Introduction
Cellular senescence is a state of stable
proliferation arrest in cells and has been linked to ageing and
ageing-related diseases (1).
Premature senescence can be induced by many stimuli, including
ionizing radiation, telomere dysfunction and reactive oxygen
species (ROS) (2). Evidence
indicates that premature senescence occurs in mouse embryonic
fibroblasts (MEFs) derived from Zmpste24
metalloproteinase-deficient mice, a progeria mouse model of
Hutchinson-Gilford Progeria Syndrome (HGPS), which is mainly caused
by the accumulation of abnormal prelamin A (also known as progerin)
(3–5). There are similar defects in the
cellular phenotypes between progeroid cells and physiological
ageing cells, such as decreased cell proliferation, increased cell
senescence, altered DNA damage responses, increased genome
instability, and dysregulated gene expression (6,7). It
is interesting that progerin is also expressed at low levels in
physiological ageing cells and is induced by telomere damage during
replicative senescence in normal human fibroblasts (8–10),
which implies a common mechanism between premature ageing and
physiological ageing. However, the link between prelamin A
accumulation and the premature senescence phenotype in
Zmpste24−/− MEFs is still poorly understood.
MicroRNAs (miRNAs/miRs) are small non-coding RNAs of
approximately 18~25 nucleotides that function as a negative
regulator of gene expression post-transcriptionally. Recently,
miRNA expression profiles and functional analyses have revealed
that miRNAs impact the premature senescence phenotype of progeroid
cells (11). For instance,
brain-specific miR-9 negatively controls lamin A and progerin
expression in neural cells and plays a neuroprotective role in the
brain (12,13). In the Zmpste24−/−
progeria mouse model, the miR-29 family is involved in the DNA
damage response in a p53-dependent manner (14). Our previous studies revealed that
miR-365 and miR-342-5p are downregulated in
Zmpste24−/− mouse embryonic fibroblasts (MEFs),
in which miR-365 serves as a negative regulator of cell
proliferation (15). Nevertheless,
the specific roles of miRNAs in the premature senescence phenotype
of progeroid cells are still largely unknown and remain to be
further studied.
miR-342-5p is an intronic miRNA hosted in the
Ena/Vasodilator-Stimulated Phosphoprotein-Like (Ena/VASP-like,
EVL) gene, which belongs to the Ena/VASP family, is involved
in actin cytoskeleton remodelling and reportedly potentiates
ERK-sustained cell proliferation (16,17).
miR-342-5p is involved in ageing-associated diseases, including
Alzheimer's disease (AD) and atherosclerosis mouse models. In AD
mouse models, miR-342-5p is upregulated and contributes to AD
axonopathy by downregulating AnkG (18). In an Apoe−/−
atherosclerosis mouse model, macrophage-derived miR-342-5p is
upregulated and promotes atherosclerosis by suppressing the
Akt1-mediated inhibition of miR-155 expression (19). As a downstream effector of Notch
signalling, miR-342-5p regulates neural stem cell proliferation and
differentiation in mice (20).
These findings suggest that miR-342-5p plays different roles in
different cell types. However, the role of miR-342-5p in the
premature senescence phenotype of Zmpste24−/−
MEFs is unclear. Here, we further investigated the function of
miR-342-5p and demonstrated that miR-342-5p modulates cell
proliferation and cell cycle by suppressing growth-arrest-specific
2 (GAS2) in Zmpste24−/− MEFs in
vitro.
Materials and methods
Cell culture
Primary MEFs were prepared from embryonic day (E)
13.5 embryos of Zmpste24−/− and wild-type (WT)
mice. All animal experiments were approved by the Committee on the
Use of Live Animals in Teaching and Research (CULATR) at the
University of Hong Kong and performed according to the regulation
of the CULATR at the University of Hong Kong. MEFs and the mouse
myoblast cell line C2C12 (obtained from Li KaShing Faculty of
Medicine of the University of Hong Kong) were grown in Dulbecco's
modified Eagle's medium (DMEM) (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% foetal bovine serum
(FBS) (Gibco; Thermo Fisher Scientific, Inc.). The cells were
passaged as they reached approximately 80~90% confluency. For the
replicative senescence analysis, early-passage WT MEFs (p2~p4)
underwent serial passages until they reached late passage
(p7~p8).
miRNA transfection
Zmpste24−/− and WT MEFs were
plated in cell culture plates at a density of approximately 60~70%
confluency and were incubated at 37°C. After a 24 h incubation, the
Zmpste24−/− MEFs were transiently transfected
with mmu-miR-342-5p Mimics (342M) or Mimics Negative Control (NC)
(50 nmol/l final), while WT MEFs were transiently transfected with
342M, NC, mmu-miR-342-5p Inhibitor (342I) or Inhibitor Negative
Control (INC) (100 nmol/l final). The Mimics, NC, Inhibitor and INC
were obtained from Ribobio Co., Ltd. (Guangzhou, China).
Transfection was performed using the Lipofectamine®
RNAiMAX Transfection Reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol.
Senescence-associated β-galactosidase
(SA-β-Gal) staining
Zmpste24−/− and WT MEFs (p4~p5)
were plated in 6-well culture plates at a density of
1.4×105 cells per well and were transfected with 342M or
342I. Serial passaging was performed until the cells reached
replicative senescence (p7~p8), and the transfection was reinforced
at every passage. SA-β-Gal activity was detected according to the
manufacturer's protocol (Beyotime Institute of Biotechnology,
Shanghai, China).
MTT assay for monitoring cell
growth
Zmpste24−/− and WT MEFs (p2~p4)
were plated in 48-well culture plates at a density of
9×104 cells per well and were transfected with 342M or
342I, and the second round of transfection was reinforced on day 3
after the first. The cells were incubated with 20 µl of MTT (5
mg/ml) (Beyotime Institute of Biotechnology) for 4 h at 37°C on
days 1, 2, 4 and 6 after the first round of transfection. The
formazan crystals in the cells were solubilized with Dimethyl
Sulphoxide (200 µl/well). The absorbance was measured at 490 nm
using a Synergy 2 microplate reader (BioTek; Winooski, VT,
USA).
EdU incorporation assay
Zmpste24−/− and WT MEFs (p2~p4)
were cultured in 24-well plates and were transfected with 342M or
342I. The cell proliferation of Zmpste24−/− and
WT MEFs (p2~p4) was evaluated by EdU incorporation assay 48 h after
the transfection using the Cell-Light™ EdU Apollo®567 In
Vitro Imaging kit (Ribobio, Guangzhou, China) following the
manufacturer's protocol. The EdU positive cells were counted from
at least 3 fields in every independent experiment using ImageJ2×
software.
Cell cycle analysis
Zmpste24−/− and WT MEFs (p2~p4)
were plated in 6-well culture plates at a density of
1.3×105 cells per well. For synchronization in the G1
stage, the cells were grown in serum-free DMEM for 24 h before
transfection. The cell cycle was analysed 72 h after the
transfection using a FACSCanto II flow cytometer (BD Biosciences,
San Jose, CA, USA). The cell cycle condition was determined using
propidium iodide staining.
Protein extraction and Western
blotting
Total protein was extracted 72 h after the
transfection using RIPA Lysis Buffer (Beyotime Institute of
Biotechnology,). The proteins were separated by SDS-polyacrylamide
gel (12%) and were transferred to polyvinylidenedifluoride
membranes (0.2 µm pore size) (EMD Millipore, Bellerica, MA, USA)
and were then detected with a rabbit anti-p21 polyclonal antibody
(sc-471, 1:600; Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA), a rabbit anti-Cdk1 monoclonal antibody (ab32384, 1:1,000;
Abcam, Cambridge, MA, USA) (used to detect dephospho-Cdk1 (Tyr15)
which refers to active Cdk1 signalling pathways) (21,22),
a mouse anti-Gas2 monoclonal antibody (M01, 1:1,000; Abnova,
Taipei, Taiwan) and a mouse anti-α-tubulin monoclonal antibody
(T5168, 1:5,000; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
The horseradish peroxidase-conjugated secondary antibodies (goat
anti-rabbit IgG or anti-mouse IgG; Beyotime Institute of
Biotechnology,) were diluted 3,000-fold, and the signals were
detected by an enhanced chemiluminescence reagent (Pierce; Thermo
Fisher Scientific, Inc.).
Construction of the luciferase
reporter vector
The WT 3′-Untranslated Regions (3′-UTR) fragments
(at least 500 bp) of mouse ABCC1, FBXW11, GAS2
and NNT, containing the putative miR-342-5p binding sites,
were amplified by polymerase chain reaction (PCR) and were cloned
into the pGL3m vector, which was kindly gifted from Prof. Shi-mei
Zhuang (23). The miR-342-5p
predicted binding seed regions in the WT 3′-UTR of FBXW11
and GAS2 were mutated (GCACCCCA→GCTTTCCA for FBXW11,
GCACCCCA→GCATTTCA for GAS2) by PCR and termed as
mutant 3′-UTR. All the constructs were confirmed by DNA
sequencing.
Dual luciferase assay
C2C12 cells were cotransfected with 40 ng of
luciferase reporter vector, 20 ng of Renilla luciferase
pRL-TK vector (Promega Corporation, Madison, WI, USA), and 342M or
NC (20 nmol/l final) using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). The Firefly
and Renilla luciferase activities were measured 48 h after
the transfection with the Dual-Luciferase Reporter Assay System
(Promega) using an FB12 Luminometer (Titertek-Berthold, Pforzheim,
Germany). The Firefly luciferase activity was normalised to
the Renilla luciferase activity.
RNA extraction and quantitative PCR
(qPCR)
Total RNA from p3 and p7 WT MEFs or the tissues from
2-month-old and 20-month-old mice or from
Zmpste24−/− and WT mice was extracted using the
Trizol Reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The RNA quality was
assessed on an agarose gel (1%), and the RNA concentration was
measured by a NanoDrop1000 spectrophotometer (NanoDrop
Technologies; Thermo Fisher Scientific, Inc., Wilmington, DE, USA).
For miRNA detection, the total RNA was reverse-transcribed using
the All-in-One™ miRNA First-Strand cDNA Synthesis kit (GeneCopoeia,
Inc., Rockville, MD, USA). qPCR was performed with All-in-One™
miRNA qPCR kit (GeneCopoeia) using a LightCycler® 96
System (Roche, Mannheim, Germany). U6 RNA was used as the internal
control. For mRNA detection, total RNA was reverse-transcribed
using the PrimeScript II 1st Strand cDNA Synthesis kit (Takara Bio,
Inc., Otsu, Japan). qPCR was performed using the SYBR-Green Master
Mix (Takara Bio, Inc.) and the following gene-specific primers:
mGAS2-PF 5′-GCCTGCCAAGACCCTACCAC-3′, mGAS2-PR
5′-GCAGAACCAGGCCTTCAGAT-3′; mEVL-PF 5′-AGCCACGATGAGTGAACAGAG-3′,
mEVL-PR 5′-TGGCAGTGTTGTGGTAGATG-3′; and mHPRT-PF
5′-AGGGATTTGAATCACGTTTG-3′, mHPRT-PR 5′-TTACTGGCAACATCAACAGG-3′.
HPRT was used as a housekeeping gene for normalization.
Relative expression levels were analysed using the
2−ΔΔCq method as described (24).
Statistical analysis
All the values were shown as the mean ± standard
deviation from at least three independent experiments unless
otherwise indicated. The non-parametric Mann-Whitney test was used
to compare the percentage of SA-β-Gal positive cells between two
groups. In other cases, statistical significance was determined
using a two-tailed Student's t-test (α=0.05).
Results
miR-342-5p overexpression ameliorated
the cellular senescence phenotype to some extent in
Zmpste24−/− MEFs
Since miR-342-5p was significantly downregulated in
premature senescent Zmpste24−/− MEFs (15), we further investigated the
expression of miR-342-5p in WT MEFs during replicative senescence
and in tissues from physiological ageing mice and
Zmpste24−/− progeroid mice. Our data showed that
miR-342-5p was downregulated in MEFs during replicative senescence
as well (at least 5-folds, P<0.01) (Fig. 1A). However, no significant
differences were observed in the expression of miR-342-5p in
several tissues from physiological ageing mice (Fig. 1B) or from
Zmpste24−/− progeroid mice (Fig. 1C). The mRNA expression of the
EVL host gene was also not consistent with the expression of
miR-342-5p in several tissues from Zmpste24−/−
mice compared with WT mice (Fig.
1D). Next, we further sought to explore whether miR-342-5p
overexpression rescued the cellular senescence phenotype in
Zmpste24−/− MEFs. As shown in Fig. 1E and F, miR-342-5p overexpression
decreased the percentage of SA-β-Gal staining positive cells (one
characteristic of cellular senescence) in
Zmpste24−/− MEFs. Moreover, the large flattened
cell morphology, another characteristic of cellular senescence, was
also improved to some degree in Zmpste24−/− MEFs
transfected with 342M (Fig. 1E).
In addition, we performed parallel experiments in WT MEFs
transfected with 342I (single-stranded antisense RNA). Nonetheless,
the cellular senescence phenotype was hardly affected in WT MEFs
when miR-342-5p was suppressed (data not shown). Meanwhile, we
detected the expression level of miR-342-5p and found that the
miR-342-5p expression level in 342M transfected group was at least
1×104-fold higher than that in NC transfected group,
while the miR-342-5p expression level in 342I transfected group was
hardly affected compared with that in INC transfected group (data
not shown). Here, the 342I could inhibit miR-342-5p without
inducing the degradation of miR-342-5p. Therefore, these results
were in line with expectations and conformed that the transfection
of 342I or 342M worked fine.
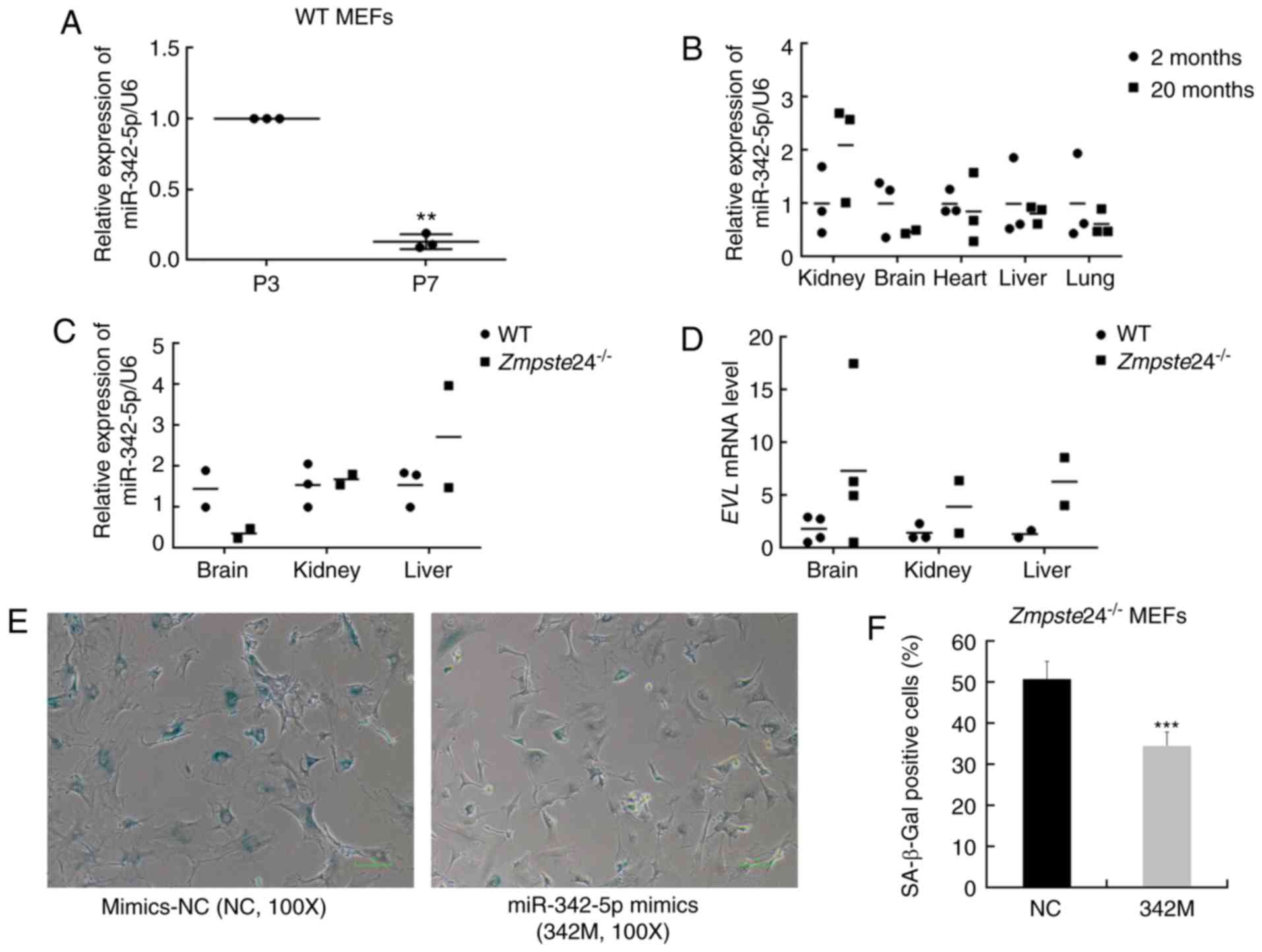 | Figure 1.Overexpression of miR-342-5p
decreased SA-β-Gal staining in Zmpste24−/− mouse
embryonic fibroblasts (MEFs). (A) qPCR was used to detect the
expression of mmu-miR-342-5p in WT MEFs during replicative
senescence. n=3 independent experiments, **P<0.01 (paired
Student's t-test). (B) The relative expression of mmu-miR-342-5p in
the kidney, brain, heart, liver and lung from 2-month-old mice
(n=3) and 20-month-old mice (n=3). (C) The relative expression of
mmu-miR-342-5p in the brain, kidney and liver from WT mice (n=3)
and Zmpste24−/− mice (n=3) (extreme outliers were
eliminated for high Cquantification cycle (Cq) values in
the qPCR). (D) EVL mRNA level in the brain, kidney and liver
from WT mice (n=4) and Zmpste24−/− mice (n=4)
(extreme outliers were eliminated for high Cq values in the qPCR).
(E) Representative images of the SA-β-Gal staining in
Zmpste24−/− MEFs. Original magnification, ×100.
(F) The percentage of SA-β-Gal positive cells is shown in the
histogram, which corresponds to the means ± standard error of the
mean (SEM) (bars) of at least 1,800 cells or 33 random fields
pooled from independent experiments, ***P<0.001 (Mann-Whitney
tests). |
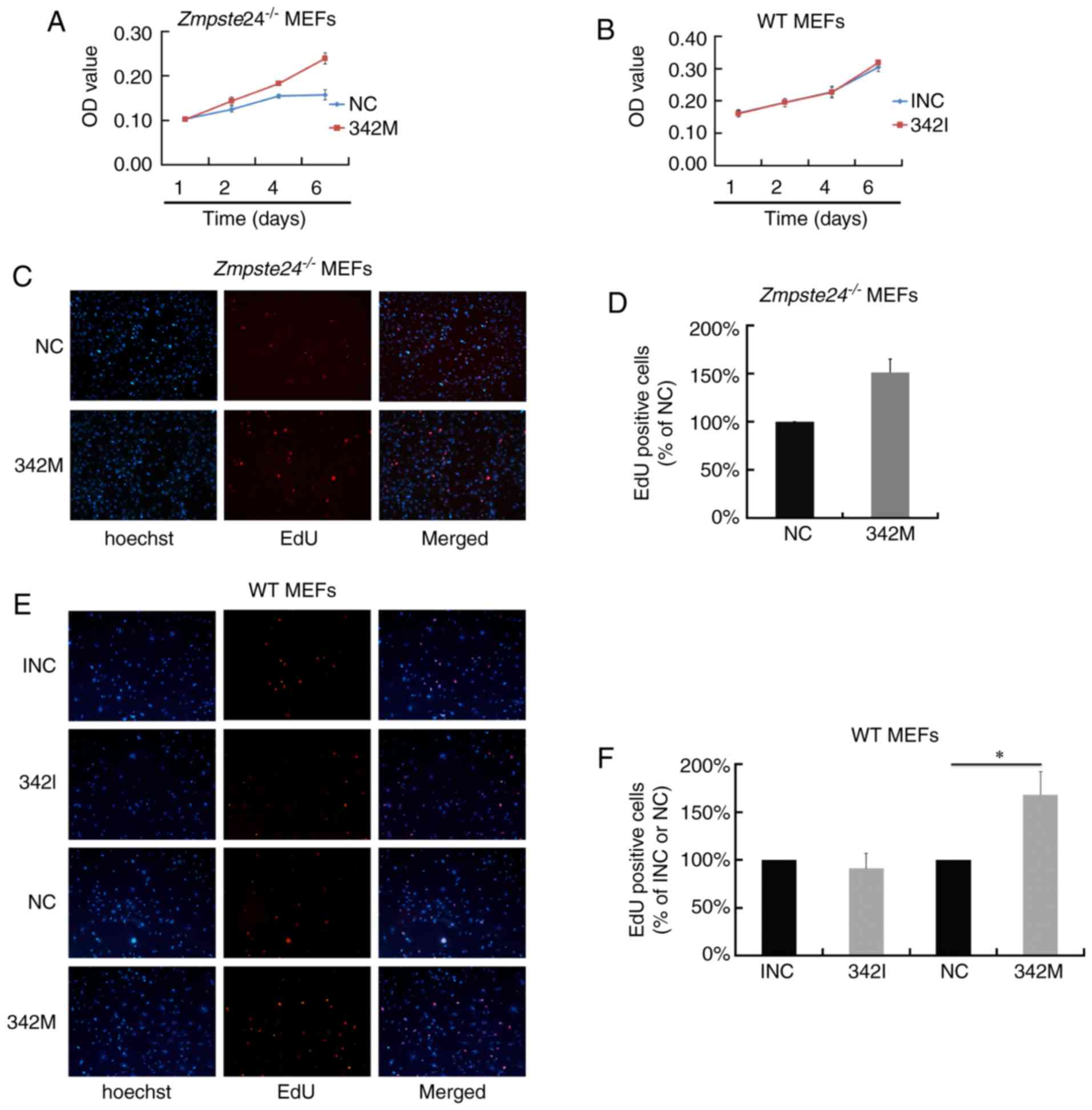
miR-342-5p overexpression promoted
cell proliferation in Zmpste24−/− and WT MEFs
To investigate the effect of miR-342-5p on cell
proliferation in Zmpste24−/− MEFs, we first
evaluated cell viability by an MTT Assay. As shown in Fig. 2A, the overexpression of miR-342-5p
increased cell viability in Zmpste24−/− MEFs.
However, cell viability was not affected in WT MEFs transfected
with 342I (Fig. 2B). Next, we
evaluated cell proliferation in Zmpste24−/− and
WT MEFs by the EdU incorporation assay. Consistent with the results
of the MTT Assay, the overexpression of miR-342-5p increased the
EdU positive cells in Zmpste24−/− and WT MEFs
(increased by ~50%, P<0.05), while the suppression of miR-342-5p
minimally affected cell proliferation in WT MEFs (Fig. 2C-F). Collectively, these results
suggest that miR-342-5p overexpression promotes
Zmpste24−/− and WT MEFs proliferation.
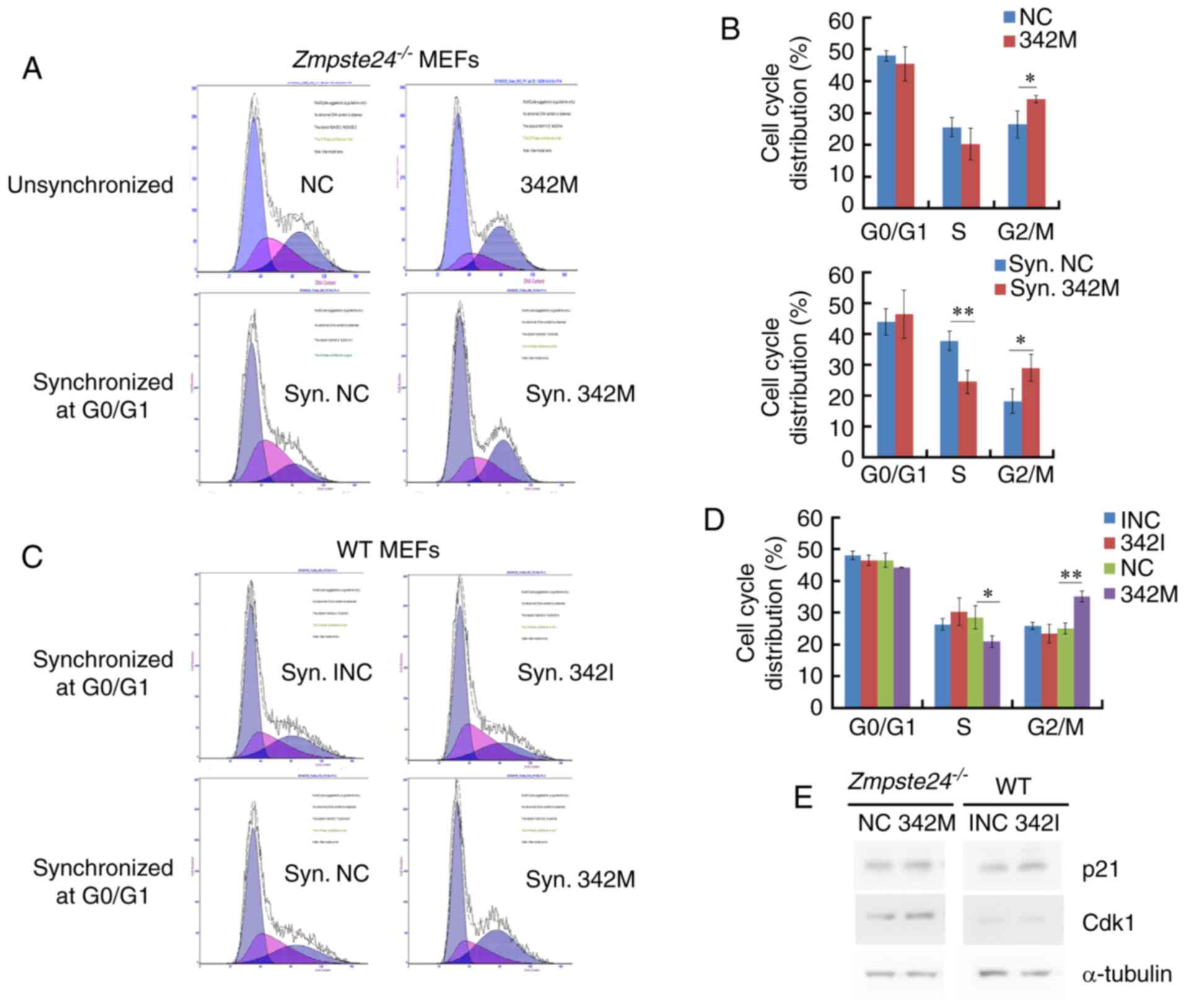
miR-342-5p overexpression increased
the G2+M cell cycle phase in Zmpste24−/− and WT
MEFs
Since miR-342-5p overexpression promotes cell
proliferation in Zmpste24−/− and WT MEFs, we
further investigated the effects of miR-342-5p on cell cycle in
Zmpste24−/− and WT MEFs. Our data showed that the
overexpression of miR-342-5p increased the G2+M cell cycle phase
and decreased the S phase in Zmpste24−/− and WT
MEFs (Fig. 3A-D). However, the
cell cycle was not affected in WT MEFs transfected with 342I
compared with INC (Fig. 3C-D).
Since p21CIP1/WAF1 and Cdk1 (cdc2) are key regulators in
the progression of the G2/M phase (22,25,26),
we further investigated the protein levels of
p21CIP1/WAF1 and Cdk1 in Zmpste24−/−
and WT MEFs. The Western blot results indicated that the
overexpression of miR-342-5p increased the protein level of Cdk1 in
Zmpste24−/− MEFs (Fig. 3E). Collectively, these results
suggested that miR-342-5p overexpression increased the G2/M phase,
likely via upregulating Cdk1 in Zmpste24−/−
MEFs.
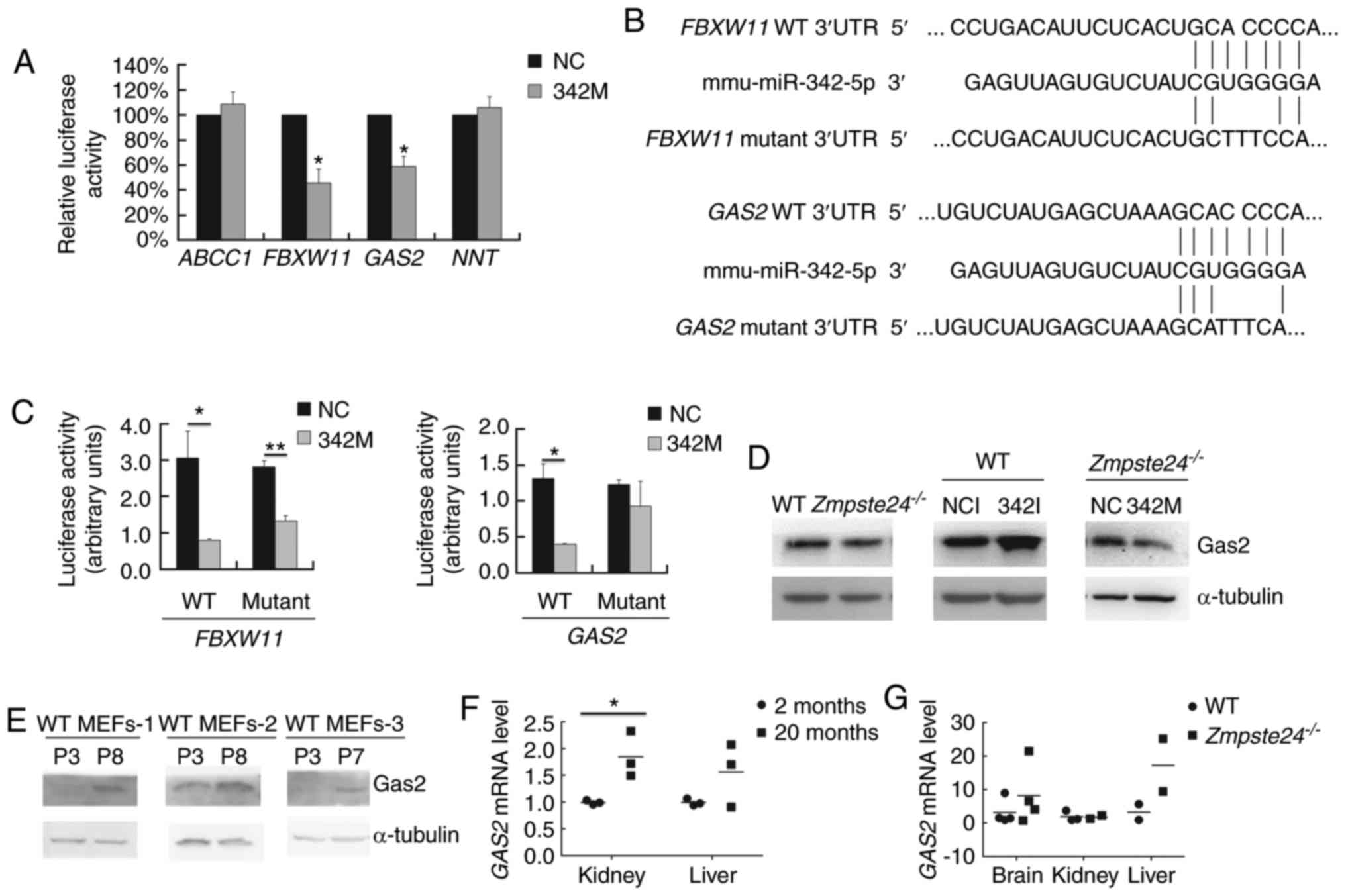
GAS2 is a target gene of
miR-342-5p
To identify the direct target genes of miR-342-5p in
Zmpste24−/− MEFs, we selected several potential
target genes via 4 target prediction algorithms in silico
(Table I). Then, we carried out
the Dual luciferase assay to assess whether miR-342-5p binds to the
3′UTR of these potential target genes in vitro. Since it was
difficult to perform the transfection with the luciferase vectors
due to the low transfection efficiency in the MEFs, we performed
the Dual luciferase assay in C2C12 cells, which is a mouse myoblast
cell line with high efficiency for gene transfection. As shown in
Fig. 4A, miR-342-5p significantly
inhibited the firefly luciferase activity of the WT 3′UTR of
FBXW11 and GAS2. Next, we mutated the seed binding
site in the WT 3′UTR of FBXW11 (GCACCCCA→GCTTTCCA)
and GAS2 (GCACCCCA→GCATTTCA) (Fig. 4B). Our data showed that the
GAS2 mutant 3′UTR restored the luciferase activity (Fig. 4C). We further checked the Gas2
protein level in WT and Zmpste24−/− MEFs (p2~p4)
transfected with 342I or 342M. As shown in Fig. 4D, the overexpression of miR-342-5p
downregulated Gas2 in Zmpste24−/− MEFs, while the
inhibition of miR-342-5p upregulated Gas2 in WT MEFs. Taken
together, these results demonstrated that miR-342-5p downregulated
GAS2 by directly binding to the 3′UTR of GAS2 mRNA in
Zmpste24−/− MEFs. We further investigated whether
GAS2 is dysregulated in WT MEFs during replicative
senescence or in tissues from physiological ageing mice or from
Zmpste24−/− progeroid mice. As shown in Fig. 4E, the Gas2 protein level was
upregulated in WT MEFs during replicative senescence. Moreover, the
GAS2 mRNA level was upregulated in the kidney of ageing mice
as well (Fig. 4F).
 | Table I.Potential target genes of
mmu-miR-342-5p predicted by target prediction algorithms. |
Table I.
Potential target genes of
mmu-miR-342-5p predicted by target prediction algorithms.
|
| TargetScan | miRanda | MicroCosm | miRDB |
|---|
| ABCC1 | √ | √ | √ | √ |
| FBXW11 | √ | √ | √ |
|
| GAS2 | √ | √ |
| √ |
| NNT | √ | √ |
| √ |
Discussion
Increasing evidence shows that miRNAs play important
roles in the premature cell senescence phenotypes of progeroid
cells; however, the functions of most miRNAs are still unclear.
Previous studies show that miR-342-5p is downregulated in
Zmpste24−/− progeroid MEFs (15). We herein revealed that miR-342-5p
overexpression was sufficient to promote
Zmpste24−/− MEFs proliferation and ameliorated
the senescence phenotype to some extent, which provides novel
insights into the role of miR-342-5p in the premature senescence
phenotypes of Zmpste24−/− MEFs.
miR-342-5p, an intronic miRNA hosted in the
EVL gene, is reportedly dysregulated in ageing-associated
diseases, such as Alzheimer's disease and atherosclerosis mouse
models (18,19). In this research, we found that
miR-342-5p was downregulated in WT MEFs during replicative
senescence (Fig. 1A), which is
consistent with the downregulation in premature senescence of
Zmpste24−/− MEFs (15). However, we did not observe a
significant dysregulation of miR-342-5p in several tissues from
physiological ageing mice or from Zmpste24−/−
progeroid mice (Fig. 1B-C), which
may be due to the small number of investigated specimens or due to
different cell backgrounds: the tissue cells are terminally
differentiated cells which is different from the MEFs, thus the
gene expression pattern of senescent MEFs (replicative senescence)
is not always consistent with that of (premature) ageing tissues.
In human colorectal cancer and inflammatory breast cancer, a
downregulation of miR-342-5p is an epigenetic silencing mechanism
due to the CpG island methylation upstream of EVL (27,28).
However, the expression of miR-342-5p was not consistent with that
of EVL mRNA in several tissues from
Zmpste24−/− mice (Fig. 1C-D). Indeed, the small sample
number is a limitation of the present study, and it is necessary to
repeat these tests with more samples in future studies.
For the cell phenotype analyses, we first tested the
effects of miR-342-5p on cellular senescence and found that
miR-342-5p overexpression ameliorated the senescence phenotype in
Zmpste24−/− MEFs to some extent (Fig. 1E-F). Hence, we speculated that
miR-342-5p might be involved in regulating cell proliferation or
cell cycle in Zmpste24−/− MEFs. Indeed,
miR-342-5p overexpression was sufficient to promoted cell
proliferation in Zmpste24−/− and WT MEFs
(Fig. 2). However, our results are
not in agreement with a recent report that miR-342-5p
overexpression inhibits endothelial cell proliferation (29). At first glance, such results may
seem contradictory, but it is worth noting that miRNAs can have
different effects in different cell types (30,31).
Next, we investigated the effects of miR-342-5p on
the cell cycle and found that miR-342-5p overexpression increased
the G2+M cell cycle phase in both Zmpste24−/− and
WT MEFs. In addition, miR-342-5p overexpression upregulated Cdk1 in
Zmpste24−/− MEFs (Fig. 3). In the cell cycle, Cdk1 is
required for the entry of all eukaryotic cells into mitosis
(22,32) and is sufficient to drive the
mammalian cell cycle (33). Hence,
miR-342-5p overexpression promotes cell proliferation probably by
upregulating Cdk1 in Zmpste24−/− MEFs. Since
p21CIP1/WAF1 serves as a negative regulator in the G2/M
transition (25,26) and plays a critical role in cellular
senescence, we speculated that miR-342-5p might upregulate Cdk1
through the inhibition of p21CIP1/WAF1. However, it was
difficult to observe a consistent suppression of
p21CIP1/WAF1 when overexpressing miR-342-5p in
Zmpste24−/− MEFs.
Of note, as we carried out a loss-of-function
analysis by transfection with the miR-342-5p Inhibitor
(single-stranded antisense siRNA) in WT MEFs, and it was difficult
to observe the opposite cell phenotypes, such as cellular
senescence, cell proliferation, and cell cycle when compared with
miR-342-5p overexpression. One possible reason may be due to the
extremely low basal expression of miR-342-5p in WT MEFs. Another
possible reason may be that there is not only one miRNA that
regulates cell phenotypes, and thus, if we suppressed all the
related miRNAs at the same time, it is possible that the
significant phenotypes of the loss-of-function could be easily
observed.
Additionally, we further identified GAS2 as
a target gene of miR-342-5p in Zmpste24−/− MEFs
(Fig. 4A-D). Gas2 was originally
identified in growth arrested mouse fibroblasts (34) and inhibits cell division in
Xenopus embryos (35). As a
p53-stabilizing protein, Gas2 is implicated in p53-induced growth
inhibition (36). In
4E-BP1−/−4E-BP2−/− double
knockout MEFs, the suppression of Gas2 by shRNA reduces the
SA-β-Gal activity and increases proliferation, demonstrating that
Gas2 expression is a prerequisite for cellular senescence (37). In this research, we found that Gas2
was upregulated in WT MEFs during replicative senescence (Fig. 4E), and GAS2 mRNA was
upregulated in kidney of ageing mice (Fig. 4F). Taken together, these results
indicate that miR-342-5p promotes Zmpste24−/−
MEFs proliferation by suppressing GAS2.
Generally, the present study mainly focused at the
cell level, thus it can not confirm that the miR-342-5p has an
effect on the cellular senescence in Zmpste24−/−
MEFs in vivo. The validation test in vivo from
animals remains to be further investigated. In conclusion, our data
suggest that downregulated miR-342-5p is involved in regulating
cell proliferation and the cell cycle via suppressing GAS2
in Zmpste24−/− MEFs in vitro, which may
have implications for the underlying mechanisms of premature
senescence in progeroid cells.
Acknowledgements
We are grateful to Professor Yousin Suh (Albert
Einstein College of Medicine, Bronx, NY, USA), Professor Brian K.
Kennedy (Buck Institute for Research on Aging, Novato, CA, USA) and
Professor Matt Kaeberlein (University of Washington, Seattle, WA,
USA) for their technical assistance and discussion. We would also
like to thank American Journal Experts for English language
editing. This study was supported by the National Natural Science
Foundation of China (nos. 81170327, 81671399, 31600976 and
81202385), the Ordinary University Innovation Team Construction
Project of Guangdong Province (no. 2015KCXTD022), and the Dongguan
International Science and Technology Cooperation (including Hong
Kong, Macao and Taiwan) Project (no. 201650812001).
Glossary
Abbreviations
Abbreviations:
|
MEFs
|
mouse embryonic fibroblasts
|
|
HGPS
|
Hutchinson-Gilford progeria
syndrome
|
|
SA-β-Gal
|
senescence-associated
β-galactosidase
|
|
GAS2
|
growth-arrest-specific 2
|
References
|
1
|
McCulloch K, Litherland GJ and Rai TS:
Cellular senescence in osteoarthritis pathology. Aging Cell.
16:210–218. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khan SY, Awad EM, Oszwald A, Mayr M, Yin
X, Waltenberger B, Stuppner H, Lipovac M, Uhrin P and Breuss JM:
Premature senescence of endothelial cells upon chronic exposure to
TNFα can be prevented by N-acetyl cysteine and plumericin. Sci Rep.
7:395012017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu B, Wang J, Chan KM, Tjia WM, Deng W,
Guan X, Huang JD, Li KM, Chau PY, Chen DJ, et al: Genomic
instability in laminopathy-based premature aging. Nat Med.
11:780–785. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eriksson M, Brown WT, Gordon LB, Glynn MW,
Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et
al: Recurrent de novo point mutations in laminA cause
Hutchinson-Gilford progeria syndrome. Nature. 423:293–298. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pendás AM, Zhou Z, Cadiñanos J, Freije JM,
Wang J, Hultenby K, Astudillo A, Wernerson A, Rodríguez F,
Tryggvason K and López-Otín C: Defective prelamin A processing and
muscular and adipocyte alterations in Zmpste24
metalloproteinase-deficient mice. Nat Genet. 31:94–99.
2002.PubMed/NCBI
|
|
6
|
Goldman RD, Shumaker DK, Erdos MR,
Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M,
Varga R and Collins FS: Accumulation of mutant lamin A causes
progressive changes in nuclear architecture in Hutchinson-Gilford
progeria syndrome. Proc Natl Acad Sci USA. 101:pp. 8963–8968. 2004;
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barthélémy F, Navarro C, Fayek R, Da Silva
N, Roll P, Sigaudy S, Oshima J, Bonne G, Papadopoulou-Legbelou K,
Evangeliou AE, et al: Truncated prelaminA expression in HGPS-like
patients: A transcriptional study. Eur J Hum Genet. 23:1051–1061.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Olive M, Harten I, Mitchell R, Beers JK,
Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, et al:
Cardiovascular pathology in Hutchinson-Gilford progeria:
Correlation with the vascular pathology of aging. Arterioscler
Thromb Vascr Biol. 30:2301–2309. 2010. View Article : Google Scholar
|
|
9
|
Scaffidi P and Misteli T: Lamin
A-dependent nuclear defects in human aging. Science. 312:1059–1063.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cao K, Blair CD, Faddah DA, Kieckhaefer
JE, Olive M, Erdos MR, Nabel EG and Collins FS: Progerin and
telomere dysfunction collaborate to trigger cellular senescence in
normal human fibroblasts. J Clin Invest. 121:2833–2844. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Arancio W, Pizzolanti G, Genovese SI,
Pitrone M and Giordano C: Epigenetic involvement in
Hutchinson-Gilford progeria syndrome: A mini-review. Gerontology.
60:197–203. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jung HJ, Coffinier C, Choe Y, Beigneux AP,
Davies BS, Yang SH, Barnes RH II, Hong J, Sun T, Pleasure SJ, et
al: Regulation of prelamin A but not lamin C by miR-9, a
brain-specific microRNA. Proc Natl Acad Sci USA. 109:pp. E423–E431.
2012; View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nissan X, Blondel S, Navarro C, Maury Y,
Denis C, Girard M, Martinat C, De Sandre-Giovannoli A, Levy N and
Peschanski M: Unique preservation of neural cells in
Hutchinson-Gilford progeria syndrome is due to the expression of
the neural-specific miR-9 microRNA. Cell Rep. 2:1–9. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ugalde AP, Ramsay AJ, de la Rosa J, Varela
I, Mariño G, Cadiñanos J, Lu J, Freije JM and López-Otín C: Aging
and chronic DNA damage response activate a regulatory pathway
involving miR-29 and p53. EMBO J. 30:2219–2232. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiong XD, Jung HJ, Gombar S, Park JY,
Zhang CL, Zheng H, Ruan J, Li JB, Kaeberlein M, Kennedy BK, et al:
MicroRNA transcriptome analysis identifies miR-365 as a novel
negative regulator of cell proliferation in Zmpste24-deficient
mouse embryonic fibroblasts. Mutat Res. 777:69–78. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bear JE, Svitkina TM, Krause M, Schafer
DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy
GG and Gertler FB: Antagonism between Ena/VASP proteins and actin
filament capping regulates fibroblast motility. Cell. 109:509–521.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tavares S, Vieira AF, Taubenberger AV,
Araújo M, Martins NP, Brás-Pereira C, Polónia A, Herbig M, Barreto
C, Otto O, et al: Actin stress fiber organization promotes cell
stiffening and proliferation of pre-invasive breast cancer cells.
Nat Commun. 8:152372017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun X, Wu Y, Gu M and Zhang Y: miR-342-5p
decreases ankyrin G levels in Alzheimer's disease transgenic mouse
models. Cell Rep. 6:264–270. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wei Y, Nazari-Jahantigh M, Chan L, Zhu M,
Heyll K, Corbalán-Campos J, Hartmann P, Thiemann A, Weber C and
Schober A: The microRNA-342-5p fosters inflammatory macrophage
activation through an Akt1- and microRNA-155-dependent pathway
during atherosclerosis. Circulation. 127:1609–1619. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao F, Zhang YF, Zhang ZP, Fu LA, Cao XL,
Zhang YZ, Guo CJ, Yan XC, Yang QC, Hu YY, et al: miR-342-5p
regulates neural stem cell proliferation and differentiation
downstream to Notch signaling in mice. Stem Cell Reports.
8:1032–1045. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Zhang Y, Xu S, Li W, Chen Z, Wang
Z, Han X, Zhao Y and Li S: Prognostic significance of G2/M arrest
signaling pathway proteins in advanced non-small cell lung cancer
patients. Oncol Lett. 9:1266–1272. 2015.PubMed/NCBI
|
|
22
|
Fesquet D, Labbé JC, Derancourt J, Capony
JP, Galas S, Girard F, Lorca T, Shuttleworth J, Dorée M and
Cavadore JC: The MO15 gene encodes the catalytic subunit of a
protein kinase that activates cdc2 and other cyclin-dependent
kinases (CDKs) through phosphorylation of Thr161 and its
homologues. EMBO J. 12:3111–3121. 1993.PubMed/NCBI
|
|
23
|
Su H, Yang JR, Xu T, Huang J, Xu L, Yuan Y
and Zhuang SM: MicroRNA-101, down-regulated in hepatocellular
carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer
Res. 69:1135–1142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta DeltaC(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Niculescu AB III, Chen X, Smeets M, Hengst
L, Prives C and Reed SI: Effects of p21(Cip1/Waf1) at both the G1/S
and the G2/M cell cycle transitions: pRb is a critical determinant
in blocking DNA replication and in preventing endoreduplication.
Mol Cell Biol. 18:629–643. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barboule N, Lafon C, Chadebech P, Vidal S
and Valette A: Involvement of p21 in the PKC-induced regulation of
the G2/M cell cycle transition. FEBS Lett. 444:32–37. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Grady WM, Parkin RK, Mitchell PS, Lee JH,
Kim YH, Tsuchiya KD, Washington MK, Paraskeva C, Willson JK, Kaz
AM, et al: Epigenetic silencing of the intronic microRNA
hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene.
27:3880–3888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Van der Auwera I, Yu W, Suo L, Van Neste
L, van Dam P, Van Marck EA, Pauwels P, Vermeulen PB, Dirix LY and
Van Laere SJ: Array-based DNA methylation profiling for breast
cancer subtype discrimination. PLoS One. 5:e126162010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yan XC, Cao J, Liang L, Wang L, Gao F,
Yang ZY, Duan JL, Chang TF, Deng SM, Liu Y, et al: miR-342-5p is a
Notch downstream molecule and regulates multiple angiogenic
pathways including Notch, vascular endothelial growth factor and
transforming growth factor β signaling. J Am Heart Assoc.
5:e0030422016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Peter M, Le Peuch C, Labbé JC, Meyer AN,
Donoghue DJ and Doree M: Initial activation of cyclin-B1-cdc2
kinase requires phosphorylation of cyclin B1. EMBO Rep. 3:551–556.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Santamaría D, Barrière C, Cerqueira A,
Hunt S, Tardy C, Newton K, Cáceres JF, Dubus P, Malumbres M and
Barbacid M: Cdk1 is sufficient to drive the mammalian cell cycle.
Nature. 448:811–815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schneider C, King RM and Philipson L:
Genes specifically expressed at growth arrest of mammalian cells.
Cell. 54:787–793. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang T, Dayanandan B, Rouiller I,
Lawrence EJ and Mandato CA: Growth-arrest-specific protein 2
inhibits cell division in Xenopus embryos. PLoS One. 6:e246982011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kondo Y, Shen L, Cheng AS, Ahmed S,
Boumber Y, Charo C, Yamochi T, Urano T, Furukawa K, Kwabi-Addo B,
et al: Gene silencing in cancer by histone H3 lysine 27
trimethylation independent of promoter DNA methylation. Nat Genet.
40:741–750. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Petroulakis E, Parsyan A, Dowling RJ,
LeBacquer O, Martineau Y, Bidinosti M, Larsson O, Alain T, Rong L,
Mamane Y, et al: p53-dependent translational control of senescence
and transformation via 4E-BPs. Cancer Cell. 16:439–446. 2009.
View Article : Google Scholar : PubMed/NCBI
|