells,
0.6 pmol/mg membrane protein) were used. For D1 dopamine receptors,
1 μM dopamine induced rapid and sustained ERK phosphorylation
during 60 min treatment with a peak at 5 min but did not induce any
change in pPEBP compared with the control treatment (Fig. 5A). For the α1A adrenergic receptor,
a Gq-coupled receptor, rapid activation of ERK and also a
significant upregulation of pPEBP was observed during 60 min of
treatment with 1 μM PE (Fig. 5B).
The ERK activation was rapid with a peak (5–6 fold of control) at 5
min and then a significant decrease (~2 fold of control) until 30
min. The phosphorylation of PEBP gradually increased following the
phosphorylation of ERK. Gö6983 preincubation completely eradicated
PE-induced PEBP phosphorylation (Fig.
5C) and significantly reduced the level of ERK phosphorylation
(Fig. 5D).
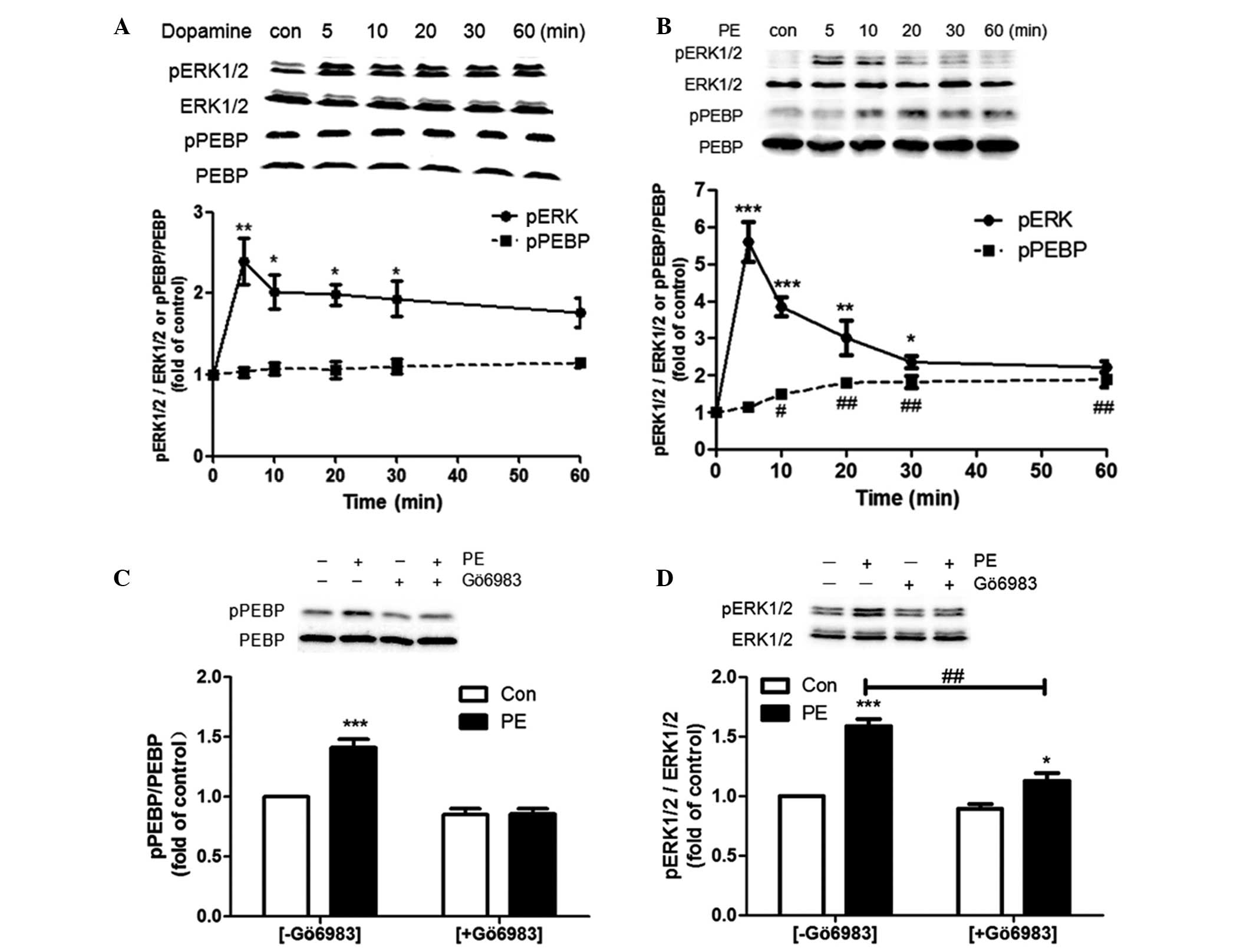 | Figure 5Effect of the activation of the
dopamine D1 receptor and adrenergic α1A receptor on ERK and PEBP
phosphorylation in HEK293 cells. (A) HEK293/D1 cells were treated
with 1 μM dopamine for the indicated time intervals. (B) HEK293/α1A
cells were treated with 1 μM PE for the indicated time intervals.
One-way analysis of variance was used followed by Dunnett’s post
hoc test. For pERK, *P<0.05, **P<0.01
and ***P<0.001, compared with the Con; for pPEBP,
#P<0.05 and ##P<0.01, compared with the
Con. (C and D) HEK293/α1A cells were pretreated with 1 μM Gö6983 or
vehicle (dimethyl sulfoxide) for 30 min and were then stimulated
with 1 μM PE for 30 min. Two-way analysis of variance was used
followed by the Bonferroni post hoc test. *P<0.05 and
***P<0.001, compared with each Con. Student’s t-test
was used for the ERK activity comparison with or without Gö6983
(##P<0.01). All data are presented as the mean ±
standard error of the mean from three independent experiments.
PEBP, phosphatidylethanolamine-binding protein; PE, phenylephrine;
ERK, extracellular signal-regulated kinase; CHO cells, Chinese
hamster ovary cells; HEK, human embryonic kidney; Con, control. |
Discussion
The crosstalk between opioid receptors and ERK has
undergone comprehensive investigation, however, the underlying
mechanism remains to be elucidated. PEBP is a small (21–23 kDa)
soluble protein, which is widely expressed in cells and tissues
(23) and was previously
identified to be a negative regulator of ERK activation (15). However, the role of PEBP in
μ-opioid receptor-mediated activation of ERK remains to be
elucidated. PKC activity appears to be involved in opioid-induced
ERK activation, due to specific evidence, which suggested that
DAMGO induced PKC activation in SH-SY5Y cells by stimulating
μ-opioid receptors (24) and
(D-Pen2, D-Pen5)-enkephalin induced PKC activation in NG-108 cells
by stimulating δ opioid receptors (25). Since PKC-induced phosphorylation of
PEBP results in the disinhibition of Raf-1 signaling, which leads
to the activation of ERK (26), it
is possible that μ-opioid receptor-mediated activation of PKC may
induce the phosphorylation of PEBP, which then regulates the
activity of ERK through Raf-1 kinase. However, the present study
demonstrated that activation of the μ-opioid receptor did not
regulate the phosphorylation of PEBP.
In the present study, two cell lines that express
different levels of μ-opioid receptors were used. CHO/μ cells were
selected as they express high levels of μ-opioid receptors, which
is an advantage for the functional study of opioids, while SH-SY5Y
cells were selected due to their similarity to the neurons that
express endogenous μ-opioid receptors (27). Morphine and DAMGO are two
well-investigated agonists of the μ-opioid receptor, characterized
by differing structure and efficacy. The intracellular signaling
mechanisms mediated by DAMGO are often largely different from that
mediated by morphine, including receptor phosphorylation,
internalization and desensitization (28–31),
therefore, these two agonists were used to activate μ-opioid
receptors in the present study.
The data presented in the present study provide
evidence that morphine and DAMGO induce a rapid activation of ERK;
however, no significant phosphorylation of PEBP was observed
following short-term opioid treatment in CHO/μ cells and SH-SY5Y
cells. Kramer et al (24)
reported a significant PKC translocation to the cell membrane of
SH-SY5Y cells after 4 h of DAMGO stimulation (~2-fold of control),
therefore, 4 h was selected as the end point of the observation.
However, a corresponding elevation of pPEBP was not observed and
the phosphorylation of ERK induced by DAMGO was only augmented in
the first 30 min and then returned to the basal level until the end
of stimulation. The present findings suggested that μ-opioid
receptor-mediated rapid ERK activation was not associated with PEBP
phosphorylation and short-term stimulation of μ-opioid receptor did
not induce the change in pPEBP levels, even with different
agonists.
Since PKC activation caused the activation of ERK,
whether μ-opioid receptor-induced rapid ERK activation involved PKC
activity was assessed in the present study. Gö6983 is a selective
inhibitor for the majority of PKC isoenzymes, including PKC α, β,
γ, δ and ξ (32), among which PKC
α, β, γ and ξ were responsible for PEBP phosphorylation at Ser153
(17). Therefore, the application
of Gö6983 may inhibit PKC-induced PEBP phosphorylation. It was
found that DAMGO and morphine-induced ERK activation in SH-SY5Y
cells was independent of Gö6983-sensitive PKC activity. A similar
result was also obtained in rat cortical astrocytes in a study by
Belcheva et al (33).
Long-term opioid treatment may cause a comprehensive
adaption of opioid receptor trafficking and signaling, including AC
superactivation. It has been reported that sustained morphine
treatment augmented forskolin-stimulated cAMP formation (34,35)
and the withdrawal using naloxone following chronic opioid
treatment led to cAMP overshoot (36). The present study found that
prolonged morphine treatment had no effect on the phosphorylation
level of ERK compared with that induced by the vehicle. However, a
significant downregulation of pERK was observed in CHO/μ cells and
SH-SY5Y cells that were precipitated with naloxone after 36 h of
morphine treatment, which was also demonstrated in previous studies
(37,38). In vivo, chronic morphine
administration resulted in differential regulation of ERK activity
in different brain regions, including a decrease in pERK in the
cerebral cortex (39) and an
increase in pERK at the spinal level (40). For a cell line, the acute elevation
of intracellular cAMP resulting from naloxone precipitation may
enhance PKA activity, acting as a negative regulator of Raf-1
kinase (10), possibly
contributing to decreased phosphorylation of ERK. After 36 h of
morphine treatment, it was observed that chronic morphine exposure
had no effect on the expression level of PEBP, as well as the
phosphorylation of PEBP. However, there are studies suggesting that
PKC activity was upregulated in vivo following chronic
morphine administration (41,42)
and in addition no significant phosphorylation of PEBP was
identified even in the cells precipitated with naloxone. The
present results indicated that PEBP possibly did not contribute to
the cellular adaptation induced by chronic morphine treatment
through the alteration in either phosphorylation or expression,
particularly in the modulation of ERK.
The possible modulation of PEBP phosphorylation by
other types of GPCR evoked our interest. Besides Gi/o-coupled
μ-opioid receptor, the Gs-coupled dopamine D1 receptor and
Gq-coupled adrenergic α1A receptor were also investigated to
examine the effect of receptor activation on PEBP phosphorylation.
It was found that the activation of the dopamine D1 receptor
induced sustained ERK activation, but failed to alter the level of
pPEBP during 60 min of dopamine treatment, indicating that PEBP
phosphorylation was not involved in the activation of ERK induced
by the Gs-coupled receptor. Lefkowitz et al (43) has described a mechanism of
Gs-dependent ERK activation: The activation of
Gs-coupled receptor induces accumulation of cAMP and
Rap-1 is activated by cAMP and B-Raf is also activated by Rap-1,
thus ERK is activated.
For the Gq-coupled adrenergic α1A receptor, however,
activation leads to the elevation of intracellular diacylglycerol
and Ca2+ (44), which
are activators of PKC, therefore, it was expected that PE induced
the phosphorylation of PEBP. It was found that PE-induced PEBP
phosphorylation was delayed compared with ERK activation, similar
to that induced by PMA in SH-SY5Y cells (data not shown),
indicating that the rapid activation of ERK mediated by the
adrenergic α1A receptor was not as a result of PEBP
phosphorylation. However, it was unknown whether successive ERK
phosphorylation following the acute phase was associated with PEBP
phosphorylation. Inhibition of PKC completely eradicated PE-induced
PEBP phosphorylation and significantly reduced the level of ERK
activation, indicating that adrenergic α1A receptor-mediated PEBP
and ERK phosphorylation are PKC activity dependent.
Taken together, the present results demonstrated
that activation of the μ-opioid receptor does not modulate the
phosphorylation of PEBP and PEBP did not contribute to
GPCR-mediated rapid activation of ERK. Thus, PEBP may have a minor
role in μ-opioid receptor-mediated ERK regulation.
Acknowledgements
The present study was supported by the Beijing
Municipal Natural Science Foundation (grant no. 7082075), the
National Natural Science Foundation of China (grant no. 30772563)
and the National Basic Research Program of China (grant no.
2009CB522008).
References
|
1
|
Lefkowitz RJ: Historical review: A brief
history and personal retrospective of seven-transmembrane
receptors. Trends Pharmacol Sci. 25:413–422. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Law PY, Wong YH and Loh HH: Molecular
mechanisms and regulation of opioid receptor signaling. Annu Rev
Pharmacol Toxicol. 40:389–430. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bailey CP and Connor M: Opioids: cellular
mechanisms of tolerance and physical dependence. Curr Opin
Pharmacol. 5:60–68. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Watts VJ: Molecular mechanisms for
heterologous sensitization of adenylate cyclase. J Pharmacol Exp
Ther. 302:1–7. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eisinger DA and Ammer H: δ-Opioid
receptors activate ERK/MAP kinase via integrin-stimulated receptor
tyrosine kinases. Cell Signal. 20:2324–2331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zheng H, Loh HH and Law PY:
β-arrestin-dependent μ-opioid receptor-activated extracellular
signal-regulated kinases (ERKs) translocate to nucleus in contrast
to G protein-dependent ERK activation. Mol Pharmacol. 73:178–190.
2008. View Article : Google Scholar :
|
|
7
|
Girault A, Valjent E, Caboche J and Herve
D: ERK2: a logical and gate critical for drug-induced plasticity?
Curr Opin Pharmacol. 7:77–85. 2007. View Article : Google Scholar
|
|
8
|
Dhillon AS, von Kriegsheim A, Grindlay J
and Kolch W: Phosphatase and feedback regulation of Raf-1
signaling. Cell Cycle. 6:3–7. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lefkowitz RJ and Shenoy SK: Transduction
of receptor signals by β-arrestins. Science. 308:512–517. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chong H, Vikis HG and Guan KL: Mechanisms
of regulating the Raf kinase family. Cell Signal. 15:463–469. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cacace AM, Ueffing M, Philipp A, et al:
PKC epsilon functions as an oncogene by enhancing activation of the
Raf kinase. Oncogene. 13:2517–2526. 1996.PubMed/NCBI
|
|
12
|
Cai H, Smola U, Wixler V, et al: Role of
diacylglycerol-regulated protein kinase C isotypes in growth factor
activation of the Raf-1 protein kinase. Mol Cell Biol. 17:732–741.
1997.PubMed/NCBI
|
|
13
|
Marais R, Light Y, Mason C, et al:
Requirement of Ras-GTP-Raf complexes for activation of Raf-1 by
protein kinase C. Science. 280:109–112. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng Y, Liu H, Coughlin J, et al:
Phosphorylation of RasGRP3 on threonine 133 provides a mechanistic
link between PKC and Ras signaling systems in B cells. Blood.
105:3648–3654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yeung KC, Steitz T, Li S, et al:
Suppression of Raf-1 kinase activity and MAP kinase signalling by
RKIP. Nature. 401:173–177. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yeung KC, Janosch P, McFerran B, et al:
Mechanism of suppression of the Raf/MEK/extracellular
signal-regulated kinase pathway by the raf kinase inhibitor
protein. Mol Cell Biol. 20:3079–3085. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Corbit KC, Trakul N, Eves EM, et al:
Activation of Raf-1 signaling by protein kinase C through a
mechanism involving Raf kinase inhibitory protein. J Biol Chem.
278:13061–13068. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wei QH, Wu N, Bian JM, et al: Involvement
of hippocampal phosphatidylethanolamine-binding protein in morphine
dependence and withdrawal. Addict Biol. 18:230–240. 2013.
View Article : Google Scholar
|
|
19
|
Wu N, Su RB, Xu B, et al: IRAS, a
candidate for I1-imidazoline receptor, mediates inhibitory effect
of agmatine on cellular morphine dependence. Biochem Pharmcol.
70:1079–1087. 2005. View Article : Google Scholar
|
|
20
|
Cao Y, Xie KQ and Zhu XZ: The enhancement
of dopamine D1 receptor desensitization by adenosine A1 receptor
activation. Eur J Pharmacol. 562:34–38. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lei B, Zhang Y and Han C: Sustained
norepinephrine stimulation induces different regulation of
expression in three α1-adrenoceptor subtypes. Life Sci. 69:301–308.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li F, Wu N, Su RB, et al: Comparison of
agmatine with moxonidine and rilmenidine in morphine dependence in
vitro: role of imidazoline I(1) receptors. Eur J Pharmacol.
612:1–8. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Theroux S, Pereira M, Casten KS, et al:
Raf kinase inhibitory protein knockout mice: Expression in the
brain and olfaction deficit. Brain Res Bull. 71:559–567. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kramer HK and Simon EJ: Role of protein
kinase C (PKC) in agonist-induced μ-opioid receptor
down-regulation: I. PKC translocation to the membrane of SH-SY5Y
neuroblastoma cells is induced by μ-opioid agonists. J Neurochem.
72:585–593. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lou LG and Pei G: Modulation of protein
kinase C and cAMP-dependent protein kinase by δ-opioid. Biochem
Biophys Res Commun. 236:626–629. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lorenz K, Lohse MJ and Quitterer U:
Protein kinase C switches the Raf kinase inhibitor from Raf-1 to
GRK-2. Nature. 426:574–579. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kazmi SM and Mishra RK: Comparative
pharmacological properties and functional coupling of mu and delta
opioid receptor sites in human neuroblastoma SH-SY5Y cells. Mol
Pharmacol. 32:109–118. 1987.PubMed/NCBI
|
|
28
|
McPherson J, Rivero G, Baptist M, et al:
μ-opioid receptors: correlation of agonist efficacy for signalling
with ability to activate internalization. Mol Pharmacol.
78:756–766. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu Y, Zhang L, Yin X, et al: Mu opioid
receptor phosphorylation, desensitization, and ligand efficacy. J
Biol Chem. 272:28869–28874. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Johnson EA, Oldfield S, Braksator E, et
al: Agonist-selective mechanisms of μ-opioid receptor
desensitization in human embryonic kidney 293 cells. Mol Pharmacol.
70:676–685. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zuo Z: The role of opioid receptor
internalization and β-arrestins in the development of opioid
tolerance. Anesth Analg. 101:728–734. 2005. View Article : Google Scholar
|
|
32
|
Gschwendt M, Dieterich S, Rennecke J, et
al: Inhibition of protein kinase C by various inhibitors.
Differentiation from protein kinase C isoenzymes. FEBS Lett.
392:77–80. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Belcheva MM, Clark AL, Haas PD, et al: μ
and κ opioid receptors activate ERK/MAPK via different protein
kinase C isoforms and secondary messengers in astrocytes. J Biol
Chem. 280:27662–27669. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yue X, Tumati S, Navratilova E, et al:
Sustained morphine treatment augments basal CGRP release from
cultured primary sensory neurons in a Raf-1 dependent manner. Eur J
Pharmacol. 584:272–277. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nevo I, Avidor-Reiss T, Levy R, et al:
Regulation of adenylyl cyclase isozymes upon acute and chronic
activation of inhibitory receptors. Mol Pharmacol. 54:419–426.
1998.PubMed/NCBI
|
|
36
|
Nevo I, Avidor-Reiss T, Levy R, et al:
Acute and chronic activation of the μ-opioid receptor with the
endogenous ligand endomorphin differentially regulates adenylyl
cyclase isozymes. Neuropharmacology. 39:364–371. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Z and Sadée W: Tolerance to morphine
at the mu-opioid receptor differentially induced by cAMP-dependent
protein kinase activation and morphine. Eur J Pharmacol.
389:165–171. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bilecki W, Zapart G, Ligęza A, et al:
Regulation of the extracellular signal-regulated kinases following
acute and chronic opioid treatment. Cell Mol Life Sci.
62:2369–2375. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ferrer-Alcón M, García-Fuster MJ, La Harpe
R, et al: Long-term regulation of signalling components of adenylyl
cyclase and mitogen-activated protein kinase in the pre-frontal
cortex of human opiate addicts. J Neurochem. 90:220–230. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cao JL, He JH, Ding HL, et al: Activation
of the spinal ERK signaling pathway contributes
naloxone-precipitated withdrawal in morphine-dependent rats. Pain.
118:336–349. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mao J, Price DD, Phillips LL, et al:
Increases in protein kinase C gamma immunoreactivity in the spinal
cord of rats associated with tolerance to the analgesic effects of
morphine. Brain Res. 677:257–267. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Smith FL, Lohmann AB and Dewey WL:
Involvement of phospholipid signal transduction pathways in
morphine tolerance in mice. Br J Pharmacol. 128:220–226. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lefkowitz RJ, Pierce K and Luttrell LM:
Dancing with different partners: protein kinase A phosphorylation
of seven membrane-spanning receptors regulates their G
protein-coupling specificity. Mol Pharmacol. 62:971–974. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rhee SG: Regulation of
phosphoinositide-specific phospholipase C. Annu Rev Biochem.
70:281–312. 2001. View Article : Google Scholar : PubMed/NCBI
|