Introduction
Leber's hereditary optic neuropathy (LHON) is a
neurodegenerative eye disorder, which is clinically characterized
by rapid, painless, bilateral central visual loss, that commonly
affects young adults (1–4). The majority of cases of LHON are the
result of one of three primary mutations:
m.11778G>A/mitochondrially encoded NADH:ubiquinone
oxidoreductase core MT-ND4, m.3460G>A/MT-ND1 or
m.14484T>C/MT-ND6 (5–7),
whereas the remaining 5% of cases are caused by rare mitochondrial
DNA (mtDNA) mutations and/or other factors. At present, ~40
mutations (www.mitomap.org/foswiki/bin/view/MITOMAP/MutationsLHON),
which mainly occur in complex I, have been associated with LHON.
Therefore, mtDNA mutations are considered the molecular basis for
LHON disease (5,8), and they often present near or at
homoplasmy. However, male bias and incomplete penetrance are
typical clinical characteristics of LHON, thus indicating that LHON
has a complex etiology (9,10). Therefore, mtDNA mutations are
considered insufficient to result in the phenotypic expression of
LHON, and other modifying determinants, including mitochondrial
haplotypes and nuclear genetic backgrounds, and environmental
factors, are likely to modulate the phenotypic manifestation of
LHON-associated common or rare mtDNA mutations (11–14).
LHON is the first disorder that was recognized to be
maternally inherited, and is the first to have been attributed to a
point mutation in mtDNA (5). In
previous investigations, besides the three primary mutations
(m.11778G>A, m.3460G>A, m.14484T>C), we identified
LHON-associated m.3394T>C/MT-ND1,
m.3635G>A/MT-ND1, m.3866T>C/MT-ND1,
m.11696G>A/MT-ND4, m.12238T>C/MT-ND5 and
m.14502T>C/MT-ND6 mutations (15–19).
In the present study, the clinical, genetic and molecular features
of two Chinese families with maternally transmitted LHON were
investigated; the m.5587T>C mutation in the mitochondrially
encoded transfer (t)RNA alanine (MT-TA) gene was detected in
the families, which lacked the three known primary mutations. To
elucidate the role of other genetic factors, including
mitochondrial haplotype, in the phenotypic expression of the
m.5587T>C mutation, 24 overlapping fragments were used to
perform polymerase chain reaction (PCR) amplification of fragments
spanning the entire mitochondrial genome and a DNA sequence
analysis was subsequently conducted.
Materials and methods
Subjects and ophthalmologic
examinations
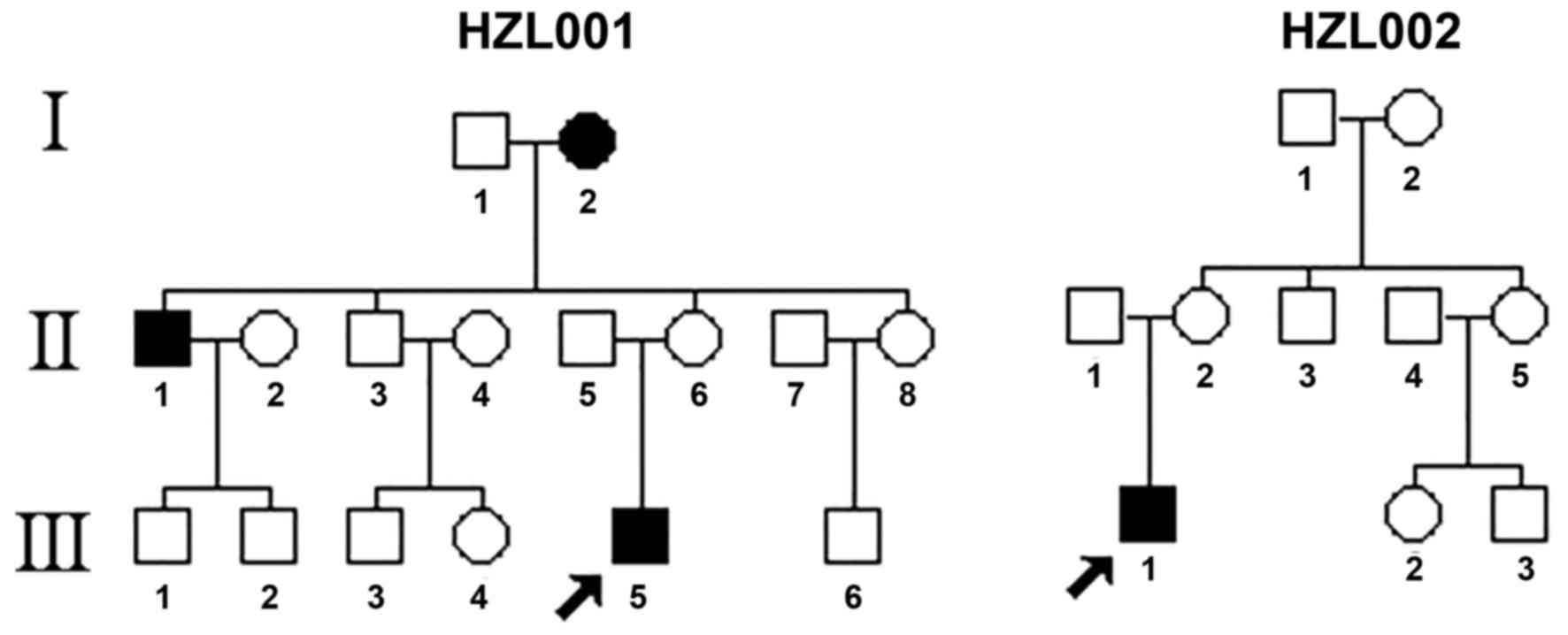
As shown in Fig. 1,
the patients from two Han Chinese families (HZL001 and HZL002) were
recruited at the School of Ophthalmology and Optometry, Wenzhou
Medical University (Wenzhou, China) and at Dongfang Hospital
(Beijing, China). A total of 376 control DNA samples were obtained
from a panel of unaffected subjects with Han Chinese ancestry from
the same region. The ophthalmic examinations of the probands and
other matrilineal relatives, including visual field test, visual
acuity examination, fundus photography, visual evoked potentials
and determination of the degree of visual impairment, as well as
other clinical evaluations, were conducted as described previously
(11–13). Blood samples were also obtained
from the participants. Written informed consent was obtained from
the probands and other affected relatives evaluated, and the
present study was approved by the Ethic Committees of Wenzhou
Medical University and Zhejiang University (Hangzhou, China).
mtDNA mutational analysis
Genomic DNA was isolated from the whole blood
samples of participants and controls using QIAamp DNA Blood Mini
kit (cat. no. 51104; Qiagen, Inc., Valencia, CA, USA). The presence
of the m.11778G>A, m.3460G>A and m.14484T>C mutations was
detected as previously described (11–13).
Briefly, DNA fragments of probands and affected members spanning
these mtDNA mutations were amplified by PCR using
oligodeoxynucleotides corresponding to mtDNA at positions 11,654 to
11,865 for the m.11778G>A mutation, 3,108 to 3,717 for the
m.3460G>A mutation, and 14,260 to 14,510 for the m.14484T>C
mutation (20). Each fragment was
purified by PCR clean-up (cat. no. AP-PCR-50; Axygen; Corning
Incorporated, Corning, NY, USA) and subsequently analyzed by direct
sequencing in an ABI 3700 automated DNA sequencer (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) using
the Big Dye Terminator Cycle sequencing reaction kit (Thermo Fisher
Scientific, Inc.). The three fragment sequence results were
compared with the updated consensus Cambridge sequence (GenBank
accession no. NC_012920) (13).
For detecting the m.5587T>C mutation in the
MT-TA gene, PCR amplification using oligodeoxynucleotides
corresponding to mtDNA at positions 5,238–6,050 was conducted. The
primer pair sequences for PCR were as follows: Forward:
5′-CTAACCGGCTTTTTGCCC-3′ and reverse 5′-ACCTAGAAGGTTGCCTGGCT-3′,
which were designed according to a previously described method
(20). Subsequently, this fragment
was purified and analyzed as aforementioned.
Haplogroup analyses and phylogenetic
analysis
The mtDNA sequences of 17 different vertebrates were
used to conduct an interspecific analysis as reported previously
(11–13). The conservation index (CI) was
calculated by comparing the human nucleotide variants with the
other 16 vertebrates. The CI was used to indicate the percentage of
species from the list of 16 different vertebrates that have the
wild-type nucleotide at the same position.
The Asian mitochondrial haplogroup of these two
probands were determined using online software (www.mitotool.org/genomeRSRS.html) or
based on the nomenclature of mitochondrial haplogroups previously
reported (21,22).
Results
Clinical presentation
Clinical data for the two Chinese probands are
summarized in Table I. The four
individuals that were affected by LHON in the two pedigrees
comprised one woman and three men. Comprehensive medical histories
of the probands confirmed that they suffered from no other clinical
abnormalities, including hearing dysfunction, muscular diseases,
diabetes and neurological diseases. In pedigree HZL001, the proband
(III-5) was first examined at the age of 17 at the Ophthalmology
Clinic of Wenzhou Medical University. He started to suffer from
bilateral visual loss at the age of 15; he observed a dark cloud in
the center of his vision and had difficulty discerning colors,
which all appeared dark grey. Visual field test detected a large
centrocecal scotomata in both eyes. His visual acuity was oculus
dexter, 0.02; and oculus sinister, 0.05. Fundus examination
demonstrated that both of his optic discs were abnormal: Vascular
tortuosity of the central retinal vessels, circumpapillary
telangiectatic microangiopathy and swelling of the retinal nerve
fiber layer were detected. These findings indicated that the
patient exhibited the typical clinical features of LHON. In
addition, his uncle (II-1) and grandmother (I-2) exhibited visual
loss, as presented in Table I. In
family HZL002, the proband (III-1) exhibited bilateral visual loss
from the age of 6. He was diagnosed with LHON by the Ophthalmology
Clinic at Beijing University of Chinese Medicine and Pharmacology
(Beijing, China). The visual field test detected a large
centrocecal scotomata in both eyes. His visual acuity was 0.1 in
both eyes. In addition, both optic discs were abnormal, as
determined by fundus examination: Vascular tortuosity of the
central retinal vessels, circumpapillary telangiectatic
microangiopathy and swelling of the retinal nerve fiber layer were
detected. These findings indicated that the patient exhibited the
typical clinical features of LHON. Conversely, none of the other
six matrilineal relatives in HZL001 exhibited visual loss.
 | Table I.Summary of clinical data for four
patients with Leber's hereditary optic neuropathy from two
pedigrees carrying the m.5587T>C mutation. |
Table I.
Summary of clinical data for four
patients with Leber's hereditary optic neuropathy from two
pedigrees carrying the m.5587T>C mutation.
| Subject | Sex | Age of test
(years) | Age of onset
(years) | Visual acuity,
righta | Visual acuity,
lefta | Level of visual
impairment |
|---|
| HZL001-III-5 | F | 17 | 15 |
0.02 | 0.05 | Severe |
| HZL001-II-1 | M | 42 | 29 |
0.01 | CF/30 cm | Profound |
| HZL001-I-2 | M | 65 | 32 | 0.2 | 0.1 | Mild |
| HZL002-III-1 | M | 11 | 6 | 0.1 | 0.1 | Moderate |
mtDNA analysis
To explore the molecular basis of visual loss in the
two pedigrees, a mutational analysis of the mitochondrial genome in
the two Chinese families was conducted. Initially, three well known
LHON-associated mtDNA mutations (m.11778G>A, m.3460G>A and
m.14484T>C) were detected by PCR amplification and restriction
enzyme digestion analysis of PCR fragments derived from each
proband (data not shown). The m.11778G>A, m.3460G>A and
m.14484T>C mutations were not detected. Subsequently, PCR
amplification of fragments spanning the entire mitochondrial genome
and a DNA sequence analysis was conducted using DNA obtained from
the two probands and the affected matrilineal relatives. The
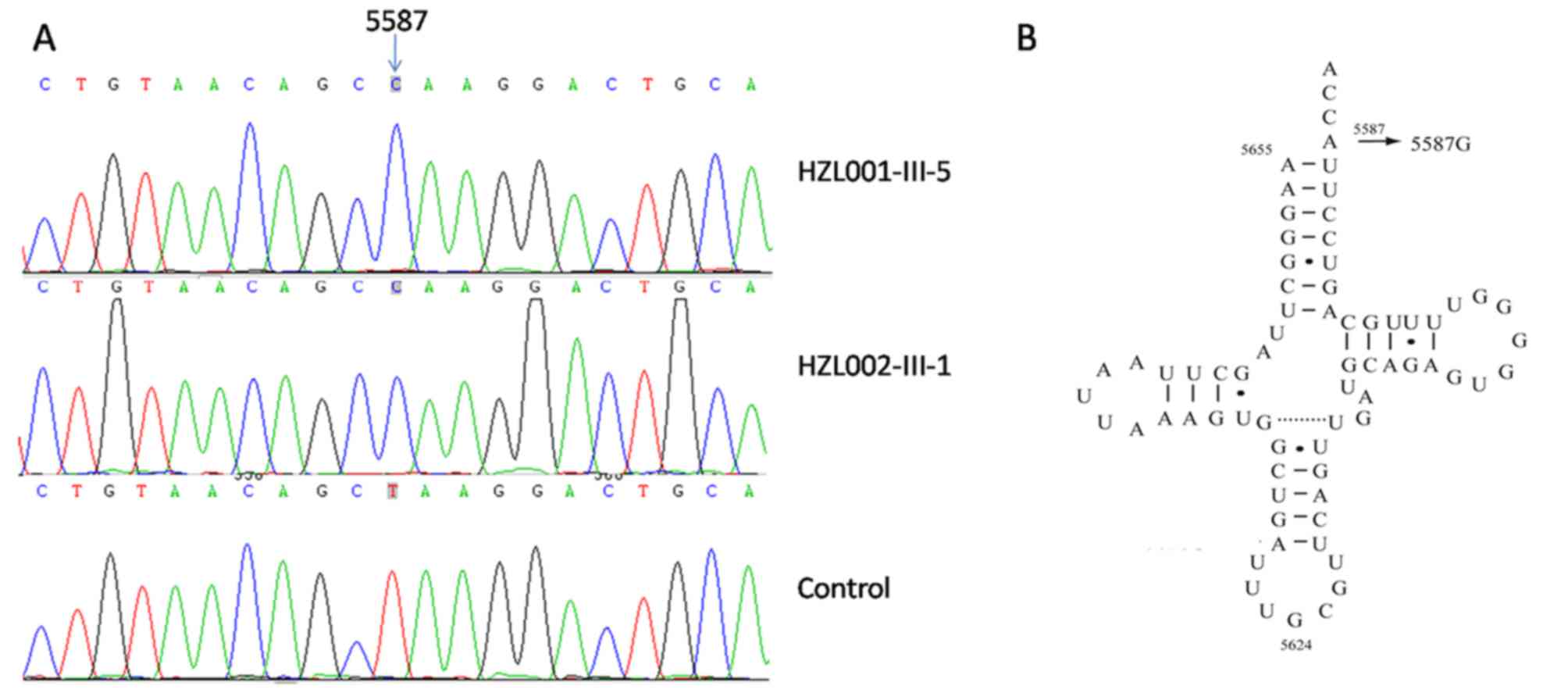
homoplasmic m.5587T>C mutation was detected (Fig. 2A). The m.5587T>C mutation refers
to a T to C transition at position 5,587 in the MT-TA gene,
which is localized in the end of the MT-TA gene (position
73; Fig. 2B), and was detected in
the two pedigrees evaluated in the present study. As shown in
Table II, this point at position
73 in the MT-TA gene is highly conserved among all 17
organisms analyzed, with the exception of Hylobates lar.
Notably, the m.5587T>C mutation has been reported to be
associated with progressive unstable gait, dysarthria, hearing
loss, muscle cramps and myalgia (23,24).
Further analysis of this gene fragment sequence, demonstrated that
the homoplasmic m.5587T>C mutation was detected in matrilineal
relatives of the two families but not in non-matrilineal relatives
(data not shown). Furthermore, none of the 376 unrelated Chinese
control subjects carried the m.5587T>C mutation.
| Table II.Alignment of the MT-TA gene
from 17 different species. Position 73 is the location of the
m.5587T>C mutation. |
Table II.
Alignment of the MT-TA gene
from 17 different species. Position 73 is the location of the
m.5587T>C mutation.
| Species | Acc-stem, pos
1 | Pos 8 | D-stem, pos 10 | D-loop, pos 14 | D-stem, pos 22 | Pos 26 | Ac-stem, Pos
27 | Anticd-loop, Pos
32 | Ac-stem, Pos
39 | V-region, Pos
44 | T-stem, Pos 49 | T-loop, Pos 54 | T-stem, Pos 61 | Acc-stem, Pos
66 | CCA tail, Pos
73 |
|---|
| Cebus
albifrons | GAGGGCT | TA | GCTT | AATTA | AAGT | A | GTTGA | TTTGCGT | TCAAT | TGAT | GCAAG | GTATAG | TTTGC | AGTCCTT | A |
| Cercopithecus
aethiops | AGGGGCT | TA | GCTT | AATTA | AAGT | G | GTTGA | TTTGCGT | TCAAT | TGAT | GCAGA | GTAGGTT | TTTGC | AGTCCTT | A |
| Colobus
guereza | AAGGGCT | TA | GCTT | AATGA | AAGT | G | ATTGA | TTTGCGT | TCAGT | TGAT | GCAGA | GTAGAGT | TTTGC | AGTCCTT | A |
| Gorilla
gorilla | AAGGGCT | TA | GCTT | AATTA | AAGT | G | GCTGA | TTTGCGT | TCAGT | TGAT | GCAGA | GTAGGGT | TTTGC | AGTCCTT | A |
| Homo
sapiens | AAGGGCT | TA | GCTT | AATTA | AAGT | G | GCTGA | TTTGCGT | TCAGT | TGAT | GCAGA | GTGGGGT | TTTGC | AGTCCTT | A |
| Hylobates
lar | AAGGGCT | TA | GCTT | AATTA | AAGT | G | ACTGA | TTTGCGT | TCGGT | TGAT | GCAAA | GTGGGC | TTTGC | AGTCCTT | G |
| Lemur
catta | GAGGATT | TA | GCTT | AATTA | AAGT | G | ATTGA | TTTGCGT | TCAGT | TGAT | GTAAG | ATATAAT | CTTGC | AGTCCTT | A |
| Macaca
mulatta | AAGGGCT | TA | GCTT | AATTA | AAGT | G | GTTGA | TTTGCGT | TCAAT | TGAT | GCAGA | GTAGGTG | TTTGC | AGTCCTT | A |
| Macaca
sylvanus | AAGGGCT | TA | GCTT | AATTA | AAGT | A | GTTGA | TTTGCGT | TCAAT | TGAT | GCAGA | GCAGGTG | TTTGC | AGTCCTT | A |
| Nycticebus
coucang | GAGGACT | TA | GCTT | AATTA | AAGT | A | ATTGA | TTTGCGT | TCAGT | TGAT | GTAGG | AGAAGT | CTTGC | AGTCCTT | A |
| Pan
paniscus | AAGGGCT | TA | GCTT | AATTA | AAGT | G | GCTGA | TTTGCGT | TCAGT | TGAT | GCAGA | GTGGGGT | TTTGC | AGTCCTT | A |
| Pan
troglodytes | AAGGGCT | TA | GCTT | AATTA | AAGT | G | GCTGA | TTTGCGT | TCAGT | TGAT | GCAGA | GTGGGGT | TTTGC | AGTCCTT | A |
| Papio
hamadryas | AAGGGCT | TA | GTTT | AATTA | AAGC | G | ATTGA | TTTGCGT | TCAGT | TGAT | GCGGA | GTAGGTG | TCTGC | AGTCCTT | A |
| Pongo
pygmaeus | GAGGGCT | TA | GCTT | AATTA | AAGT | G | GCTGA | TTTGCGC | TCAGT | TGAT | GCAAA | GTGGGGT | TTTGC | AGTCCTT | A |
| Pongo pygmaeus
abelii | GAGGGCT | TA | GCTT | AATTA | AAGT | G | GCTGG | TTTGCGT | TCAGT | TGAT | GCAGA | GCGGGGC | TTTGC | AGTCCTT | A |
| Tarsius
bancanus | GAGGACT | TA | GCTT | AAGTTA | AAGT | A | GCTAA | TTTGCAG | TTAGT | TGAT | GTAGA | GTGAGTC | TTTGC | AGTCCTT | A |
| Trachypithecus
bscurus | AAGGGCT | TA | GCTT | AATTA | AAGT | A | ACTGG | TTTGCGT | TCAGT | TGAT | GCAGA | ATGAGAT | TCTGT | AGTCCTT | A |
As presented in Tables
II and III, in addition to
the m.5587T>C mutation, which exhibited evolutionary
conservation among 17 organisms, there are many mtDNA polymorphic
loci in the two families investigated in the present study. Of the
other identified nucleotide alterations in these two mitochondrial
genomes, there were 15 known variants in the D-loop, two known
variants in the 12S ribosomal (r)RNA gene, one known variant in the
16S rRNA gene, 23 silent variants (one novel) in protein-encoding
genes, and seven missense mutations in protein-encoding genes. The
missense mutations were as follows: m.8860A>G(p.T112A) in the
mitochondrially encoded ATP synthase 6 gene, m.10609T>C(p.M47T)
in the MT-ND4 L gene, m.12406G>A(p.V24I) and
m.13928G>C(p.S531T) in the MT-ND5 gene,
m.14766C>T(p.T7I), m.15024G>A (p.C93H) and
m.15326A>G(p.T194A) in the mitochondrially encoded cytochrome B
(MT-CYB) gene. Subsequently, these RNA and polypeptide
variants were further assessed by phylogenetic analysis of these
variants and sequences from other organisms, including mice
(25), cows (26) and Xenopus laevis (27). The mtDNA variant
m.15024G>A(p.C93H) in the MT-CYB gene exhibited
evolutionary conservation among the four organisms. However, none
of the other variants exhibited evolutionary conservation,
suggesting that these variants may not be functionally significant.
Based on the nomenclature of mitochondrial haplogroups, the mtDNA
sequence variations among two Chinese probands were used to
establish the haplogroup affiliation of each mtDNA (23,24).
In the present study, the mtDNAs of the two pedigrees belonged to
haplogroup F1.
| Table III.mtDNA variants in two Chinese
families with Leber's hereditary optic neuropathy. |
Table III.
mtDNA variants in two Chinese
families with Leber's hereditary optic neuropathy.
| Gene | Position | Replacement | Conservation
(H/B/M/X) | CRS | HZL001 | HZL002 | Previously
reporteda |
|---|
| D-loop | 73 | A-G |
| A | G | G | Yes |
|
| 150 | C-T |
| C |
| T | Yes |
|
| 195 | T-C |
| T |
| C | Yes |
|
| 204 | T-C |
| T | C |
| Yes |
|
| 207 | G-A |
| G | A |
| Yes |
|
| 215 | A-G |
| A | G |
| Yes |
|
| 249 | delA |
| A | del | del | Yes |
|
| 263 | A-G |
| A | G | G | Yes |
|
| 310 | T-CTC |
| T | CTC | CTC | Yes |
|
| 522 | Del C |
| C | Del |
| Yes |
|
| 523 | Del A |
| A | Del |
| Yes |
|
| 16,183 | A-C |
| A | C |
| Yes |
|
| 16,189 | T-C |
| T | C |
| Yes |
|
| 16,304 | T-C |
| T | C | C | Yes |
|
| 16,519 | T-C |
| T | C | C | Yes |
| MT-RNR1 | 750 | A-G | A/A/A/- | A | G | G | Yes |
|
| 1,438 | A-G | A/A/A/G | A | G | G | Yes |
| MT-RNR2 | 2,706 | A-G | A/G/A/A | A | G | G | Yes |
| MT-ND1 | 3,970 | C-T |
| C | T | T | Yes |
| MT-ND2 | 4,769 | A-G |
| A | G | G | Yes |
|
| 5,201 | T-C |
| T | C |
| Yes |
| MT-TA | 5,587 | T-C | T/T/-/T | T | C | C | Yes |
| MT-CO1 | 6,182 | G-A |
| G |
| A | Yes |
|
| 6,392 | T-C |
| T | C | C | Yes |
|
| 6,962 | G-A |
| G | A | A | Yes |
|
| 7,028 | C-T |
| C | T | T | Yes |
| MT-CO2 | 8,149 | A-G |
| A |
| G | Yes |
|
| 8,152 | G-A |
| G | A |
| Yes |
| MT-ATP6 | 8,860 | A-G(Thr-Ala) | T/A/A/T | A | G | G | Yes |
|
| 9,165 | T-C |
| T |
| C | Yes |
| MT-CO3 | 10,310 | G-A |
| G | A | A | Yes |
| MT-ND4L | 10,490 | T-C |
| T | C |
| Yes |
|
| 10,609 | T-C(Met-Thr) | M/T/T/T | T | C | C | Yes |
| MT-ND4 | 11,471 | C-T |
| C |
| T | Yes |
|
| 11,719 | G-A |
| G | A | A | Yes |
| MT-ND5 | 12,406 | G-A(Val-Ile) | V/F/S/F | G | A | A | Yes |
|
| 12,882 | C-T |
| C | T | T | Yes |
|
| 13,707 | G-A |
| G | A |
| Yes |
|
| 13,928 | G-C(Ser-Thr) | S/T/S/T | G | C | C | Yes |
| MT-CYB | 14,766 | C-T(Thr-Ile) | T/S/T/S | C | T | T | Yes |
|
| 15,024 | G-A(Cys-His) | C/C/C/C | G | A | A | No |
|
| 15,326 | A-G(Thr-Ala) | T/M/I/I | A | G | G | Yes |
Discussion
In the present study, the genetic, clinical and
molecular features of two Chinese pedigrees with LHON were
reported. The primary characteristic of LHON is bilateral visual
loss, which was only present in the maternal lineage of the
pedigrees evaluated, thus suggesting that mtDNA mutations are the
molecular basis for this disorder. Sequence analysis of the
complete mitochondrial genomes of these pedigrees demonstrated that
the three primary mutations associated with LHON were not present;
however, distinct mtDNA polymorphisms and the m.5587T>C mutation
in the MT-TA gene were detected in both pedigrees. Notably,
this homoplasmic mutation was only present in the maternal lineage
of the pedigrees, but not in the other members of these families.
Position 73 is evolutionarily conserved in the MT-TA gene.
This mutation may influence the 3′ end sequences of the amino acid
arm of tRNA; amino acids linked affect the tRNA structure and infer
structural changes. Therefore, this mutation may affect the
efficiency of amino acid translation, hinder protein synthesis and
induce mitochondrial dysfunction; in particular it may affect
encoding of the compound enzyme that initiates mitochondrial
oxidative phosphorylation required for respiratory chain and enzyme
activity. In response to abnormal oxidative phosphorylation, a
series of pathological alterations may be induced, including
production of oxygen free radicals and a reduction in the use of
nitric oxide. In addition, this mutation may have a potential
modifying role in deafness by worsening m.7505T>C
mutation-induced mitochondrial dysfunction (23). Furthermore, in a previous study, a
28-year-old woman that carried this mutation presented with a
16-year history of progressive unstable gait, dysarthria, hearing
loss, muscle cramps and myalgia (24). Notably, in the present study, the
presence of the m.5587T>C mutation in the two genetically
different pedigrees, both influenced by visual loss, indicated that
this mutation may be associated with the pathogenesis of visual
loss.
In these two families, the age of onset for visual
impairment ranged between 6 and 32 years old (average, 20.5 years
old). Previous studies confirmed that the age of onset for visual
impairment in the two pedigrees harboring the m.5587T>C mutation
was similar to that in other Chinese families with LHON carrying
the m.11778G>A mutation (28–32).
In contrast, a number of Chinese subjects carrying the
m.11778G>A mutation, which exhibited profound visual loss, the
affected subjects carrying the m.5587T>C mutation suffered from
mild to profound visual impairment, similar to that exhibited in
patients carrying m.3394T>C, m.3635G>A, m.3866T>C,
m.11696G>A, m.12238T>C and m.14502T>C mutations (15–19,33).
Furthermore, there is a high penetrance of visual loss in some
Chinese subjects harboring the m.11778G>A mutation (28,29,31–34);
however, the penetrance of visual impairment was very low in the
two Chinese pedigrees carrying the m.5587T>C mutation. In
addition, the m.5587T>C mutation was not detected in the 376
Chinese control subjects. Similar to the three common mutations,
and m.3394T>C, m.33635G>A, m.3866T>C, m.11696G>A,
m.12238T>C and m.14502T>C mutations (15–19),
the incomplete penetrance of visual loss associated with the
m.5587T>C mutation, and the lack of the mutation in any of the
376 control subjects, indicated that the m.5587T>C mutation is
itself insufficient to result in the clinical phenotype. These
findings suggested that other modifying factors, including
mitochondrial haplotype, environmental factors and nuclear
background are required for the phenotypic manifestation of the
m.5587T>C mutation. In particular, mitochondrial haplotypes have
been reported to influence the penetrance and expressivity of
visual loss associated with primary mtDNA mutations. Therefore,
secondary LHON mutations, such as m.4216T>C and m.13708G>A,
have been implicated to increase the penetrance of LHON-associated
primary m.11778G>A or m.14484T>C mutations (35). In addition, in a large cohort of
subjects with European ancestry, the mitochondrial haplogroup J was
able to affect the phenotypic manifestation of LHON-associated
m.11778G>A and m.14484T>C mutations (36,37).
Furthermore, the m.11696G>A, m.14502T>C, m.15951A>G and
m.4435A>G mutations have been reported to increase LHON
penetrance in Chinese subjects (28,31,38).
In the two pedigrees evaluated in the present study, the mtDNA
variants p.C93H in the MT-CYB gene exhibited evolutionary
conservation; however, other known mutations were not detected in
the LHON pedigrees. This mtDNA variant may have a potential
modifying role in the development of visual impairment associated
with the m.5587T>C mutation in these two pedigrees.
It has been suggested that mitochondrial haplotypes
may affect the penetrance and expressivity of LHON associated with
the three primary mutations (m.11778G>A, m.3460G>A and
m.14484T>C). In European pedigrees, mitochondrial haplogroup J
may raise the risk of LHON-associated m.11778G>A or
m.14484T>C mutations (14,39),
whereas haplogroup K may aggrandize the penetrance of primary
LHON-associated m.3460G>A mutation (14). Conversely, haplogroup H may reduce
the risk of LHON-associated m.11778G>A mutation (14). In addition, the haplogroup M7b1′2
significantly increases the occurrence of LHON, whereas the
haplogroup M8a has a protective effect in Chinese pedigrees
(40). In the two families
analyzed in the present study, the mitochondrial genomes of the
HZL001 and HZL002 pedigrees belonged to the Eastern Asian
haplogroup F1. However, since data from only two families were
analyzed, and due to the lack of a large data analysis, the role
the mitochondrial haplotype serves in the two families cannot be
fully explained. In addition, nuclear modifier genes, including the
tyrosyl-tRNA synthetase 2 gene in Chinese families (11), or environmental factors, may have
an important role in the phenotypic expression of the
LHON-associated m.5587T>C mutation in the two Chinese pedigrees.
Thus, the results of the present study may provide novel insights
into the understanding of clinical diagnosis and valuable
information on the management of LHON.
Acknowledgements
The present study was supported by grants from the
Natural Science Foundation of China (grant nos. 31471191, 81400434
and 31601025); the College Students' Science and Technology
Innovation (Xinmiao Talents Program) in Zhejiang province (grant
no. 2015R401217); and a general financial grant from China
Postdoctoral Science Foundation (grant no. 2016M591986).
References
|
1
|
Rasool N, Lessell S and Cestari DM: Leber
hereditary optic neuropathy: Bringing the lab to the clinic. Semin
Ophthalmol. 31:107–116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kirches E: LHON: Mitochondrial mutations
and more. Curr Genomics. 12:44–54. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu-Wai-Man P, Griffiths PG, Hudson G and
Chinnery PF: Inherited mitochondrial optic neuropathies. J Med
Genet. 46:145–158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carelli V, La Morgia C, Valentino ML,
Barboni P, Ross-Cisneros FN and Sadun AA: Retinal ganglion cell
neurodegeneration in mitochondrial inherited disorders. Biochim
Biophys Acta. 1787:518–528. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wallace DC, Singh G, Lott MT, Hodge JA,
Schurr TG, Lezza AM, Elsas LJ II and Nikoskelainen EK:
Mitochondrial DNA mutation associated with Leber's hereditary optic
neuropathy. Science. 242:1427–1430. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huoponen K, Vilkki J, Aula P,
Nikoskelainen EK and Savontaus ML: A new mtDNA mutation associated
with Leber hereditary optic neuroretinopathy. Am J Hum Genet.
48:1147–1153. 1991.PubMed/NCBI
|
|
7
|
Johns DR, Neufeld MJ and Park RD: An ND-6
mitochondrial DNA mutation associated with Leber hereditary optic
neuropathy. Biochem Biophys Res Commun. 187:1551–1557. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Howell N: LHON and other optic nerve
atrophies: The mitochondrial connection. Dev Ophthalmol. 37:94–108.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu-Wai-Man P, Griffiths PG and Chinnery
PF: Mitochondrial optic neuropathies-disease mechanisms and
therapeutic strategies. Prog Retin Eye Res. 30:81–114. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qu J, Li R, Tong Y, Hu Y, Zhou X, Qian Y,
Lu F and Guan MX: Only male matrilineal relatives with Leber's
hereditary optic neuropathy in a large Chinese family carrying the
mitochondrial DNA G11778A mutation. Biochem Biophys Res Commun.
328:1139–1145. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang P, Jin X, Peng Y, Wang M, Liu H, Liu
X, Zhang Z, Ji Y, Zhang J, Liang M, et al: The exome sequencing
identified the mutation in YARS2 encoding the mitochondrial
tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic
manifestation of Leber's hereditary optic neuropathy-associated
mitochondrial DNA mutation. Hum Mol Genet. 25:584–596. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiang P, Liang M, Zhang J, Gao Y, He Z, Yu
H, Zhao F, Ji Y, Liu X, Zhang M, et al: Prevalence of mitochondrial
ND4 mutations in 1281 Han Chinese subjects with leber's hereditary
optic neuropathy. Invest Ophthalmol Vis Sci. 56:4778–4788. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liang M, Jiang P, Li F, Zhang J, Ji Y, He
Y, Xu M, Zhu J, Meng X, Zhao F, et al: Frequency and spectrum of
mitochondrial ND6 mutations in 1218 Han Chinese subjects with
leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci.
55:1321–1331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hudson G, Carelli V, Spruijt L, Gerards M,
Mowbray C, Achilli A, Pyle A, Elson J, Howell N, La Morgia C, et
al: Clinical expression of Leber hereditary optic neuropathy is
affected by the mitochondrial DNA-haplogroup background. Am J Hum
Genet. 81:228–233. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liang M, Guan M, Zhao F, Zhou X, Yuan M,
Tong Y, Yang L, Wei QP, Sun YH, Lu F, et al: Leber's hereditary
optic neuropathy is associated with mitochondrial ND1 T3394C
mutation. Biochem Biophys Res Commun. 383:286–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang J, Jiang P, Jin X, Liu X, Zhang M,
Xie S, Gao M, Zhang S, Sun YH, Zhu J, et al: Leber's hereditary
optic neuropathy caused by the homoplasmic ND1 m.3635G>A
mutation in nine Han Chinese families. Mitochondrion. 18:18–26.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou X, Qian Y, Zhang J, Tong Y, Jiang P,
Liang M, Dai X, Zhou H, Zhao F, Ji Y, et al: Leber's hereditary
optic neuropathy is associated with the T3866C mutation in
mitochondrial ND1 gene in three Han Chinese families. Invest
Ophthalmol Vis Sci. 53:4586–4594. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou X, Wei Q, Yang L, Tong Y, Zhao F, Lu
C, Qian Y, Sun Y, Lu F, Qu J and Guan MX: Leber's hereditary optic
neuropathy is associated with the mitochondrial ND4 G11696A
mutation in five Chinese families. Biochem Biophys Res Commun.
340:69–75. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao F, Guan M, Zhou X, Yuan M, Liang M,
Liu Q, Liu Y, Zhang Y, Yang L, Tong Y, et al: Leber's hereditary
optic neuropathy is associated with mitochondrial ND6 T14502C
mutation. Biochem Biophys Res Commun. 389:466–472. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Andrews RM, Kubacka I, Chinnery PF,
Lightowlers RN, Turnbull DM and Howell N: Reanalysis and revision
of the Cambridge reference sequence for human mitochondrial DNA.
Nat Genet. 23:1471999. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kong QP, Bandelt HJ, Sun C, Yao YG, Salas
A, Achilli A, Wang CY, Zhong L, Zhu CL, Wu SF, et al: Updating the
East Asian mtDNA phylogeny: A prerequisite for the identification
of pathogenic mutations. Hum Mol Genet. 15:2076–2086. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tanaka M, Cabrera VM, González AM, Larruga
JM, Takeyasu T, Fuku N, Guo LJ, Hirose R, Fujita Y, Kurata M, et
al: Mitochondrial genome variation in eastern Asia and the peopling
of Japan. Genome Res. 14:1832–1850. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang X, Li R, Zheng J, Cai Q, Zhang T,
Gong S, Zheng W, He X, Zhu Y, Xue L, et al: Maternally inherited
hearing loss is associated with the novel mitochondrial tRNA Ser
(UCN) 7505T>C mutation in a Han Chinese family. Mol Genet Metab.
100:57–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Crimi M, Sciacco M, Galbiati S, Bordoni A,
Malferrari G, Del Bo R, Biunno I, Bresolin N and Comi GP: A
collection of 33 novel human mtDNA homoplasmic variants. Hum Mutat.
20:4092002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bibb MJ, Van Etten RA, Wright CT, Walberg
MW and Clayton DA: Sequence and gene organization of mouse
mitochondrial DNA. Cell. 26:167–180. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gadaleta G, Pepe G, De Candia G,
Quagliariello C, Sbisà E and Saccone C: The complete nucleotide
sequence of the Rattus norvegicus mitochondrial genome: Cryptic
signals revealed by comparative analysis between vertebrates. J Mol
Evol. 28:497–516. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roe BA, Ma DP, Wilson RK and Wong JF: The
complete nucleotide sequence of the Xenopus laevis mitochondrial
genome. J Biol Chem. 260:9759–9774. 1985.PubMed/NCBI
|
|
28
|
Qu J, Li R, Zhou X, Tong Y, Lu F, Qian Y,
Hu Y, Mo JQ, West CE and Guan MX: The novel A4435 G mutation in the
mitochondrial tRNAMet may modulate the phenotypic expression of the
LHON-associated ND4 G11778A mutation. Invest Ophthalmol Vis Sci.
47:475–483. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qu J, Li R, Zhou X, Tong Y, Yang L, Chen
J, Zhao F, Lu C, Qian Y, Lu F and Guan MX: Cosegregation of the ND4
G11696A mutation with the LHON-associated ND4 G11778A mutation in a
four generation Chinese family. Mitochondrion. 7:140–146. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qu J, Zhou X, Zhang J, Zhao F, Sun YH,
Tong Y, Wei QP, Cai W, Yang L, West CE and Guan MX: Extremely low
penetrance of Leber's hereditary optic neuropathy in 8 Han Chinese
families carrying the ND4 G11778A mutation. Ophthalmology.
116:558–564. e3. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li R, Qu J, Zhou X, Tong Y, Hu Y, Qian Y,
Lu F, Mo JQ, West CE and Guan MX: The mitochondrial tRNA(Thr)
A15951G mutation may influence the phenotypic expression of the
LHON-associated ND4 G11778A mutation in a Chinese family. Gene.
376:79–86. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xie S, Zhang J, Sun J, Zhang M, Zhao F,
Wei QP, Tong Y, Liu X, Zhou X, Jiang P, et al: Mitochondrial
haplogroup D4j specific variant m.11696G>A(MT-ND4) may increase
the penetrance and expressivity of the LHON-associated
m.11778G>A mutation in Chinese pedigrees. Mitochondrial DNA A
DNA Mapp Seq Anal. 28:434–441. 2017.PubMed/NCBI
|
|
33
|
Liu XL, Zhou X, Zhou J, Zhao F, Zhang J,
Li C, Ji Y, Zhang Y, Wei QP, Sun YH, et al: Leber's hereditary
optic neuropathy is associated with the T12338C mutation in
mitochondrial ND5 gene in six Han Chinese families. Ophthalmology.
118:978–985. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou X, Zhang H, Zhao F, Ji Y, Tong Y,
Zhang J, Zhang Y, Yang L, Qian Y, Lu F, et al: Very high penetrance
and occurrence of Leber's hereditary optic neuropathy in a large
Han Chinese pedigree carrying the ND4 G11778A mutation. Mol Genet
Metab. 100:379–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Torroni A, Petrozzi M, D'Urbano L,
Sellitto D, Zeviani M, Carrara F, Carducci C, Leuzzi V, Carelli V,
Barboni P, et al: Haplotype and phylogenetic analyses suggest that
one European-specific mtDNA background plays a role in the
expression of Leber hereditary optic neuropathy by increasing the
penetrance of the primary mutations 11778 and 14484. Am J Hum
Genet. 60:1107–1121. 1997.PubMed/NCBI
|
|
36
|
Brown MD, Starikovskaya E, Derbeneva O,
Hosseini S, Allen JC, Mikhailovskaya IE, Sukernik RI and Wallace
DC: The role of mtDNA background in disease expression: A new
primary LHON mutation associated with Western Eurasian haplogroup
J. Hum Genet. 110:130–138. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Howell N, Oostra RJ, Bolhuis PA, Spruijt
L, Clarke LA, Mackey DA, Preston G and Herrnstadt C: Sequence
analysis of the mitochondrial genomes from Dutch pedigrees with
Leber hereditary optic neuropathy. Am J Hum Genet. 72:1460–1469.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jiang P, Liang M, Zhang C, Zhao X, He Q,
Cui L, Liu X, Sun YH, Fu Q, Ji Y, et al: Biochemical evidence for a
mitochondrial genetic modifier in the phenotypic manifestation of
Leber's hereditary optic neuropathy-associated mitochondrial DNA
mutation. Hum Mol Genet. 25:3613–3625. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brown MD, Sun F and Wallace DC: Clustering
of Caucasian Leber hereditary optic neuropathy patients containing
the 11778 or 14484 mutations on an mtDNA lineage. Am J Hum Genet.
60:381–387. 1997.PubMed/NCBI
|
|
40
|
Ji Y, Zhang AM, Jia X, Zhang YP, Xiao X,
Li S, Guo X, Bandelt HJ, Zhang Q and Yao YG: Mitochondrial DNA
haplogroups M7b1′2 and M8a affect clinical expression of leber
hereditary optic neuropathy in Chinese families with the
m.11778G>a mutation. Am J Hum Genet. 83:760–768. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bailey IL and Lovie JE: New design
principles for visual acuity letter charts. Am J Optom Physiol Opt.
53:740–745. 1976. View Article : Google Scholar : PubMed/NCBI
|