Introduction
Werner Syndrome (WS; OMIM entry no. 277700;
https://omim.org/entry/277700) is a rare
progeroid syndrome associated with a number of aging phenotypes. WS
often occurs in consanguineous families and affects ~1 in 1 million
individuals in the general population (1–3).
Patients with WS often exhibit adult-onset progeria and have an
increased risk of developing cancer (4). At birth, patients with WS do not
present any clinical symptoms; the lack of pubertal growth spurts
are generally the first symptom identified (5). Over time, the typical characteristics
of WS become evident, including a ‘bird-like’ appearance,
cataracts, canities, ulcerations around the ankles and certain
patients develop hypogonadism with atrophic genitalia and
infertility (6). The average life
span of affected individuals is 54 years (5).
WS is caused by mutations in the Werner syndrome
RecQ like helicase gene (WRN), which was first cloned in
1996 (7). WRN is located on
chromosome 8p11-p12, spanning ~250 kb and consists of 35 exons, 34
of which are protein coding (7).
WRN is generally considered to follow an autosomal-recessive
pattern of inheritance (8).
WRN, coding a 180 kDa multifunctional nuclear protein,
belongs to the family of RecQ type helicases (9). Sequence analysis and subsequent
biochemical analysis have revealed that human WRN possesses
helicase and exonuclease functions (10). It serves a role in DNA replication,
transcription, repair, recombination and heterochromatin
maintenance (including telomere maintenance), indicating that one
of the major causes of WS pathogenesis may be associated with
genomic instability (11). In the
absence of functioning WRN, cells accumulate potentially toxic DNA
intermediates or critically short telomeres, which induce genetic
instability, misexpression and mutagenesis (12–14).
In addition, they may drive cell loss and produce tissue-specific
defects (15). Compromised cell or
tissue structure and function leads to two seemingly divergent
outcomes: Senescence and neoplasms (16,17).
To date, the majority of disease-inducing mutations in WRN
are truncating mutations (5).
In addition to WRN, a small number of
heterozygous mutations in the lamin A/C gene (LMNA) have
been revealed to produce similar phenotypes to WS, which suggests
that LMNA may be an underlying disease-inducing gene in WS
(3,18,19).
LMNA encodes nuclear intermediate filaments, lamin A and
lamin C (1). A previous study
demonstrated that WS patients with LMNA mutations exhibited
a younger onset when compared to patients with classical WS
(5). In addition, a number of
studies have suggested that barrier to autointegration factor 1
(BANF1), zinc metallopeptidase STE24 (ZMPSTE24) and
DNA polymerase Δ1 (POLD1) may be involved in progeroid
syndrome (15,20,21).
The present study investigated the potential
causative gene in a consanguineous family with WS from Northwest
China. A novel homozygous splice-site mutation (c.IVS28+2T>C)
was identified in intron 28 of WRN in the proband and
co-segregated with the affected WS family members. To the best of
our knowledge, this mutation has not been reported in previous
studies, nor was it identified in our previous control cohorts or
the single nucleotide polymorphism (dbSNP) database (https://www.ncbi.nlm.nih.gov/projects/SNP/) and the
Exome Variant Server database (http://evs.gs.washington.edu/EVS/).
Materials and methods
Patients
A consanguineous family from Northwest China (Gansu
province) consisting of 11 living members across four generations
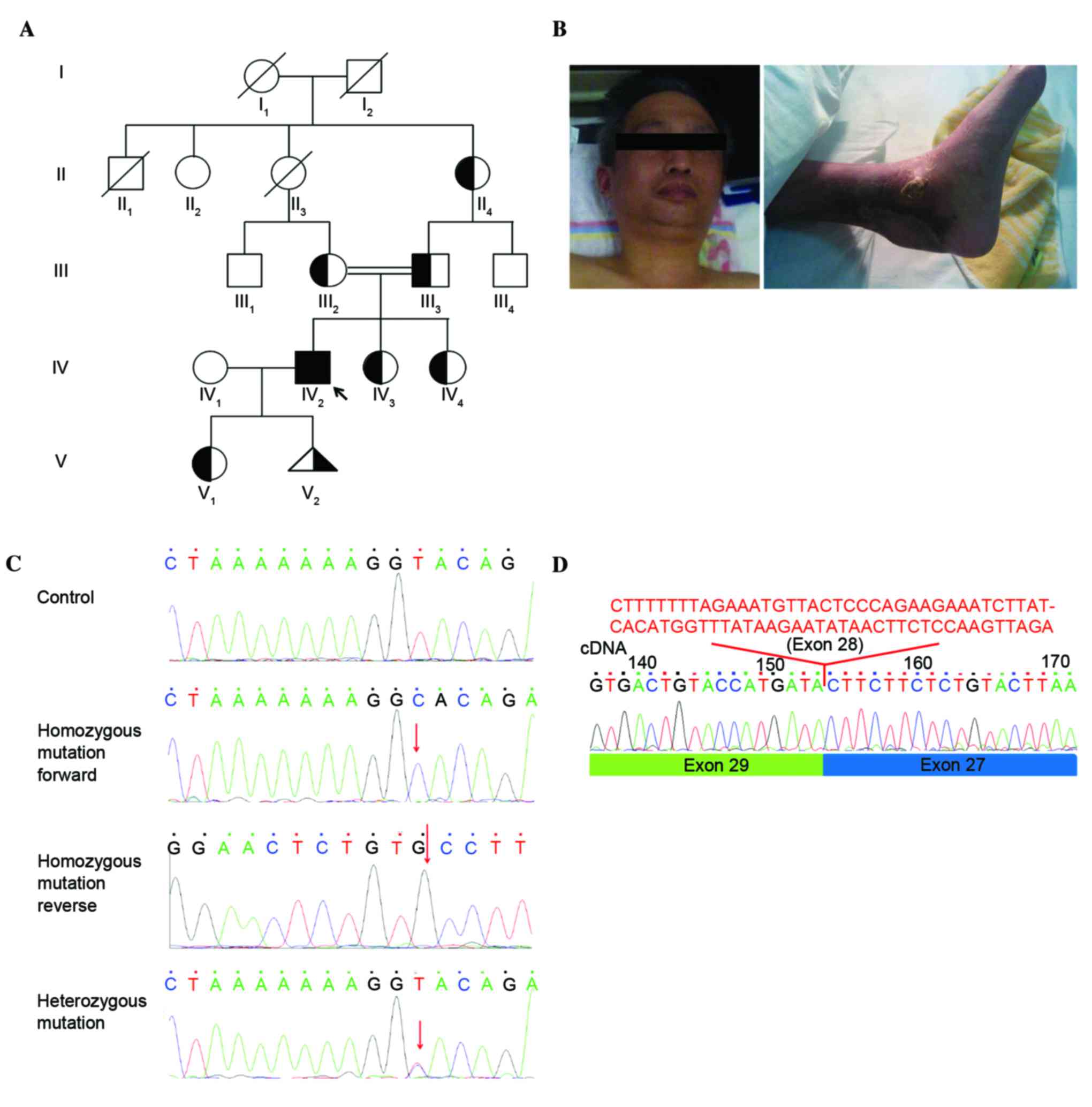
participated in the present study (Fig. 1A). The proband, family member 2
from the 4th generation (IV:2), was diagnosed with WS. The
remaining 10 members (II:2, II:4, III:1, III:2, III:3, III:4, IV:1,
IV:3, IV:4 and V1) were phenotypically normal. The proband was
admitted to Xiangya Hospital in April 2015 (Changsha, China) for
treatment of an ankle ulcer. The 38-year-old patient (gender, male)
had a ‘bird-like’ face, gray hair, a husky voice and a recurrent
ulceration around his ankle which first presented 12 years
previously (Fig. 1B). The Review
Board of Xiangya Hospital of the Central South University (Hunan,
China) approved this research and all family members involved gave
written informed consent.
DNA extraction
Genomic DNA was extracted from the peripheral blood
of the patient and the other family members using a DNeasy Blood
& Tissue kit (Qiagen, Inc., Valencia, CA, USA) on the QIAcube
automated DNA extraction robot (Qiagen, Inc.).
Mutation sequencing
The entire coding regions, including the flanking
intronic sequences of WRN [Refseq (https://www.ncbi.nlm.nih.gov/refseq/), NM_000553],
LMNA (NM_170,707), BANF1 (NM_003860), ZMPSTE24
(NM_005857) and POLD1 (NM_001256849) were amplified by
polymerase chain reaction (PCR; primer sequences are available upon
request). PCR product sequences were determined using the ABI 3100
Genetic Analyzer (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
as previously described (22).
Multiple sequence alignments and
bioinformatic prediction of mutation
The multiple WRN protein sequences across mammals
were aligned using the multiple sequence comparison by
log-expectation program (version 3.6; https://www.ncbi.nlm.nih.gov, and the MUSCLE software)
(23,24).
RNA extraction and reverse
transcription for verification
Total RNA was extracted from mononuclear cells from
the peripheral blood of the patient using the Nuclearspin RNA II
kit (Macherey-Nagel GmbH, Düren, Germany) and DNase (DNase I,
RNase-free (1 U/µl); Thermo Fisher Scientific, Inc.) treated
(25). Reverse transcription
(RT)-qPCR was performed to convert extracted total RNA into cDNA
using the PrimeScript RT reagent kit (Takara Bio, Inc., Otsu,
Japan) according to the manufacturer's instructions (26). cDNA was then sequenced following
amplification by PCR. The PCR was conducted in a 25 µl reaction
mixture, which consisted of 0.3 mM deoxyribonucleotide
triphosphates, 1X PCR buffer (10 mM Tris-HCl pH 9.0, 50 mM KCl,
0.1% Triton X-100, and 0.01% w/v gelatin), 2.0 mM MgCl2, 0.5 µM of
each primer (forward and reverse), 1.5 U of Taq polymerase, and 50
ng genomic DNA. The thermal cycling consisted of an initial
denaturation at 95°C for 4 min, followed by 35 cycles of
amplification consisting of denaturation at 95°C for 1 min, primer
annealing at 55–61°C for 30 sec and primer extension at 72°C for 1
min. A final extension step was performed at 72°C for 7 min. The
results were compared to the normal control for variant analysis
via multiple sequence alignment using the MUSCLE software (24).
Results
The present study investigated the case of a patient
with WS (IV:2) born to consanguineous parents (family members III:2
and III:3) in a family comprised of four generations with 11 living
members. The proband, with a ‘bird-like’ face, husky voice,
canities and ankle ulcers, conforms to the typical phenotypes
associated with WS. The present study investigated the potential
causative genes among all family members. Sequence analysis of
WRN, LMNA, BANF1, ZMPSTE24 and
POLD1, identified a previously unreported homozygous
splice-site mutation in intron 28 (c.IVS28+2T>C) of the
WRN gene in the proband (IV:2) and co-segregated with the
affected family members (Fig. 1C).
No further relevant mutations were identified by direct sequencing
of the genes for LMNA, BANF1, ZMPSTE24 and
POLD1. In addition, the cDNA sequencing results from the
proband identified deletion of WRN exon 28 (74 nucleotides;
Fig. 1D). This newly discovered
c.IVS28+2T>C mutation was not identified in the 200 control
cohorts that our group studied previously (23). In addition, this mutation was not
present in the dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/) or Exome
Variant Server databases (http://evs.gs.washington.edu/EVS).
Discussion
The WRN gene encodes a nuclear protein which
belongs to the family of RecQ type helicases and comprises of five
functional domains including an exonuclease region, a helicase
region, a RecQ C-terminus consensus region, an RNase D consensus
region and a nuclear localization signal region (NLS) (27). According to previous studies,
homology-dependent recombination repair (HDR) may be used to repair
DNA damage while suppressing gene loss or rearrangement. In
addition, WRN appears to serve a role late in the HDR process when
recombinant molecules are topologically disentangled for
segregation to daughter cells (28,29),
as well as in the maintenance of telomere length and the
suppression of telomere sister-chromatid exchanges (30,31).
WRN may also be involved in non-homologous DNA-end joining,
base-excision repair, DNA-damage signaling and transcription
(15). Zhang et al
(11) revealed that the
progressive heterochromatin disorganization observed in
WRN-deficient mesenchymal stem cells underlies cellular aging
(32). Thus, loss of WRN may
disrupt genetic stability, lead to cell aging and death. Therefore,
it may be pivotal in human aging and the development of WS.
The present study revealed that the novel mutation
(c.IVS28+2T>C) causes a change in the splicing pattern, leading
to a deletion of 74 nucleotides in the mRNA. It suggested that the
substitution and subsequent skipping of exon 28 may produce a
frameshift transcript, resulting in the absence of the NLS domain.
The NLS domain is essential for WRN protein targeting to the
nucleus via the nuclear pore complex (33). In addition, the majority of
previously reported causative WRN mutations also give rise to a
lack of NLS at the C-terminus of the protein (Fig. 2) (5,34).
During this research, the wife of the proband (IV:1)
was pregnant. It was speculated that the infant (V:2) may be a
carrier without any pathological phenotype. However, amniocentesis
or shotgun sequencing of maternal plasma DNA is required to produce
accurate and convincing results to verify this. As cancer
predisposition is a key feature in WS, the proband may have a
higher risk of cancer development and should therefore have regular
health examinations.
In conclusion, the present study identified a novel
homozygous splice-site mutation (c.IVS28+2T>C) in a four
generation family with WS. The present identification of a novel
mutation expands the spectrum of known WRN mutations (only 87
mutations were identified as of February 2015; http://www.hgmd.cf.ac.uk/ac/search.php)
and it may contribute to novel approaches to genetic diagnosis and
counseling of families with WS.
Acknowledgements
The authors would like to thank the State Key
Laboratory of Medical Genetics of China (Hunan, China) for their
technical assistance. The present study was supported by the
National Natural Science Foundation of China (Beijing, China; grant
no. 81370394) and the National Basic Research Program of China (973
Program; Beijing, China; grant no. 2012CB517900).
References
|
1
|
Masala MV, Scapaticci S, Olivieri C,
Pirodda C, Montesu MA, Cuccuru MA, Pruneddu S, Danesino C and
Cerimele D: Epidemiology and clinical aspects of Werner's syndrome
in North Sardinia: Description of a cluster. Eur J Dermatol.
17:213–216. 2007.PubMed/NCBI
|
|
2
|
Hasty P, Campisi J, Hoeijmakers J, van
Steeg H and Vijg J: Aging and genome maintenance: Lessons from the
mouse? Science. 299:1355–1359. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen L, Lee L, Kudlow BA, Dos Santos HG,
Sletvold O, Shafeghati Y, Botha EG, Garg A, Hanson NB, Martin GM,
et al: LMNA mutations in atypical Werner's syndrome. Lancet.
362:440–445. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Muftuoglu M, Oshima J, von Kobbe C, Cheng
WH, Leistritz DF and Bohr VA: The clinical characteristics of
Werner syndrome: Molecular and biochemical diagnosis. Hum Genet.
124:369–377. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oshima J and Hisama FM: Search and
insights into novel genetic alterations leading to classical and
atypical Werner syndrome. Gerontology. 60:239–246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Friedrich K, Lee L, Leistritz DF, Nürnberg
G, Saha B, Hisama FM, Eyman DK, Lessel D, Nürnberg P, Li C, et al:
WRN mutations in Werner syndrome patients: Genomic rearrangements,
unusual intronic mutations and ethnic-specific alterations. Hum
Genet. 128:103–111. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama
F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, et al:
Positional cloning of the Werner's syndrome gene. Science.
272:258–262. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sinha JK, Ghosh S and Raghunath M:
Progeria: A rare genetic premature ageing disorder. Indian J Med
Res. 139:667–674. 2014.PubMed/NCBI
|
|
9
|
Choudhary S, Sommers JA and Brosh RM Jr:
Biochemical and kinetic characterization of the DNA helicase and
exonuclease activities of werner syndrome protein. J Biol Chem.
279:34603–34613. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Orren DK, Theodore S and Machwe A: The
Werner syndrome helicase/exonuclease (WRN) disrupts and degrades
D-loops in vitro. Biochemistry. 41:13483–13488. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang W, Li J, Suzuki K, Qu J, Wang P,
Zhou J, Liu X, Ren R, Xu X, Ocampo A, et al: Aging stem cells. A
Werner syndrome stem cell model unveils heterochromatin alterations
as a driver of human aging. Science. 348:1160–1163. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Laud PR, Multani AS, Bailey SM, Wu L, Ma
J, Kingsley C, Lebel M, Pathak S, De Pindo RA and Chang S: Elevated
telomere-telomere recombination in WRN-deficient, telomere
dysfunctional cells promotes escape from senescence and engagement
of the ALT pathway. Gen Dev. 19:2560–2570. 2005. View Article : Google Scholar
|
|
13
|
Eller MS, Liao X, Liu S, Hanna K, Bäckvall
H, Opresko PL, Bohr VA and Gilchrest BA: A role for WRN in
telomere-based DNA damage responses. Proc Natl Acad Sci USA.
103:pp. 15073–15078. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dhillon KK, Sidorova J, Saintigny Y, Poot
M, Gollahon K, Rabinovitch PS and Monnat RJ Jr: Functional role of
the Werner syndrome RecQ helicase in human fibroblasts. Aging Cell.
6:53–61. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kudlow BA, Kennedy BK and Monnat RJ Jr:
Werner and Hutchinson-Gilford progeria syndromes: Mechanistic basis
of human progeroid diseases. Nat Rev Mol Cell Biol. 8:394–404.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Monnat RJ Jr and Saintigny Y: Werner
syndrome protein-unwinding function to explain disease. Sci Aging
Knowledge Environ. 2004:re32004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kipling D, Davis T, Ostler EL and Faragher
RG: What can progeroid syndromes tell us about human aging?
Science. 305:1426–1431. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Csoka AB, Cao H, Sammak PJ, Constantinescu
D, Schatten GP and Hegele RA: Novel lamin A/C gene (LMNA) mutations
in atypical progeroid syndromes. J Med Genet. 41:304–308. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Renard D, Fourcade G, Milhaud D, Bessis D,
Esteves-Vieira V, Boyer A, Roll P, Bourgeois P, Levy N and De
Sandre-Giovannoli A: Novel LMNA mutation in atypical Werner
syndrome presenting with ischemic disease. Stroke. 40:e11–e14.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lessel D, Hisama FM, Szakszon K, Saha B,
Sanjuanelo AB, Salbert BA, Steele PD, Baldwin J, Brown WT, Piussan
C, et al: POLD1 germline mutations in patients initially diagnosed
with Werner syndrome. Human mutation. 36:1070–1079. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Barcena C, Osorio FG and Freije JM:
Detection of nuclear envelope alterations in senescence. Methods
Mol Biol. 965:243–251. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tan ZP, Huang C, Xu ZB, Yang JF and Yang
YF: Novel ZFPM2/FOG2 variants in patients with double outlet right
ventricle. Clin Genet. 82:466–471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xiang R, Fan LL, Huang H, Cao BB, Li XP,
Peng DQ and Xia K: A novel mutation of GATA4 (K319E) is responsible
for familial atrial septal defect and pulmonary valve stenosis.
Gene. Oct 26–2013.(Epub ahead of print).
|
|
24
|
Edgar RC: MUSCLE: Multiple sequence
alignment with high accuracy and high throughput. Nucleic Acids
Res. 32:1792–1797. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kadari A, Mekala S, Wagner N, Malan D,
Köth J, Doll K, Stappert L, Eckert D, Peitz M, Matthes J, et al:
Robust generation of cardiomyocytes from human iPS cells requires
precise modulation of BMP and WNT signaling. Stem Cell Rev.
11:560–569. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gotoh A, Hamada Y, Shiobara N, Kumagai K,
Seto K, Horikawa T and Suzuki R: Skew in T cell receptor usage with
polyclonal expansion in lesions of oral lichen planus without
hepatitis C virus infection. Clin Exp Immunol. 154:192–201. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oshitari T, Kitahashi M, Mizuno S, Baba T,
Kubota-Taniai M, Takemoto M, Yokote K, Yamamoto S and Roy S: Werner
syndrome with refractory cystoid macular edema and
immunohistochemical analysis of WRN proteins in human retinas. BMC
Ophthalmol. 14:312014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Prince PR, Emond MJ and Monnat RJ Jr: Loss
of Werner syndrome protein function promotes aberrant mitotic
recombination. Genes Dev. 15:933–938. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Saintigny Y, Makienko K, Swanson C, Emond
MJ and Monnat RJ Jr: Homologous recombination resolution defect in
werner syndrome. Mol Cell Biol. 22:6971–6978. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chang S, Multani AS, Cabrera NG, Naylor
ML, Laud P, Lombard D, Pathak S, Guarente L and DePinho RA:
Essential role of limiting telomeres in the pathogenesis of Werner
syndrome. Nat Genet. 36:877–882. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Crabbe L, Verdun RE, Haggblom CI and
Karlseder J: Defective telomere lagging strand synthesis in cells
lacking WRN helicase activity. Science. 306:1951–1953. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jones B: Ageing: Heterochromatin
disorganization associated with premature ageing. Nat Rev Genet.
16:3182015. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Floch AG, Tareste D, Fuchs PF, Chadrin A,
Naciri I, Léger T, Schlenstedt G, Palancade B and Doye V: Nuclear
pore targeting of the yeast Pom33 nucleoporin depends on
karyopherin and lipid binding. J Cell Sci. 128:305–316. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Oshima J, Yu CE, Piussan C, Klein G,
Jabkowski J, Balci S, Miki T, Nakura J, Ogihara T, Ells J, et al:
Homozygous and compound heterozygous mutations at the Werner
syndrome locus. Hum Mol Genet. 5:1909–1913. 1996. View Article : Google Scholar : PubMed/NCBI
|