Introduction
Hepatic fibrosis is a common response to chronic
hepatic injury of varying etiology, and is associated with the
aberrant deposition of extracellular matrix components in the
liver. An event of critical importance during its progression is
the activation of hepatic stellate cells (HSCs) (1,2).
Transforming growth factor (TGF)-β1 is a major fibrogenic factor in
the liver, which has been reported to contribute to the activation
and proliferation of HSCs (2).
Preventing the activation of HSCs or inhibiting the activity of
TGF-β1 have been demonstrated to reverse the progression of
fibrosis (3).
The TGF-β superfamily comprises three different
isoforms in mammals, namely TGF-β1, TGF-β2 and TGF-β3, each
participating in distinct biological functions (4). TGF-β1 has traditionally been
considered a key fibrogenic and proliferative stimulus in HSCs,
whereas TGF-β3 has an antagonistic effect on the actions of TGF-β1.
Recombinant TGF-β3 has been reported to inhibit the mRNA and
protein expression of TGF-β1, suppress collagen synthesis and
upregulate the expression of matrix metalloproteinase-9 in HSCs.
Furthermore, the expression of type I collagen was revealed to be
decreased in pcDNA3.1(+)-TGF-β3 and pcDNA3.1(+)-TGF-β1
co-transfected HSCs compared with pcDNA3.1(+)-TGF-β1 transfected
HSCs, a finding that may indicate that TGF-β3 inhibited TGF-β1
signaling. In addition, recombinant adeno-associated virus 2-TGF-β3
treatment was reported to reduce the histopathological damage
associated with liver fibrosis in rats treated with carbon
tetrachloride in vivo (5–7).
However, the mechanism underlying the antagonistic effects of
TGF-β3 on TGF-β1-induced liver fibrosis has yet to be
elucidated.
Activation of the TGF-β1/Smad signaling pathway is
implicated in the response to hepatic fibrosis. In this pathway,
TGF-β1 binds to the TGF-β receptor (R) II, triggering the
phosphorylation of TGF-βRI, which results in the activation of
downstream receptor-regulated Smad proteins (R-Smads), including
Smad2 and Smad3. Phosphorylated R-Smads oligomerize with Smad4 to
form a transcriptional complex, which translocates to the nucleus
to activate the transcription of target genes. Inhibitory Smads
(I-Smads), including Smad6 and Smad7, are negative regulators of
this pathway (8). Therefore, it
may be hypothesized that TGF-β3 can inhibit hepatic fibrosis via
regulating the TGF-β1/Smad signaling pathway in HSCs.
Our previous study demonstrated that cAMP-responsive
element binding protein (CREB) 1 is a critical transcription factor
implicated in TGF-β3 autoregulation in HSCs (9). CREB1 is expressed in numerous cell
types and acts as a transcription factor to regulate promoter
activity via binding to cAMP-responsive elements (CREs). Previous
studies have suggested that CREB1 may be involved in fibrogenic
processes in various tissues, including the heart and lungs;
however, the exact role of CREB1 in fibrosis, as well as its
implication in hepatic fibrosis, have yet to be elucidated
(10–12). Notably, CREB1 has been reported to
cooperate with bone morphogenetic protein (BMP)-stimulated Smad
signaling to enhance activation of the Smad6 promoter in
chondrocytes (13). Therefore, it
may be hypothesized that the regulatory effects of CREB1 on I-Smads
contribute to the inhibitory action of TGF-β3 on fibrogenic
processes in HSCs.
The present study demonstrated that TGF-β3 induced
Smad7 expression in HSCs. CREB1 and Smad3 are required for this
induction, with Smad3 acting as the key regulator and CREB1 acting
as a co-regulator. These results suggested that this mechanism may
underlie the antagonizing effects of TGF-β3 on hepatic
fibrosis.
Materials and methods
Materials
The phenotypically activated rat HSC-T6 cell line
was obtained from the Hepatopathy Institute of Shanghai University
of Traditional Chinese Medicine (Shanghai, China). TGF-β3 and
TGF-β1 were purchased from PeproTech, Inc. (Rocky Hill, NJ, USA)
and their purity was >98%, as assessed via SDS-PAGE.
pGenesil-1.1-short hairpin (sh)RNA-CREB1
(3′-CGGUGUCUAACGGUGUAAU-5′) was purchased from Wuhan GeneSil
Biotechnology Co., Ltd. (Wuhan, China). pRSV-CREB1 expression
vector (9) was obtained from Dr
Michael Greenberg (Department of Neurobiology, Harvard Medical
School, Boston, MA, USA). Small interfering (si)RNA-Smad3 was
purchased from Qiagen China Co., Ltd. (SI00255983; Shanghai,
China). SP600125, a c-Jun N-terminal kinase (JNK) inhibitor;
SB203580, a p38 inhibitor; PD98059, a mitogen-activated protein
kinase kinase (MEK) inhibitor; and H89, a protein kinase A (PKA)
inhibitor, were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). TRIzol® reagent, primers and SYBR
Green I were purchased from Invitrogen (Thermo Fisher Scientific,
Inc., Waltham, MA, USA).
Cell culture
HSCs were cultured in Dulbecco's modified Eagle's
medium (DMEM) supplemented with 10% fetal bovine serum (both from
Thermo Fisher Scientific, Inc.) and maintained at 37°C in a 5%
CO2 atmosphere. All experiments were conducted when
cells were at the exponential phase of growth. Cells were seeded
into 25 cm2 plastic culture flasks or 6-well plates
until 70–80% confluent, and treated with exogenous TGF-β3 or TGF-β1
(10 ng/ml) at 37°C for various durations. Cells that did not
receive TGF-β3 or TGF-β1 served as the control group. HSCs were
cultured in 6-well plates and treated with the JNK inhibitor
SP600125 (20 µM), the p38 inhibitor SB203580 (20 µM), the ERK
inhibitor PD98059 (20 µM) or the PKA inhibitor H89 (5 µM) for 30
min, then stimulated with exogenous TGF-β3 for an additional 2 h at
37°C. In all experiments, control cells received a PBS vehicle
treatment. Total RNA was extracted from cells belonging to all
treatment groups for reverse transcription-quantitative polymerase
chain reaction (RT-qPCR).
Transient transfection
siRNA-Smad3 was utilized to silence Smad3 expression
in HSCs, pGenesil-1.1-shRNA-CREB1 was used to silence CREB1
expression via RNA interference, and pRSV-CREB1 expression vector
was used to induce CREB1 expression. HSCs were seeded in 6-well
plates and grown until 80–90% confluent, then transiently
transfected with siRNA-Smad3, pGenesil-1.1-shRNA-CREB1 or
pRSV-CREB1, or AllStars Negative Control siRNA (Qiagen China Co.,
Ltd.) or pGenesil-1.1-shRNA-KB (2 µg/well; Wuhan GeneSil
Biotechnology Co., Ltd.), using Lipofectamine™ 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) as the delivery agent. Each well
contained 5 µl Lipofectamine 2000, 250 µl Opti-MEM® I
Reduced Serum Medium (Gibco; Thermo Fisher Scientific, Inc.) and 2
ml DMEM. A total of 5 h post-transfection, the culture medium was
replaced with fresh DMEM and cells were incubated for an additional
17 h at 37°C. Subsequently, 10 ng/ml exogenous TGF-β3/TGF-β1 was
added to each well and HSCs were incubated for 2 h at 37°C. Total
RNA was extracted from cells belonging to all treatment groups and
CREB1, Smad3 and Smad7 mRNA expression levels were assessed using
RT-qPCR.
RT-qPCR
Total RNA was extracted from HSCs, which were
treated as aforementioned, using TRIzol® reagent,
according to the manufacturer's protocol. Total RNA was reverse
transcribed into cDNA using a PrimeScript RT reagent kit with gDNA
Eraser (Takara Biotechnology, Co., Ltd., Dalian, China). The
residual genomic DNA was cleared by incubating at 42°C for 2 min
with the gDNA Eraser enzyme. The pretreated total RNA was mixed
with the buffer containing Oligo dT Primer and RT Enzyme, and was
subsequently reverse transcribed into cDNA. qPCR was performed
using SYBR Premix Ex Taq II (Tli RNaseH Plus; Takara Biotechnology,
Co., Ltd.). Rat specific forward and reverse primer sequences
(Table I) were designed using the
Primer Premier software version 5.0 (PREMIER Biosoft, Palo Alto,
CA, USA). The total PCR reaction volume of each sample was 20 µl,
containing 1.6 µl of each specific primer (10 µM), 10 µl 2X SYBR
Premix Ex Taq II reaction mix and 0.8 µl of Rox Reference Dye
(50x). The final cDNA concentration in each PCR reaction was
<100 ng. Amplification was performed using the ABI StepOne
system (Applied Biosystems; Thermo Fisher Scientific, Inc.), under
the following conditions: 1 cycle at 95°C for 10 min, followed by
40 cycles at 95°C for 5 sec, and at 60°C for 60 sec. Experiments
were performed in triplicate. The relative expression levels of
each gene were normalized to GAPDH and were calculated using the
2−∆∆Cq method (14).
 | Table I.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction.
| Name | Chain | Sequence | Gene ID |
|---|
| TGF- βRI | F |
5′-CACCGCGTACCAAATGAAGA-3′ | NM_012775.2 |
|
| R |
5′-TGGTGCCCTCTGAAATGAAAG-3′ |
|
| TGF-βRII | F |
5′-GACAACTGCGCCATCATCCT-3′ | NM_031132.3 |
|
| R |
5′-ATGTTGTTGGCGCACGTAGA-3′ |
|
| Smad3 | F |
5′-GGGCCTGCTGTCCAATGT-3′ | NM_013095.3 |
|
| R |
5′-AATGTGCCGCCTTGTAAGCT-3′ |
|
| Smad4 | F |
5′-CCCGAGACAGAGCATCAAAGA-3′ | NM_019275.2 |
|
| R |
5′-GAGCTCGGTGGAGGTGAATC-3′ |
|
| Smad6 | F |
5′-CCACTGGATCTGTCCGATTCTAC-3′ | NM_001109002.2 |
|
| R |
5′-GAGCAGTGATGAGGGAGTTGGT-3′ |
|
| Smad7 | F |
5′-TGGATGGCGTGTGGGTTTA-3′ | NM_030858.1 |
|
| R |
5′-TGGCGGACTTGATGAAGATG-3′ |
|
| Smurf1 | F |
5′-CTGAAGGCTACGAGCAAAGGA-3′ | NM_001109598.1 |
|
| R |
5′-CAGTCTGCGTGTGCAAAAAGTAA-3′ |
|
| Smurf2 | F |
5′-GCCCACGGCTCTTTACCATA-3′ | NM_025481.2 |
|
| R |
5′-GGGCTTTTGGCAGGTTGTT-3′ |
|
| GAPDH | F |
5′-GTATGACTCTACCCACGGCAAGT-3′ | NM_017008.4 |
|
| R |
5′-TTCCCGTTGATGACCAGCTT-3′ |
|
Western blot analysis
Total protein (30–60 µg) was extracted as previously
described (9). Proteins were
quantified using a bicinchoninic acid assay. Equal amounts (20–40
µg) of extracted protein samples were separated by 12% SDS-PAGE and
subsequently transferred to nitrocellulose membranes. The membranes
were blocked with 5% non-fat milk for 1 h and incubated with
anti-Smad7 (1:1,000; MAB2029; R&D Systems, Inc., Minneapolis,
MN, USA), anti-CREB1 (1:2,000, cat no. 9197), anti-phosphorylated
(p)-CREB1 (1:1,000; cat no. 9198), anti-Smad3 (1:2,000; cat no.
9523) (all from CST Biological Reagents Co., Ltd., Shanghai, China)
or anti-GAPDH (1:5,000; cat no. 2118; Cell Signaling Technology,
Inc., Danvers, MA, USA) primary antibodies overnight at 4°C.
Membranes were then incubated with horseradish
peroxidase-conjugated goat anti-rabbit secondary antibody
(1:10,000; cat no. sc-2004; Santa Cruz Biotechnology, Inc.) for 2 h
at room temperature. The protein bands were visualized using an
enhanced chemiluminescence detection kit (Thermo Fisher Scientific,
Inc.).
Statistical analysis
Statistical analysis was performed using SPSS
software version 13.0 (SPSS, Inc., Chicago, IL, USA). Data are
expressed as the mean ± standard deviation. Statistical differences
between groups were assessed using a t-test or a Mann-Whitney U
test. When multiple groups were compared, one-way analysis of
variance followed by Tukey's post hoc test was performed. P<0.05
was considered to indicate a statistically significant
difference.
Results
TGF-β3 increases Smad7 expression in
HSCs
To determine the mechanism underlying the
implication of TGF-β3 in hepatic fibrosis, the proteins
participating in the TGF-β1/Smad signaling pathway were
investigated in HSCs treated with or without exogenous TGF-β3.
TGF-β3 significantly increased the mRNA expression levels of Smad6
and Smad7 in HSCs (Fig. 1A), by
1.5-fold and 3.6-fold, respectively (P<0.01). Conversely, TGF-β3
had no effect on the mRNA levels of Smad3, Smad4, TGF-βRI,
TGF-βRII, Smad specific E3 ubiquitin protein ligase (Smurf) 1 and
Smurf2 (P>0.05). Smad7 is a prominent I-Smad in the TGF-β1/Smad
signaling pathway, and its mRNA levels appeared higher compared
with Smad6; therefore, the mRNA and protein expression levels of
TGF-β3-induced Smad7 were examined in HSCs treated with exogenous
TGF-β3 at various time-points. TGF-β3 appeared to rapidly increase
Smad7 mRNA levels (Fig. 1B and C),
which peaked within 1 h following stimulation (4.1-fold higher
compared with control). Induction of Smad7 protein expression
appeared to decrease within 2 h following stimulation. The present
results indicated that TGF-β3 increased Smad7 expression in
HSCs.
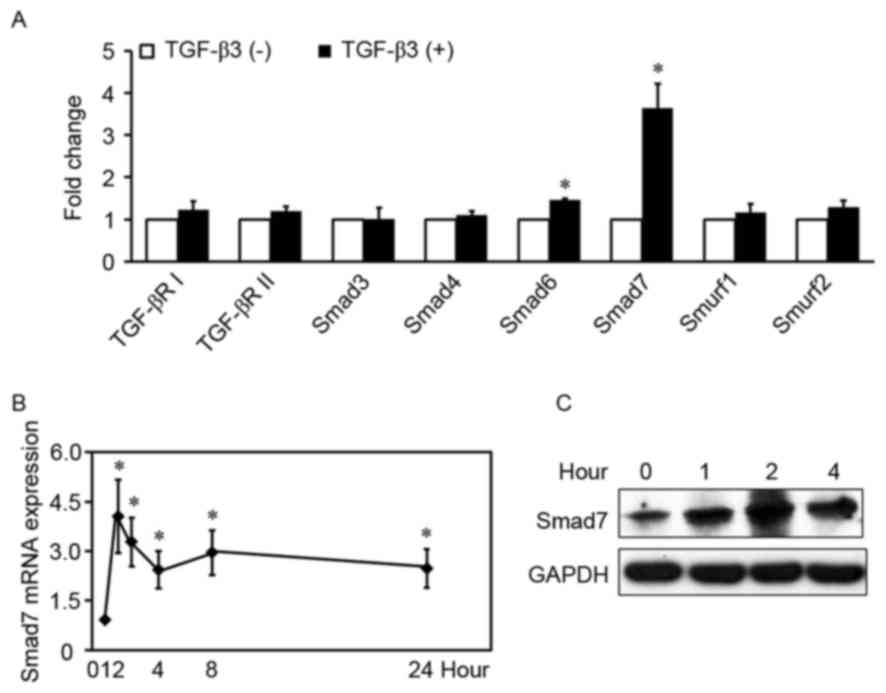 | Figure 1.TGF-β3 increases Smad7 expression in
HSCs. (A) HSCs were treated with or without exogenous TGF-β3 (10
ng/ml) for 2 h, total RNA was extracted and TGF-βRI, TGF-βRII,
Smad3, Smad4, Smad6, Smad7, Smurf1 and Smurf2 mRNA expression
levels were detected by RT-qPCR. The expression of each gene in the
control group is defined as 1 (n=5). (B) RT-qPCR analysis of Smad7
mRNA expression levels in HSCs treated with exogenous TGF-β3 (10
ng/ml) at various time-points. Control is defined as 1 (n=6). Data
are expressed as the mean ± standard deviation. (C) Western blot
analysis of Smad7 protein expression levels in HSCs treated with
exogenous TGF-β3 (10 ng/ml) at various time-points. Three
independent experiments were performed and a representative blot is
presented. *P<0.01, compared with control under basal
unstimulated conditions. TGF, transforming growth factor; HSC,
hepatic stellate cell; R, receptor; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; Smurf, Smad
specific E3 ubiquitin protein ligase. |
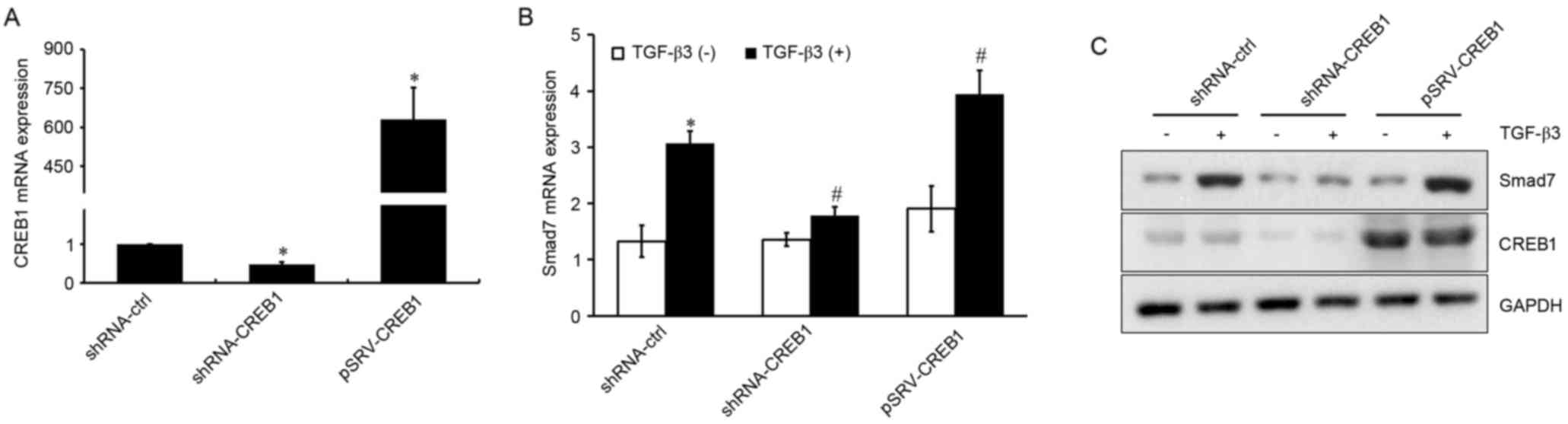
CREB1 is involved in TGF-β3-induced
Smad7 expression
Since the transcription factor CREB1 is a downstream
target in the TGF-β3 signaling pathway, its involvement in
TGF-β3-induced Smad7 expression was investigated. shRNA-CREB1 and
pRSV-CREB1 were used to silence or overexpress CREB1, respectively,
in HSCs treated with or without exogenous TGF-β3, and mRNA and
protein expression levels of Smad7 were assessed using RT-qPCR and
western blot analysis. As presented in Fig. 2A, CREB1 expression was
significantly suppressed in shRNA-CREB1-transfected HSCs compared
with in control cells (P<0.05), whereas it was upregulated in
cells transfected with pRSV-CREB1 compared with in control cells
(P<0.05). CREB1 downregulation was revealed to significantly
inhibit TGF-β3-induced Smad7 expression (P<0.05), whereas its
overexpression enhanced the TGF-β3-induced Smad7 upregulation
(P<0.05; Fig. 2B and C).
However, CREB1 inhibition or overexpression had no effect on Smad7
expression under basal, unstimulated conditions (P>0.05). These
results suggested that CREB1 may be implicated in TGF-β3-induced
Smad7 expression, but may not be required for Smad7 expression when
TGF-β3 stimulation is absent.
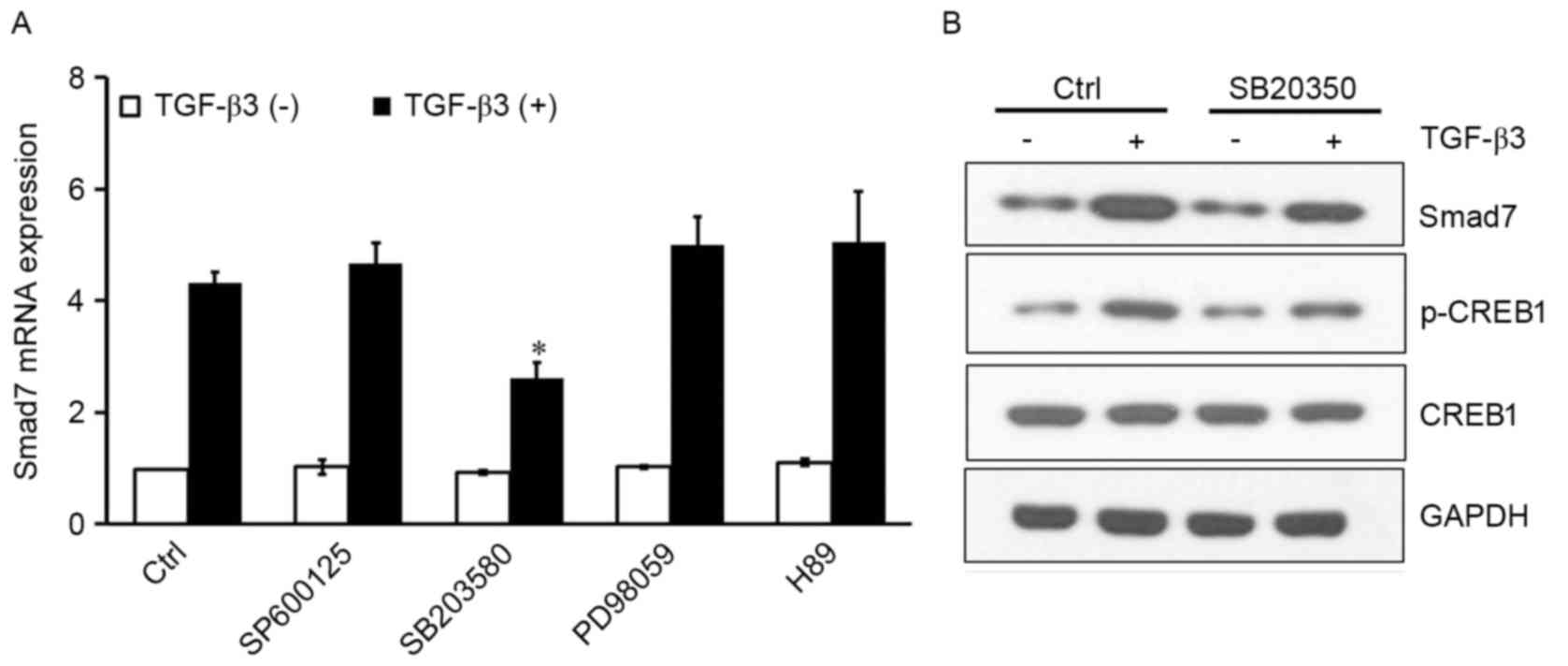
TGF-β3 activates CREB1 via p38 to
induce Smad7
Mitogen-activated protein kinases (MAPKs) and PKA
are kinases that translocate to the nucleus, where they
phosphorylate CREB1 and facilitate its binding to the consensus CRE
DNA site (15–17). To investigate whether JNK, ERK, p38
or PKA were implicated in CREB1 activation resulting in
TGF-β3-induced Smad7 expression, the following inhibitors were
used: SP600125, a selective JNK inhibitor; SB203580, a selective
p38 inhibitor; PD98059, a selective MEK inhibitor; and H89, a
selective PKA inhibitor. The inhibitors had no effect on Smad7
expression under basal conditions. SB203580 significantly inhibited
the TGF-β3-induced Smad7 expression (P<0.05); however, the other
inhibitors produced no effect (Fig.
3A). In addition, western blot analysis of protein expression
levels revealed that SB203580 reduced p-CREB1 levels, and inhibited
the TGF-β3-induced Smad7 upregulation (Fig. 3B). These results suggested that
TGF-β3 may activate CREB1 through the p38 signaling pathway,
resulting in potentiated Smad7 expression in HSCs.
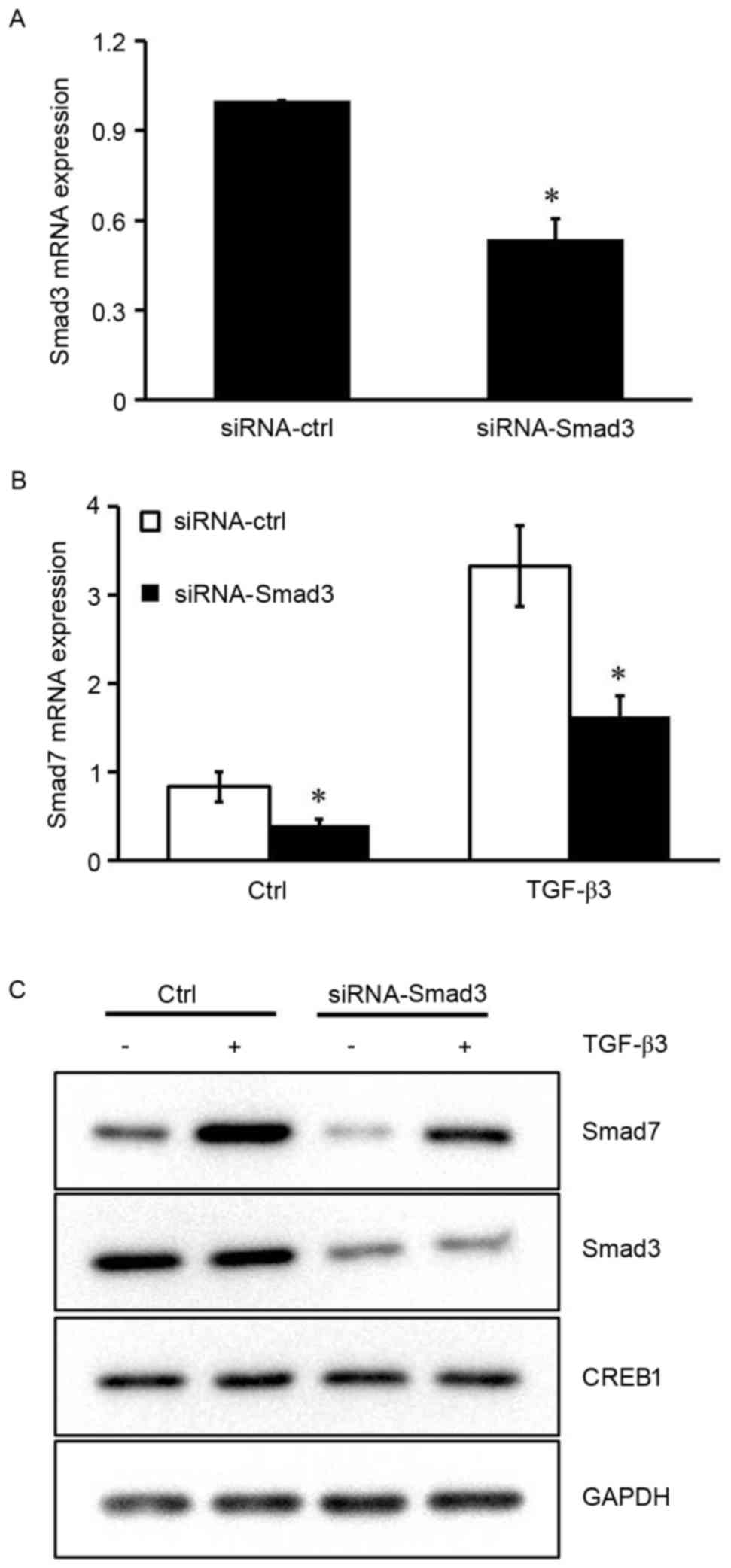
Smad3 is required for TGF-β3-induced
Smad7 expression
The results of the present study indicated that
CREB1 may not be required for Smad7 expression in the absence of
TGF-β3 stimulation, suggesting that other transcription factors are
implicated in TGF-β3-induced Smad7 expression. It has previously
been reported that the Smad7 promoter contains a Smad binding
element (SBE), and Smad3 has been demonstrated to induce its
activity via binding to SBE in HEK293 cells (18). To investigate the role Smad3 serves
in TGF-β3-induced Smad7 expression in HSCs, siRNA was used to
silence the Smad3 gene. The inhibitory efficiency of siRNA-Smad3
was ~50% (Fig. 4A). Silencing the
expression of Smad3 resulted in a marked reduction in the mRNA and
protein expression levels of Smad7 under basal conditions and
following TGF-β3 stimulation (P<0.05; Fig. 4B and C). These results suggested
that Smad3 may be implicated in the TGF-β3-induced Smad7 expression
in HSCs.
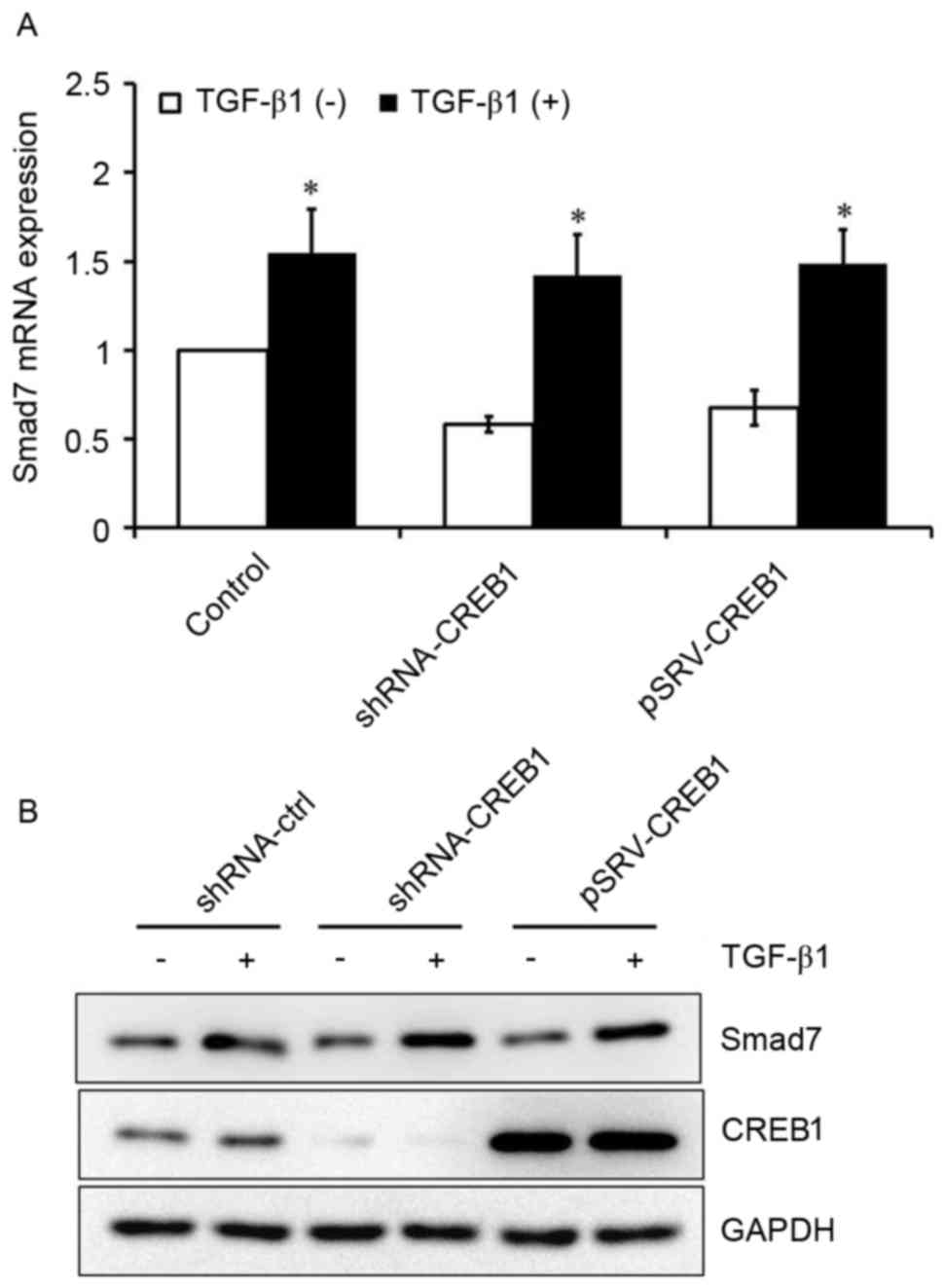
CREB1 has no effect on TGF-β1-induced
Smad7 expression in HSCs
It has previously been reported that Smad3 is an
important downstream factor of TGF-β1 (8). To investigate whether TGF-β1 may also
be able to induce Smad7 expression in HSCs, HSCs were cultured with
or without exogenous TGF-β1. Exogenous TGF-β1 was revealed to
induce Smad7 expression in HSCs (P<0.05; Fig. 5A). Furthermore, HSCs were
transfected with shRNA-CREB-1 or pRSV-CREB1 expression vector, and
subsequently treated with or without exogenous TGF-β1, in order to
investigate the role of CREB1 in TGF-β1-induced Smad7 expression.
Notably, the inhibition or overexpression of CREB1 exerted no
influence on TGF-β1-induced Smad7 expression (Fig. 5).
Discussion
Smad6 and Smad7 belong to the I-Smad family, whose
members have been reported to participate in the regulation of the
signal transduction pathways of TGF-β cytokines (19). It has previously been demonstrated
that Smad7 inhibited TGF-βR- and Activin receptor-mediated
signaling pathways, whereas Smad6 has been reported to inhibit BMP
signaling (20,21). Smad7 is able to antagonize TGF-β
signaling through various mechanisms. It has been revealed that
Smad7 interacted with TGF-βRII to inhibit the phosphorylation of
R-Smads and the subsequent formation of hetero-complexes between
R-Smads and Smad4 (22). Smad7 has
also been revealed to mediate the degradation of the activated type
I receptor ALK5/TβRI via recruiting HECT-type E3 ubiquitin ligases,
such as Smurf1 and Smurf2 (23).
Furthermore, Smad7 is able to bind the MH2 DNA domain and disrupt
the formation of functional Smad-DNA complexes (24). TGF-β1 serves a key role in
fibrogenic processes in various tissues, including skin, liver,
kidney, eye and lung, via inducing the Smad3-dependent
transcription of fibrillar collagen types. Increased TGF-β1 and
decreased Smad7 expression is often observed in fibrotic tissues,
whereas Smad7 overexpression is able to inhibit fibrotic responses
in various tissues via antagonizing the TGF-β1/Smad3 signaling
pathway (25–27).
It has previously been reported that TGF-β1 and
TGF-β3 serve opposite roles in liver fibrosis (1–7).
Although TGF-β1 has been demonstrated to regulate the expression of
Smad7, the role of TGF-β3 has yet to be elucidated. In the present
study, exogenous TGF-β1 and TGF-β3 were revealed to increase the
expression of Smad7 in HSCs; however, TGF-β3-mediated induction of
Smad7 appeared more potent than the TGF-β1-mediated induction,
suggesting that different signaling pathways may be involved in
these processes.
To explore the mechanism underlying TGF-β3-induced
Smad7 expression, the implication of CREB1 in this pathway was
investigated. CREB1 is a key downstream transcription factor in the
TGF-β3 autoregulation signaling pathway (9), which has been reported to participate
in the development of fibrosis (10,11).
Notably, the inhibition or overexpression of CREB1 produced no
effect on Smad7 expression under basal conditions in HSCs that did
not receive treatment, thus indicating that CREB1 may not be
required for the physiological expression of Smad7. Conversely, the
inhibition of CREB1 in vitro significantly decreased the
TGF-β3-induced Smad7 expression, whereas CREB1 overexpression
enhanced the Smad7-stimulating effect of exogenous TGF-β3
application. These results suggested that CREB1 may be implicated
in TGF-β3-induced Smad7 expression, where it acts as a
co-regulator. In addition, p38 was revealed to be a key kinase
upstream of CREB1 that is activated in response to TGF-β3
stimulation. CREB1 did not appear to exert an effect on
TGF-β1-induced Smad7 expression.
As a member of the TGF-β superfamily, TGF-β3 can
also activate the downstream factor Smad3, through phosphorylation
of the TGF-βR (28). In order to
characterize the role of Smad3 in TGF-β3-induced Smad7 expression,
Smad3 siRNA was used to silence the Smad3 gene in HSCs. In the
absence of Smad3, the expression of Smad7 was significantly reduced
in HSCs treated with or without exogenous TGF-β3, therefore
indicating that Smad3 may be critical in TGF-β3-induced Smad7
expression.
Various transcription factors have been reported to
contribute to the induction of Smad7 transcription. The Smad7
promoter includes an SBE, to which R-Smads or an R-Smad/Smad4
complex can bind to activate the Smad7 promoter (18,29).
However, for Smad7 transcription to be potently induced, the
involvement of other transcription factors or cofactors, such as
stimulating protein-1, activator protein 1, transcription factor
E3, activating transcription factor 2, p300 and forkhead box H1, is
required (30–33). Notably, more than one CRE site in
the Smad7 promoter has been reported, some of which lie at a close
proximity to the SBE site, thereby suggesting that CREB1 and Smad3
may both bind to the Smad7 promoter (29). The present results suggested that
SBE may be an important site for Smad7 promoter activation, and the
CRE site is near the SBE. Therefore, it may be hypothesized that
CREB1 could act as a co-factor during the TGF-β3-activated Smad7
transcription by binding with Smad3.
In conclusion, the present study demonstrated that
TGF-β3 induced Smad7 expression in HSCs, and CREB1 and Smad3 are
implicated in the mechanism of induction, Smad3 is the key
regulator while CREB-1 acts as a co-regulator. Furthermore, it may
be hypothesized that CREB1 can cooperate with Smad3 to mediate a
maximal induction of Smad7 transcription following stimulation by
TGF-β3. However, further experiments are required to investigate
and validate this hypothesis.
Acknowledgements
The present study was funded by the National Natural
Foundation of China (grant no. 3087 1153). The authors would like
to thank the Institute of Liver Diseases of Shanghai University of
Traditional Chinese Medicine for providing the HSCs cell line, and
Dr Michael E. Greenberg (Department of Neurobiology, Harvard
Medical School) for providing the pRSV-CREB-1 expression
vector.
Glossary
Abbreviations
Abbreviations:
|
BMP
|
bone morphogenetic protein
|
|
CRE
|
cAMP responsive element
|
|
CREB1
|
cAMP responsive element binding
protein 1
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
HSC
|
hepatic stellate cell
|
|
JNK
|
c-Jun N-terminal kinase
|
|
TGF-β
|
transforming growth factor-β
|
|
PKA
|
protein kinase A
|
References
|
1
|
Wallace K, Burt AD and Wright MC: Liver
fibrosis. Biochem J. 411:1–18. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wells RG: Cellular sources of
extracellular matrix in hepatic fibrosis. Clin Liver Dis.
12:759–768, viii. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Povero D, Busletta C, Novo E, di Bonzo LV,
Cannito S, Paternostro C and Parola M: Liver fibrosis: A dynamic
and potentially reversible process. Histol Histopathol.
25:1075–1091. 2010.PubMed/NCBI
|
|
4
|
Leask A and Abraham DJ: TGF-beta signaling
and the fibrotic response. FASEB J. 18:816–827. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li Q, Zhou X, Yu J, Qian W and Xu KS:
Influence of recombinant transforming growth factor-beta3 on
collagen synthesis and deposition: Experiment with rat cell model
of liver fibrosis. Zhonghua Yi Xue Za Zhi. 88:1273–1278. 2008.(In
Chinese). PubMed/NCBI
|
|
6
|
Zhou X, Yu J, Li Q, Qian W and Xu KS:
Effects of transforming growth factor-beta 3 gene transfer on type
I collagen synthesis of hepatic stellate cells. Zhonghua Gan Zang
Bing Za Zhi. 16:43–48. 2008.(In Chinese). PubMed/NCBI
|
|
7
|
Zhang Y, Liu P, Gao X, Qian W and Xu K:
rAAV2-TGF-β(3) decreases collagen synthesis and deposition in the
liver of experimental hepatic fibrosis rat. Dig Dis Sci.
55:2821–2830. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Massagué J: How cells read TGF-beta
signals. Nat Rev Mol Cell Biol. 1:169–178. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deng L, Li Y, Huang JM, Zhou Gy, Qian W
and Xu K: Effects of p-CREB-1 on transforming growth factor-beta3
auto-regulation in hepatic stellate cells. J Cell Biochem.
112:1046–1054. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Han S, Ritzenthaler JD, Rivera HN and
Roman J: Peroxisome proliferator-activated receptor-gamma ligands
suppress fibronectin gene expression in human lung carcinoma cells:
Involvement of both CRE and Sp1. Am J Physiol Lung Cell Mol
Physiol. 289:L419–L428. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chan EC, Dusting GJ, Guo N, Peshavariya
HM, Taylor CJ, Dilley R, Narumiya S and Jiang F: Prostacyclin
receptor suppresses cardiac fibrosis: Role of CREB phosphorylation.
J Mol Cell Cardiol. 49:176–185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barlow CA, Barrett TF, Shukla A, Mossman
BT and Lounsbury KM: Asbestos-mediated CREB phosphorylation is
regulated by protein kinase A and extracellular signal-regulated
kinases 1/2. Am J Physiol Lung Cell Mol Physiol. 292:L1361–L1369.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ionescu AM, Drissi H, Schwarz EM, Kato M,
Puzas JE, McCance DJ, Rosier RN, Zuscik MJ and O'Keefe RJ: CREB
Cooperates with BMP-stimulated Smad signaling to enhance
transcription of the Smad6 promoter. J Cell Physiol. 198:428–440.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shaywitz AJ and Greenberg ME: CREB: A
stimulus-induced transcription factor activated by a diverse array
of extracellular signals. Annu Rev Biochem. 68:821–861. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nishihara H, Hwang M, Kizaka-Kondoh S,
Eckmann L and Insel PA: Cyclic AMP promotes cAMP-responsive
element-binding protein-dependent induction of cellular inhibitor
of apoptosis protein-2 and suppresses apoptosis of colon cancer
cells through ERK1/2 and p38 MAPK. J Biol Chem. 279:26176–26183.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gustin JA, Pincheira R, Mayo LD, Ozes ON,
Kessler KM, Baerwald MR, Korgaonkar CK and Donner DB: Tumor
necrosis factor activates CRE-binding protein through a p38
MAPK/MSK1 signaling pathway in endothelial cells. Am J Physiol Cell
Physiol. 286:C547–C555. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nagarajan RP, Zhang J, Li W and Chen Y:
Regulation of Smad7 promoter by direct association with Smad3 and
Smad4. J Biol Chem. 274:33412–33418. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yan X, Liu Z and Chen Y: Regulation of
TGF-beta signaling by Smad7. Acta Biochim Biophys Sin (Shanghai).
41:263–272. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakao A, Afrakhte M, Morén A, Nakayama T,
Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH
and ten Dijke P: Identification of Smad7, a TGFbeta-inducible
antagonist of TGF-beta signalling. Nature. 389:631–635. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takase M, Imamura T, Sampath TK, Takeda K,
Ichijo H, Miyazono K and Kawabata M: Induction of Smad6 mRNA by
bone morphogenetic proteins. Biochem Biophys Res Commun. 244:26–29.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hayashi H, Abdollah S, Qiu Y, Cai J, Xu
YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA Jr, Wrana JL
and Falb D: The MAD-related protein Smad7 associates with the
TGFbeta receptor and functions as an antagonist of TGFbeta
signaling. Cell. 89:1165–1173. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Itoh S and ten Dijke P: Negative
regulation of TGF-beta receptor/Smad signal transduction. Curr Opin
Cell Biol. 19:176–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang S, Fei T, Zhang L, Zhang R, Chen F,
Ning Y, Han Y, Feng XH, Meng A and Chen YG: Smad7 antagonizes
transforming growth factor beta signaling in the nucleus by
interfering with functional Smad-DNA complex formation. Mol Cell
Biol. 27:4488–4499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Flanders KC: Smad3 as a mediator of the
fibrotic response. Int J Exp Pathol. 85:47–64. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang W, Koka V and Lan HY: Transforming
growth factor-beta and Smad signalling in kidney diseases.
Nephrology (Carlton). 10:48–56. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lan HY: Smad7 as a therapeutic agent for
chronic kidney diseases. Front Biosci. 13:4984–4992. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu G, Ding W, Neiman J and Mulder KM:
Requirement of Smad3 and CREB-1 in mediating transforming growth
factor-beta (TGF beta) induction of TGF beta 3 secretion. J Biol
Chem. 281:29479–29490. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stopa M, Anhuf D, Terstegen L, Gatsios P,
Gressner AM and Dooley S: Participation of Smad2, Smad3 and Smad4
in transforming growth factor beta (TGF-beta)-induced activation of
Smad7. The TGF-beta response element of the promoter requires
functional Smad binding element and E-box sequences for
transcriptional regulation. J Biol Chem. 275:29308–29317. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brodin G, Ahgren A, ten Dijke P, Heldin CH
and Heuchel R: Efficient TGF-beta induction of the Smad7 gene
requires cooperation between AP-1, Sp1, and Smad proteins on the
mouse Smad7 promoter. J Biol Chem. 275:29023–29030. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hua X, Miller ZA, Benchabane H, Wrana JL
and Lodish HF: Synergism between transcription factors TFE3 and
Smad3 in transforming growth factor-beta-induced transcription of
the Smad7 gene. J Biol Chem. 275:33205–33208. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Uchida K, Suzuki H, Ohashi T, Nitta K,
Yumura W and Nihei H: Involvement of MAP kinase cascades in Smad7
transcriptional regulation. Biochem Biophys Res Commun.
289:376–381. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gohla G, Krieglstein K and Spittau B:
Tieg3/Klf11 induces apoptosis in OLI-neu cells and enhances the
TGF-beta signaling pathway by transcriptional repression of Smad7.
J Cell Biochem. 104:850–861. 2008. View Article : Google Scholar : PubMed/NCBI
|