Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most
common global cause of chronic liver disease and is caused by fat
deposition (steatosis) in hepatocytes, which includes simple
steatosis and nonalcoholic steatohepatitis (NASH). Steatosis is
defined as the presence of hepatic triglyceride (TG) droplets in
>5% of hepatocytes (1). The
accumulation of free cholesterol in NAFLD and NASH has also been
reported (2). The incidence of
NAFLD is rapidly increasing. It has a complex pathophysiology, is
closely associated with metabolic syndrome, and is associated with
metabolic risk factors, including obesity, type 2 diabetes mellitus
and dyslipidemia (3,4). NASH is considered to be a hepatic
manifestation of metabolic syndrome (5), which can lead to hepatic injury,
fibrosis, cirrhosis and hepatocellular carcinoma (1). It is also associated with metabolic
impairment and the dysregulation of endoplasmic reticulum (ER)
homeostasis.
Glycoprotein 78 (gp78), also identified as autocrine
motility factor receptor (AMFR), is an ER membrane-anchored E3
ligase (6), which may be critical
in protecting cultured cells against the disruption of ER
homeostasis (7). Several studies
have shown that genetic disruption of gp78 in aged mice induces
hepatic steatosis fatty liver, inflammation and spontaneous
hepatocellular cancer (8–10). However, conflicting results have
been reported following the observation that liver-specific
gp78-knockout mice were lean, and had lower blood and tissue lipid
levels, with evidence to suggest that gp78 ubiquitinates HMG-CoA
reductase, an enzyme involved in regulating the rate of cholesterol
production, and is a metabolic regulator of genes involved in lipid
metabolism (11). Further
investigations involving different mouse strains and in
vitro cell cultures may assist in improving current
understanding of the role of gp78 in hepatic steatosis.
Cell death-inducing DFFA-like effector c (cidec), a
human homologue of the murine fat-specific protein 27 (FSP27) is an
adipocyte lipid droplet protein, which is important in lipid
droplet formation (12,13). It is only expressed in mature
adipocytes and is associated with adipocyte differentiation
(14,15), and loss of cidec can impede
adipocyte maturation (16). High
expression levels have been identified in fatty liver syndrome, but
not in the normal healthy liver (17,18).
In addition, obesity caused by a high fat diet can be prevented in
FSP27-knockout mice, with these animals exhibiting a lean phenotype
(19,20). However, the exact mechanism
underlying the effect of cidec in the regulation of adipocyte
differentiation remains to be fully elucidated.
In the present study, the role of gp78 and cidec in
hepatic steatosis were examined by application of an in
vitro hepatocyte cell culture model.
Materials and methods
Plasmid construction
Total RNA was extracted from the AM12 cells using
RNAiso Plus (Takara Biotechnology Co., Ltd., Dalian, China) and
cDNA was synthesized using an RT reagent kit (RR047Q; Takara
Biotechnology Co., Ltd.). Using cDNA as the template, the gp78
length of the target fragment was amplified by reverse
transcription-quantitative polymerase chain reaction (RT-PCR). The
eukaryotic expression plasmid, pCMV5-HA (Biovector, Beijing,
China), was digested with NdeI and XbaI, and the gp78
fragment was inserted into the pCMV5-HA plasmid by overnight
incubation with T4 DNA ligase at 16°C. The pCMV5-HA-gp78 construct
was extracted using 10 mg/ml agarose gel electrophoresis and
transformed into the host bacteria DH5α (18265017; Invitrogen,
Beijing, China). The plasmid constructs were extracted using a
Plasmid Midi kit according to the manufacturer's protocol (12843;
Qiagen, Inc., Valencia, CA, USA) and enzyme digestion, and sent to
Beijing Aoke Biological Technology Co. Ltd. (Beijing, China), where
the plasmid construct was sequenced.
Design and verification of the small
interfering (si) RNA gp78 interference sequence and control
sequence
The following sequences were used for verification
of the gp78 interference sequence: siRNA gp78, sense
5′-GCAUGCACACCUUGGCUUUTT-3′ and antisense
5′-AAAGCCAAGGUGUGCAUGCTT-3′; scramble RNA of gp78 (negative
control), sense 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense
5′-ACGUGACACGUUCGGAGAATT-3′. The interference RNA and negative
control RNA were transfected into the AML12 mouse hepatocyte cells,
and the cells were collected following incubation at 37°C for 48 h.
The total RNA was extracted, and the mRNA expression of gp78 was
assessed using reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) analysis. The effect of siRNA on gp78 was also
assessed.
Cell culture and transfection
The AML12 cells (CRL-2254; American Type Culture
Collection, Manassas, VA, USA) were maintained in DMEM:F-12 medium
in 1:1 ratio (DF12) and supplemented with 10% fetal bovine serum
(Sigma-Aldrich; Merck Millipore, Darmstadt, Germany), 0.005 mg/ml
insulin (Kehao Biological Engineering Co. Ltd., Xi'an, China),
0.005 mg/ml transferrin (Kehao Biological Engineering Co. Ltd.),
0.04 mg/ml hexadecadrol (Kehao Biological Engineering Co. Ltd.),
and incubated at 37°C in an atmosphere of 5% CO2. The
cells were transfected with plasmids using Lipofectamine® 2000
transfection reagent (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), according to the manufacturer's protocol.
Oil Red O staining
The AM12 cells were stimulated with 400 µM oleic
acid, as previously described (21). Lipid accumulation in the AML12
cells was assessed using Oil Red O staining. In brief, the cells
were fixed onto slides at a concentration of 2×106 cells/ml with 4%
polyformaldehyde for at least 30 min, washed with
phosphate-buffered saline (PBS) and immersed in freshly prepared 2%
Oil Red O (Sigma-Aldrich; Merck Millipore) dye at room temperature
for 25 min. The slides were then washed in 60% isopropanol
(Invitrogen; Thermo Fisher Scientific, Inc.) followed by distilled
water. A phase contrast microscope was used to visualize
staining.
Western blot analysis
The AML12 cells were seeded at a concentration of
3×106 cells/ml into 60 mm dishes and, once confluent, were lysed in
ice-cold lysis buffer containing 1% NP-40, 50 mM Tris-HCl, 0.1%
SDS, 1% sodium deoxycholate and 150 mM NaCl (pH 7.4; Tianjin
Chemical Reagent Factory, Tianjin, China) and centrifuged at 12,000
× g at 4°C for 3 min. The protein content was determined using
Quick Start™ Bradford kit (5000202EDU; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), using bovine serum albumin (BSA) as the
standard. Subsequently, 15 µl of the proteins were separated by
SDS-PAGE on 12% acrylamide gels, following which proteins were
transferred onto a PVDF membrane (EMD Millipore, Bethesda, MA,
USA). The membrane was then incubated overnight in a blocking
buffer containing appropriate diluent (ab64211; Abcam, Cambridge,
UK) of primary antibodies against gp78 (ab54787; 1:1,000, Abcam),
cidec (ab77115; 1:1,000, Abcam), PPAR-γ (ab41928; 1:1,000, Abcam)
and β-actin (ab8226; 1:1,000, Abcam) at 4°C. The proteins were
detected by incubation with horseradish peroxidase-conjugated
secondary antibody (ab6789; 1:2,000, Abcam) in diluent (ab64211;
Abcam, Cambridge, UK) at room temperature for 1 h. Following
extensive washing with Tris-buffered saline (pH 7.2) containing
0.05% Tween 20, the bands were visualized using enhanced
chemiluminescence and autoradiography.
Lipid extraction and triglyceride (TG)
content determination
To measure the total TG levels, lipids were
extracted from the cells using the Folch method, as previously
described (22). Briefly, the
cells at a concentration of 3×106 cells/ml were washed with PBS,
scraped in 1 ml PBS and transferred into a 15-ml tube. Intermixture
was added (8 ml of n-hexane: dimethyl carbinol in a 3:2 ratio) and
centrifuged (4°C, 12,000 × g, 5 min). The supernatant was removed
into a glass tube, and 0.1 ml 2% Tritonx-100 was added using a dry
nitrogen-blowing instrument, followed by shock mixing storage at
−20°C. TG content was quantified using Bio-Rad QuantityOne software
version 4.62 (Bio-Rad Laboratories, Inc.).
RNA isolation and RT-qPCR
analysis
Total RNA was extracted from the AML12 cells using
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) Total RNA was
converted into complementary DNA using avian myeloblastosis virus
reverse transcriptase (Takara Bio, Inc., Otsu, Japan). The primers
(forward, 5′-CCTGGCTAGAACAAGACACC-3′ and reverse,
5′-ATCCGAGACCCATCGAAA.T-3′) were synthesized by Aoke Company
(Beijing, China). RT-qPCR was used to quantify the complementary
DNA template. Quantitative gene expression analysis was performed
using the SYBR® Premix Ex Taq™ GC (RR071Q; Takara Bio, Inc.) and
normalized relative to the β-actin mRNA control band. The reaction
system included 12.5 µl of SYBR Premix Ex Taq GC, 0.5 µl of forward
primer, 0.5 µl of reverse primer, 2 µl of template and 9.5 µl of
H2O. The reactions were incubated in 96-well optical
plates for initial denaturation at 95°C for 3 min followed by 35
cycles of denaturation at at 95°C for 15 sec, annealing at 60°C for
30 sec, extension at 72°C for 30 sec then followed by a final
extension at 72°C for 10 min.
Immunofluorescence assay
The AML12 cells were seeded onto sterile coverslips
and, following incubation at 37°C for 24 h, the cells were
transfected with pCMV-Myc-CIDE-3 and pCMV5-HA-gp78 for 48 h, and in
5% BSA for 30 min. The cells were then incubated with primary
antibodies specific for gp78 (ab54787; dilutions 1:1,000, Abcam) or
cidec (ab213693; dilutions 1:1,000, Abcam) overnight at 4°C,
followed by incubation with secondary antibodies conjugated with
fluorescein isothiocyanate for gp78 (ab6785; dilutions 1:3,000,
Abcam) and with tetraethyl rhodamine isothiocyanate for cidec
(ab6718; 1:3,000, Abcam) for 1 h at 37°C. Prior to imaging, the
cells were counterstained with DAPI (Invitrogen; Thermo Fisher
Scientific, Inc.) for 10 min at 37°C to stain the nuclei, and were
visualized with an E1000 digital camera (Nikon Corporation, Tokyo,
Japan) with SimplePCI software version 65 (Meyer Instruments,
Houston, TX, USA).
Coimmunoprecipitation assay
The cells were cultured in 60-mm dishes at a
concentration of 3×106 cells/ml and were cotransfected with 5 µg of
pCMV-Myc-CIDE-3 and pCMV5-HA-gp78. The cells were lysed with RIPA
buffer containing 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate,
0.1% SDS and 50 mM Tris-HCl (pH 7.5; Tianjin Chemical Reagent
Factory). The cells were incubated for 10 min on ice and, following
brief sonication, the lysate was centrifuged at 12,000 × g for 10
min at 4°C. An aliquot of the lysates was removed for western blot
analysis. Antibody was coupled to Dynabeads® Protein G (Dynal;
thermo Fisher Scientific, Inc.) using dimethylpimelimidate coupling
according to the manufacturer's protocol. Equal quantities of
cellular protein were incubated for 2 h with the antibody-linked
beads at 4°C with continuous agitation. The lysates and
coimmunoprecipitates were then separated by SDS-PAGE using 12% gels
and transferred onto PVDF membranes for western blot analysis.
Statistical analysis
All data are analyzed using SPSS 12.0 software (SPSS
Inc., Chicago, IL, USA). Data are expressed as the means ± standard
error of the mean. Two-tailed Student's t test was used to compare
the values between two groups. One-way analysis of variance was
used to compare values between multiple groups. P≤0.05 was
considered to indicate a statistically significant difference.
Results
Elevated expression levels of gp78 in
the process of hepatic steatosis
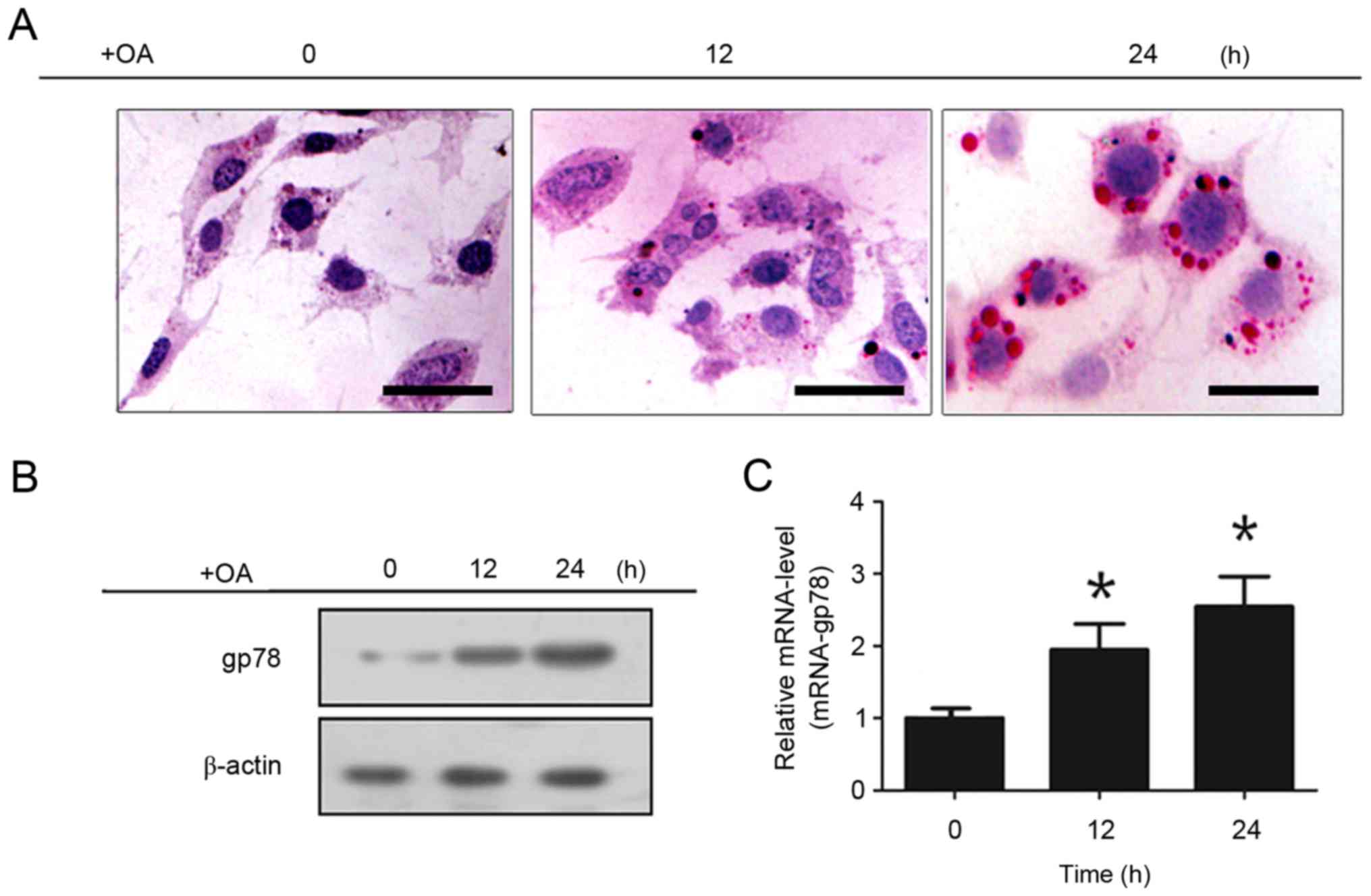
In the present study, steatosis was induced by oleic
acid in AML12, and the cells were collected at 0, 12 and 24 h for
Oil Red O staining (Fig. 1A) and
phase contrast microscopy (Fig.
1B). Increased lipid accumulation in liver cells was observed
with time. The protein and mRNA expression levels of gp78 were
confirmed using western blot and RT-qPCR analyses, respectively
(Fig. 1C and D). Compared with the
control group, the hepatocytes in the steatosis group showed
increased expression of gp78 with time.
Effects of the overexpression and
knockdown of gp78 in hepatic steatosis
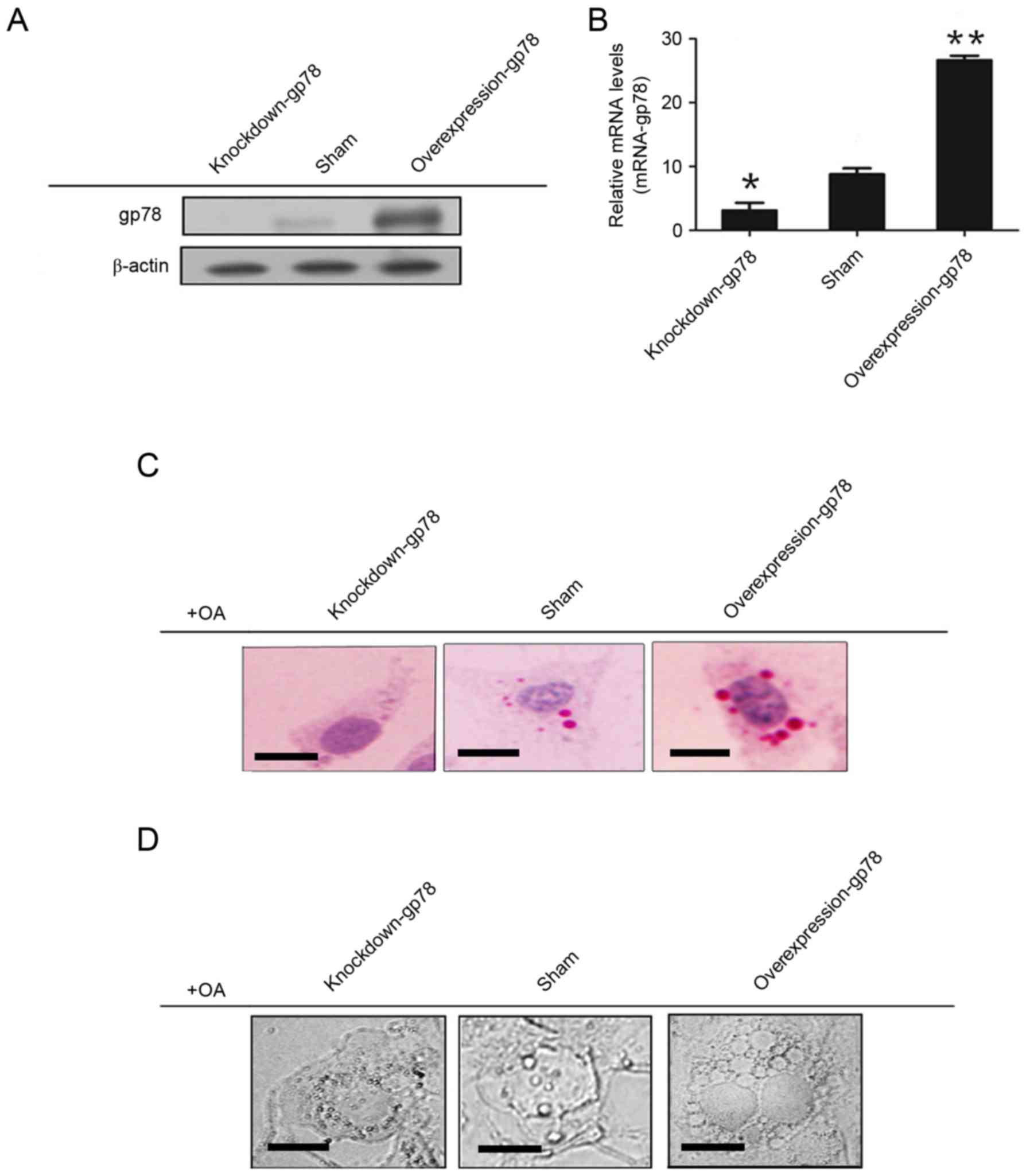
At 48 h post-transfection, the following three
groups of cells were collected: Control group; gp78-overexpression
group; and gp78-knockdown group. Western blot and RT-qPCR analyses
were used to confirm the protein and mRNA expression of gp78,
respectively (Fig. 2A and B). The
lipid accumulation in heptocytes was observed using Oil Red O
staining (Fig. 2C) and phase
contrast microscopy (Fig. 2D).
Compared with the control group, an increased number and volume of
lipid droplets were observed in the gp78-overexpression group,
whereas a decreased number and volume of lipid droplets were
observed in the gp78-knockdown group.
Roles of gp78 and cidec in hepatic
steatosis
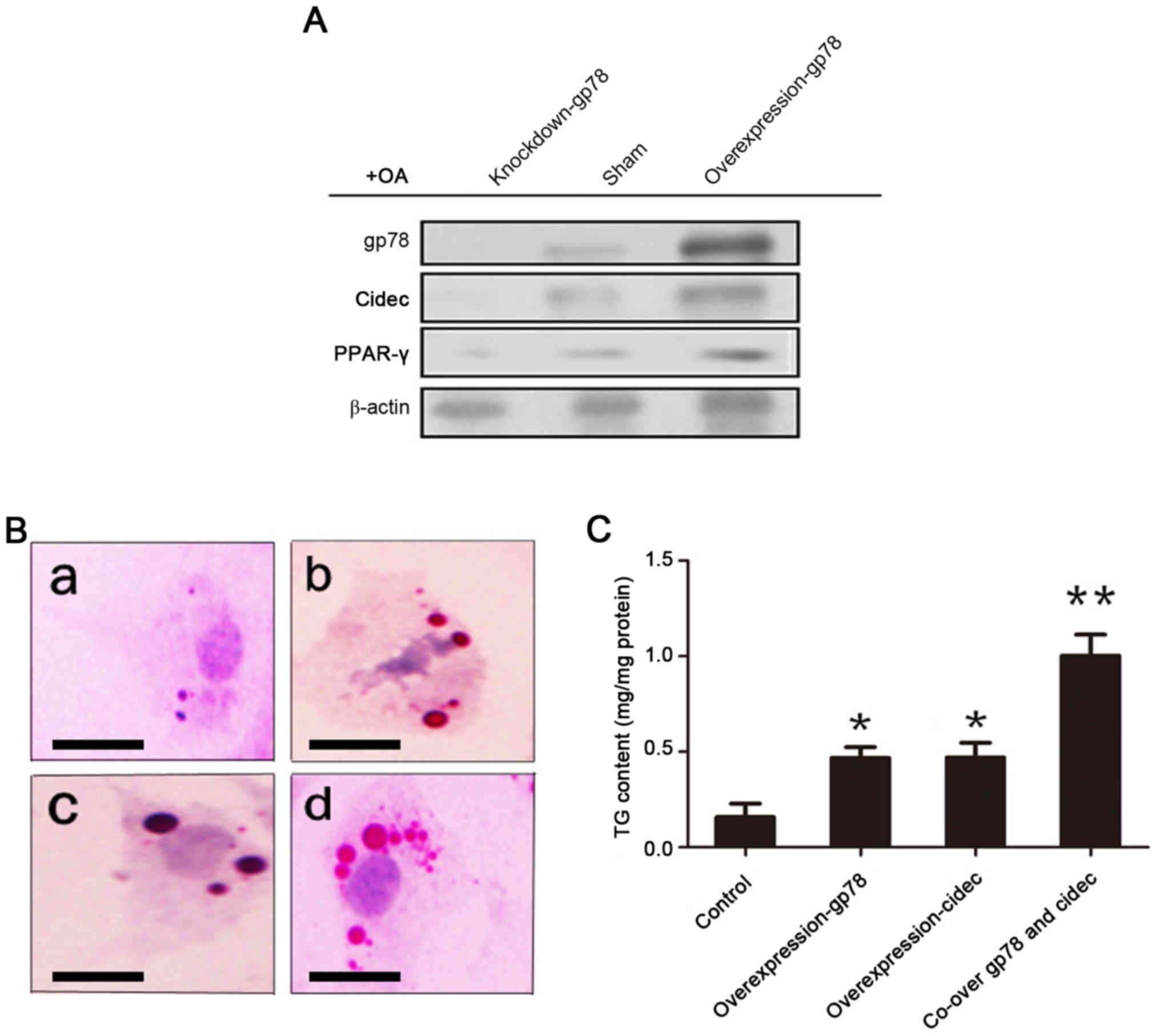
The expression levels of cidec and peroxisome
proliferator-activated receptor (PPAR)-γ were upregulated, which
was observed consistently with the overexpression of gp78 (Fig. 3A). The results suggested that the
interaction between gp78 and cidec promoted lipid accumulation
(Fig. 3B and C).
Association between gp78 and cidec in
hepatic steatosis
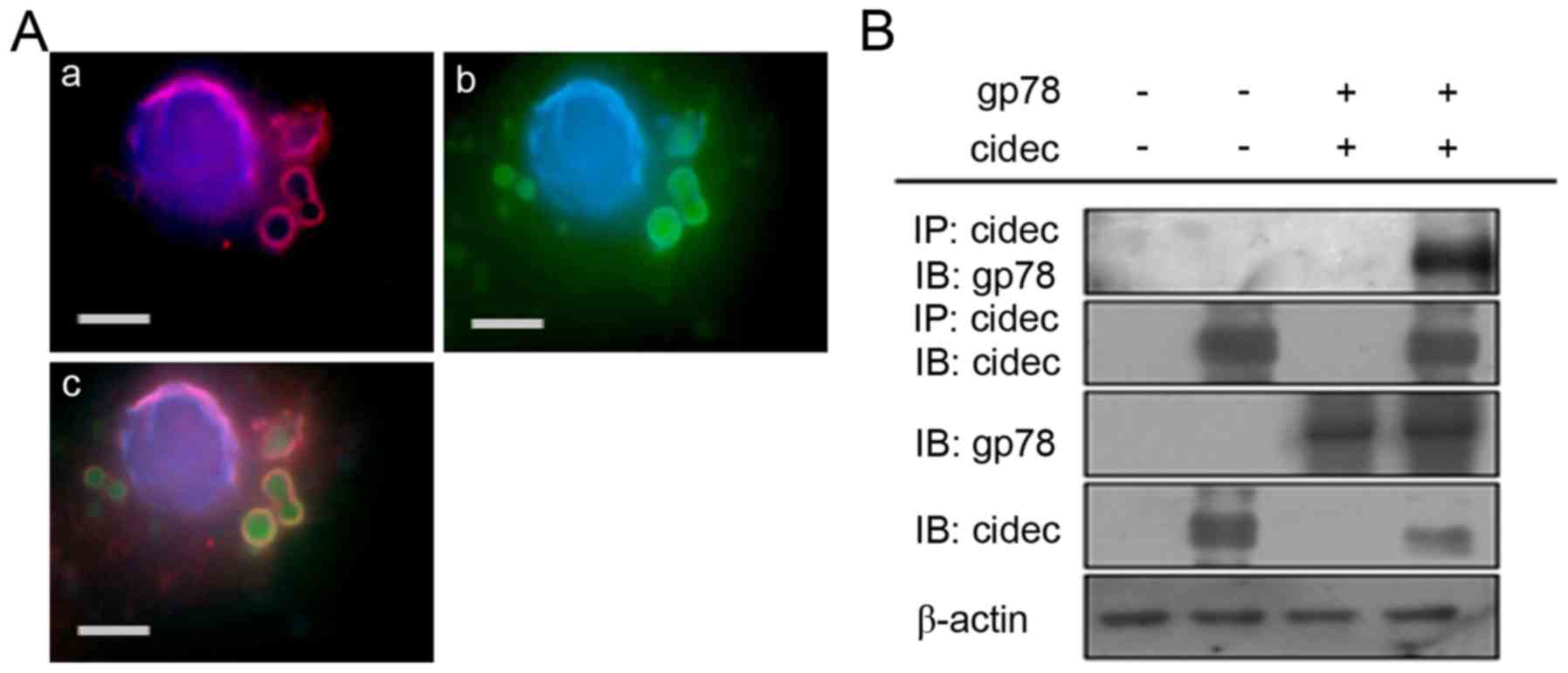
The present study found that the interaction between
gp78 and cidec promoted lipid accumulation using
coimmunoprecipitation and immunofluorescence confocal microscopy
analyses (Fig. 4A and B), which
indicated that gp78 and cidec had the same localization in the
AML12 cells.
Discussion
Although NAFLD is a commonly occurring liver
disorder in industrialized countries (23), the majority of patients present
with few or no symptoms. NAFLD is considered to be the most common
cause of chronic liver disease, cirrhosis and liver failure
(24,25). Furthermore, dysregulated
cholesterol metabolism may contribute to disease severity in NAFLD
and NASH (2,26). Cholesterol is synthesized from
acetyl-CoA through a cascade of enzymatic reactions (27), and hepatic gp78 has been reported
to be essential in regulating lipid and energy metabolism in
animals. However, the exact mechanism by which this regulation
takes place remains controversial (10,11,28,29).
In the present study, the expression of gp78 was
examined in hepatic steatosis and it was observed that the
hepatocytes in the steatosis group showed increased expression of
gp78 expression over time. Furthermore, the overexpression of gp78
induced lipid accumulation in hepatocytes, whereas the knockdown of
gp78 led to a reduction in lipid accumulation, which indicates its
potential role in the biosynthesis of cholesterol and fatty acids
in the liver (11). However,
further investigations are required to determine the exact
mechanism underlying the role of gp78 in hepatic steatosis.
The cell death-inducing DNA fragmentation factor
45-like effector proteins are important in lipid metabolism
(30). Cidec is expressed at high
levels in white adipose tissue and increases during adipogenesis in
mice (12,15). In addition, it has been
demonstrated that cidec induces apoptosis in hepatocellular
carcinoma (31). PPAR-γ is
primarily present in adipose tissue, and it regulates fatty acid
storage and glucose metabolism (32). The genes activated by PPAR-γ
stimulate lipid uptake and adipogenesis by fat cells, and
PPAR-γ-knockout mice fail to generate adipose tissue when fed a
high fat diet (32). In the
present study, it was found that the overexpression of gp78
upregulates the expression of cidec and PPAR-γ, whereas knocking
down gp78 had a suppressive effect. The interaction between gp78
and cidec induced lipid accumulation in hepatocytes.
In conclusion, the present study is the first, to
the best of our knowledge, to demonstratef an association between
gp78 and cidec, and show their combined effect on hepatic
steatosis. However, the involvement of gp78 and cidec in NAFLD
requires further experimental investigation.
Acknowledgements
This study was supported by Grants from National
Natural Science Foundation of China (grant nos. 81170798, 81000171
and 31400722.).
References
|
1
|
Cohen JC, Horton JD and Hobbs HH: Human
fatty liver disease: Old questions and new insights. Science.
332:1519–1523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Puri P, Baillie RA, Wiest MM, Mirshahi F,
Choudhury J, Cheung O, Sargeant C, Contos MJ and Sanyal AJ: A
lipidomic analysis of nonalcoholic fatty liver disease. Hepatology.
46:1081–1090. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nascimbeni F, Pais R, Bellentani S, Day
CP, Ratziu V, Loria P and Lonardo A: From NAFLD in clinical
practice to answers from guidelines. J Hepatol. 59:859–871. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chalasani N, Younossi Z, Lavine JE, Diehl
AM, Brunt EM, Cusi K, Charlton M and Sanyal AJ: The diagnosis and
management of non-alcoholic fatty liver disease: Practice guideline
by the American association for the study of liver diseases,
American college of gastroenterology and the American
gastroenterological association. Hepatology. 55:2005–2023. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marchesini G, Bugianesi E, Forlani G,
Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N,
Melchionda N and Rizzetto M: Nonalcoholic fatty liver,
steatohepatitis, and the metabolic syndrome. Hepatology.
37:917–923. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fairbank M, St-Pierre P and Nabi IR: The
complex biology of autocrine motility factor/phosphoglucose
isomerase (AMF/PGI) and its receptor, the gp78/AMFR E3 ubiquitin
ligase. Mol Biosyst. 5:793–801. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Song BL, Javitt NB and DeBose-Boyd RA:
Insig-mediated degradation of HMG CoA reductase stimulated by
lanosterol, an intermediate in the synthesis of cholesterol. Cell
Metab. 1:179–189. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song BL, Sever N and DeBose-Boyd RA: Gp78,
a membrane-anchored ubiquitin ligase, associates with Insig-1 and
couples sterol-regulated ubiquitination to degradation of HMG CoA
reductase. Mol Cell. 19:829–840. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen Y, Ballar P, Apostolou A, Doong H and
Fang S: ER stress differentially regulates the stabilities of ERAD
ubiquitin ligases and their substrates. Biochem Biophys Res Commun.
352:919–924. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang T, Kho DH, Wang Y, Harazono Y,
Nakajima K, Xie Y and Raz A: Gp78, an E3 ubiquitin ligase acts as a
gatekeeper suppressing nonalcoholic steatohepatitis (NASH) and
liver cancer. PLoS One. 10:e01184482015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu TF, Tang JJ, Li PS, Shen Y, Li JG,
Miao HH, Li BL and Song BL: Ablation of gp78 in liver improves
hyperlipidemia and insulin resistance by inhibiting SREBP to
decrease lipid biosynthesis. Cell Metab. 16:213–225. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Puri V, Konda S, Ranjit S, Aouadi M,
Chawla A, Chouinard M, Chakladar A and Czech MP: Fat-specific
protein 27, a novel lipid droplet protein that enhances
triglyceride storage. J Biol Chem. 282:34213–34218. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Min J, Zhang W, Gu Y, Hong L, Yao L, Li F,
Zhao D, Feng Y, Zhang H and Li Q: CIDE-3 interacts with
lipopolysaccharide-induced tumor necrosis factor, and
overexpression increases apoptosis in hepatocellular carcinoma. Med
Oncol. 28 Suppl 1:S219–S227. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Danesch U, Hoeck W and Ringold GM: Cloning
and transcriptional regulation of a novel adipocyte-specific gene,
FSP27. CAAT-enhancer-binding protein (C/EBP) and C/EBP-like
proteins interact with sequences required for
differentiation-dependent expression. J Biol Chem. 267:7185–7193.
1992.PubMed/NCBI
|
|
15
|
Kim JY, Liu K, Zhou S, Tillison K, Wu Y
and Smas CM: Assessment of fat-specific protein 27 in the adipocyte
lineage suggests a dual role for FSP27 in adipocyte metabolism and
cell death. Am J Physiol Endocrinol Metab. 294:E654–E667. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li F, Gu Y, Dong W, Li H, Zhang L, Li N,
Li W, Zhang L, Song Y, Jiang L, et al: Cell death-inducing
DFF45-like effector, a lipid droplet-associated protein, might be
involved in the differentiation of human adipocytes. FEBS J.
277:4173–4183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Toh SY, Gong J, Du G, Li JZ, Yang S, Ye J,
Yao H, Zhang Y, Xue B, Li Q, et al: Up-regulation of mitochondrial
activity and acquirement of brown adipose tissue-like property in
the white adipose tissue of fsp27 deficient mice. PLoS One.
3:e28902008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guillen N, Navarro MA, Arnal C, Noone E,
Arbonés-Mainar JM, Acín S, Surra JC, Muniesa P, Roche HM and Osada
J: Microarray analysis of hepatic gene expression identifies new
genes involved in steatotic liver. Physiol Genomics. 37:187–198.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Puri V and Czech MP: Lipid droplets: FSP27
knockout enhances their sizzle. J Clin Invest. 118:2693–2696.
2008.PubMed/NCBI
|
|
20
|
Matsusue K, Kusakabe T, Noguchi T,
Takiguchi S, Suzuki T, Yamano S and Gonzalez FJ: Hepatic steatosis
in leptin-deficient mice is promoted by the PPARgamma target gene
Fsp27. Cell Metab. 7:302–311. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jiang L, Gu Y, Ye J, Liu F, Zhao Y, Wang
C, Xu Y, Cao X, Zhang L, Dong W, et al: Resveratrol prevents
hepatic steatosis induced by hepatitis C virus core protein.
Biotechnol Lett. 34:2205–2212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Folch J, Lees M and Stanley GH Sloane: A
simple method for the isolation and purification of total lipides
from animal tissues. J Biol Chem. 226:497–509. 1957.PubMed/NCBI
|
|
23
|
Shaker M, Tabbaa A, Albeldawi M and
Alkhouri N: Liver transplantation for nonalcoholic fatty liver
disease: New challenges and new opportunities. World J
Gastroenterol. 20:5320–5330. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moore JB: Non-alcoholic fatty liver
disease: The hepatic consequence of obesity and the metabolic
syndrome. Proc Nutr Soc. 69:211–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
White DL, Kanwal F and El-Serag HB:
Association between nonalcoholic fatty liver disease and risk for
hepatocellular cancer, based on systematic review. Clin
Gastroenterol Hepatol. 10:1342–1359.e2. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Min HK, Kapoor A, Fuchs M, Mirshahi F,
Zhou H, Maher J, Kellum J, Warnick R, Contos MJ and Sanyal AJ:
Increased hepatic synthesis and dysregulation of cholesterol
metabolism is associated with the severity of nonalcoholic fatty
liver disease. Cell Metab. 15:665–674. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goldstein JL, DeBose-Boyd RA and Brown MS:
Protein sensors for membrane sterols. Cell. 124:35–46. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Travers KJ, Patil CK, Wodicka L, Lockhart
DJ, Weissman JS and Walter P: Functional and genomic analyses
reveal an essential coordination between the unfolded protein
response and ER-associated degradation. Cell. 101:249–258. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen Z, Ballar P, Fu Y, Luo J, Du S and
Fang S: The E3 ubiquitin ligase gp78 protects against ER stress in
zebrafish liver. J Genet Genomics. 41:357–368. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gong J, Sun Z and Li P: CIDE proteins and
metabolic disorders. Curr Opin Lipidol. 20:121–126. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Greene ME, Blumberg B, McBride OW, Yi HF,
Kronquist K, Kwan K, Hsieh L, Greene G and Nimer SD: Isolation of
the human peroxisome proliferator activated receptor gamma cDNA:
Expression in hematopoietic cells and chromosomal mapping. Gene
Expr. 4:281–299. 1995.PubMed/NCBI
|
|
32
|
Jones JR, Barrick C, Kim KA, Lindner J,
Blondeau B, Fujimoto Y, Shiota M, Kesterson RA, Kahn BB and
Magnuson MA: Deletion of PPARgamma in adipose tissues of mice
protects against high fat diet-induced obesity and insulin
resistance. Proc Natl Acad Sci USA. 102:6207–6212. 2005. View Article : Google Scholar : PubMed/NCBI
|