Introduction
Enlarged vestibular aqueduct (EVA) syndrome, most
frequently found by radiological examination in patients with
nonsyndromic hearing loss (NSHL), is typically characterized by
congenital, bilateral profound sensorineural hearing loss (SHL) or
progressive hearing loss (HL) (1,2). EVA
in SHL is often accompanied with or without Pendred syndrome (PS;
MIM: 274600), which is often caused by mutation of the pendrin
protein encoded by the SLC26A4 gene. Autosomal recessive
NSHL with SLC26A4 mutations has been frequently reported to
present with inner ear malformations, including EVA, which can be
associated with Mondini dysplasia (MD), and abnormality of cochlear
spirals detected using computed tomography (CT) and magnetic
resonance imaging (MRI) (3–5).
SLC26A4 comprises 21 exons and produces an 86 kDa protein
containing 780 amino acids. The SLC26A4 protein, a member of the
solute carrier family 26A and also known as pendrin, is mapped on
chromosome 7q31 (6). Pendrin, the
gene product of SLC26A4, is important in the regulation of
endolymphatic pH and the maintenance of endocochlear potential by
mediating the exchange of fluid and ions, including formate,
bicarbonate, chloride and iodide ions (7). SLC26A4 mutation can cause PS
and autosomal recessive nonsyndromic hearing impairment locus 4
(MIM: 600791)-type NSHL. To date, >200 SLC26A4 mutations
have been reported in patients with nonsyndromic EVA or PS
(http://www.healthcare.uiowa.edu/labs/pendredandbor/slcMutations.htm).
In addition, 98.9% of the SLC26A4 mutants detected in
patients with EVA are from the Chinese population (8). A previous study reported that
children with PS and EVA showed improved performance in speech
perception, compared with a reference group with an unknown cause
of hearing impairment following cochlear implantation (9). Expanding the SLC26A4 mutation
spectrum may be beneficial for molecular assessment, enabling early
diagnosis and planning effective clinical strategies. Through the
use of targeted sequence capture and direct sequencing, the present
study investigated 20 coding exons of the SLC26A4 gene in 52
NSHL patients diagnosed with EVA by CT and MRI. The results
provided the first evidence, to the best of our knowledge, of the
compound heterozygous mutation p.R549Kfs*15 and p.H723R in
SLC26A4, which expands on the SLC26A4 mutation
spectrum in the Chinese population.
Patients and methods
Subjects and clinical
investigations
All Chinese individuals enrolled in the present
study were recruited from the Ear, Nose and Throat Department of
Drum Tower Hospital Affiliated to Nanjing University (Nanjing,
China). A total of 60 normal-hearing Chinese individuals underwent
pure-tone audiometry (PTA) testing and otoscopy at the Drum Tower
Hospital (male/female, 32/28; age, 7.86±4.71 years). Those
individuals with normal tympanum morphology and an average
threshold of PTA (250–8,000 Hz) <25 dB HL were considered as
normal-hearing controls. A total of 52 NSHL patients with EVA
(male/female, 30/22; age, 3.52±6.13 years) and their parents
underwent systematic clinical examinations and audiometric
evaluations prior to the patients receiving a cochlear implant at
Drum Tower Hospital. The average age of the 52 NSHL patients with
EVA was significantly lower, compared with that of the normal
controls, determined using Student's t-test (P<0.001). As the
majority of the normal controls were inpatients suffering from
adenoid hypertrophy and chronic tonsillitis, there were 37
pre-language HL patients in the group of 52 NSHL patients with EVA
in the present study. In addition to PTA and otoscopy, the patients
underwent tympanometry, auditory brainstem response (ABR),
distortion products otoacoustic emissions (DPOAE) and imaging
examinations. Imaging of the ear was performed by CT and MRI. The
patients or their parents were also interviewed to obtain a
detailed medical history and family history, details of the
mother's health during pregnancy, the patient's clinical history,
including history of head or brain injury, infection and the use of
medicines, including aminoglycoside antibiotics. The data
collected, including characteristics and age at diagnosis, are
listed in Table I.
 | Table I.Clinicopathological characteristics of
52 patients with NSHL and 60 normal controls. |
Table I.
Clinicopathological characteristics of
52 patients with NSHL and 60 normal controls.
| Characteristic | Patients with NSHL
and EVA n (%) | Normal controls n
(%) | P-value |
|---|
| Agea (years) | 3.52±6.13 | 7.86±4.71 | <0.0001 |
| Sex |
|
|
|
| Male | 30 (57.69) | 32 (53.33) | 0.644 |
|
Female | 22 (42.31) | 28 (46.67) |
|
| Family history |
|
|
|
| Yes | 14 (26.92) | NA |
|
| No | 38 (73.08) | NA |
|
| Hearing loss |
|
|
|
|
Pre-language | 37 (71.15) | NA |
|
|
Post-language | 15 (28.85) | NA |
|
| Modini
dysmorphia |
|
|
|
|
With | 2 (3.85) | NA |
|
|
Without | 50 (96.15) | NA |
|
| c.2168A> G |
|
|
|
|
Positive | 9
(17.31) | 0 |
|
|
Negative | 43 (82.69) | 60 |
|
|
c.1644_1645insA |
|
|
|
|
Positive | 2 (3.85) | 0 |
|
|
Negative | 50 (96.15) | 60 |
|
All participants and the parents of all minors
recruited in the present study provided written informed consent,
and the study protocol was performed in accordance with
institutional bioethics guidelines approved by the Research and
Ethics Committee of Drum Tower Hospital (201601502).
DNA isolation, polymerase chain
reaction (PCR) and sequence data analyses
Blood samples were collected following the provision
of written informed consent. Total genomic DNA was extracted from
the peripheral blood using a gDNA Isolation Micro kit (Watson
Biotechnologies, Shanghai, China). All 20 coding exons of the
SLC26A4 gene were amplified using PCR with primers designed
using Primer3 software (http://primer3.ut.ee/; Table II). The PCR mixture contained 250
µm dNTP Mixture, 10X Ex Taq buffer and Ex Taq (5 U/µl) polymerase
(Takara Biotechnology Co., Ltd., Dalian, China), and underwent 30
cycles of amplification with denaturation at 96°C for 20 sec,
annealing at 62°C for 20 sec, extension at 72°C for 60 sec, and
extension at 72°C for 5 min in a PTC-200 PCR thermal cycler
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The PCR amplified
products were purified and then sequenced using Sanger sequencing
with an ABI 3730XL genetic analyzer (Beijing Genomics Institute,
Beijing, China). The sequence data were analyzed by comparing them
with the sequence of SLC26A4 (NM_000441.1) of the National
Center for Biotechnology Information (NCBI) using DNAStar 5.0
software (DNAStar, Inc., Madison, WI, USA). In addition, the 60
normal controls were checked at the same locus.
| Table II.Specific primers used to amplify the
coding exons of SLC26A4. |
Table II.
Specific primers used to amplify the
coding exons of SLC26A4.
| Exon | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Product size
(bp) |
|---|
| 2 |
GGGGACTGGGTGGAACT |
GCCCGAGACTGATGGAG | 678 |
| 3 |
GCAAATTGGTTGTGACTGAG |
GAAGGGTAAGCAACCATCTGTCAC | 294 |
| 4 |
GTTGGGCAAAATAATCTAACGCA |
GCAGGCAAAACACTGAAATCC | 575 |
| 5 |
GGTCCGGCTCAGCTTCTT |
GCACCTGACCTAAAACAACGT | 481 |
| 6 |
AGGAAGGGGAGTGATAGGGT |
GTCTCAAACTCCTGGGCTCA | 497 |
|
7–8 |
CATGGTTTTTCATGTGGGAAGATTC |
AGACTGACTTACTGACTTAATGT | 502 |
| 9 |
GAGGACAAAGAAATCAGCCAGT |
CCCCTTCTTTAGCTGACACC | 456 |
| 10 |
ATCAGGTGCTATTTCTTG |
TTTCAGGTGAGGGAGTG | 526 |
| 11–12 |
TGTTCAGTTTTGTGGCTTGAG |
TCACATGGAAGACTTCATGGC | 663 |
| 13 |
TGTTTGTGGATCATTGA |
GCACAGCAGTAGAGGAC | 527 |
| 14 |
ACCTTTCAGGGTTATGG |
TTTCTCCCTTTGGCTAC | 491 |
| 15 |
TTGAGTGCTGCTACCC |
TTCTCATTGCCCTACA | 346 |
| 16 |
CCCTTTGAGAAATAGCC |
TTGCCAAGAAATACACT | 587 |
| 17 |
CTACCCACCATAGAAGG |
GCAATACTGGACAACCC | 626 |
| 18 |
ATTTAGCACCTCCACG |
CCACAGTCCCAGATAG | 375 |
| 19 |
TTTCAAATCTGGGTCAC |
CTACCAGGTAATTTCCTAT | 674 |
| 20 |
AGAAGCACCAGGAAAGC |
AGGAAGGTCATAGGGTT | 570 |
| 21 |
GGCAACAGTGAGTGAGATTCA |
AGCTTACCCTGGACGCTG | 462 |
Statistical analysis
Statistical analysis was performed using SPSS 22.0
for Windows (IBM SPSS, Armonk, NY, USA). The difference in age
between the NSHL patients with EVA and the normal controls was
determined using Student's t-test. The sex differences were
analyzed using Pearson's χ2 test. The statistical
associations of the prevalence of the novel compound heterozygous
mutation of SLC26A4 were analyzed using Fisher's exact test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Clinical and audiological
evaluation
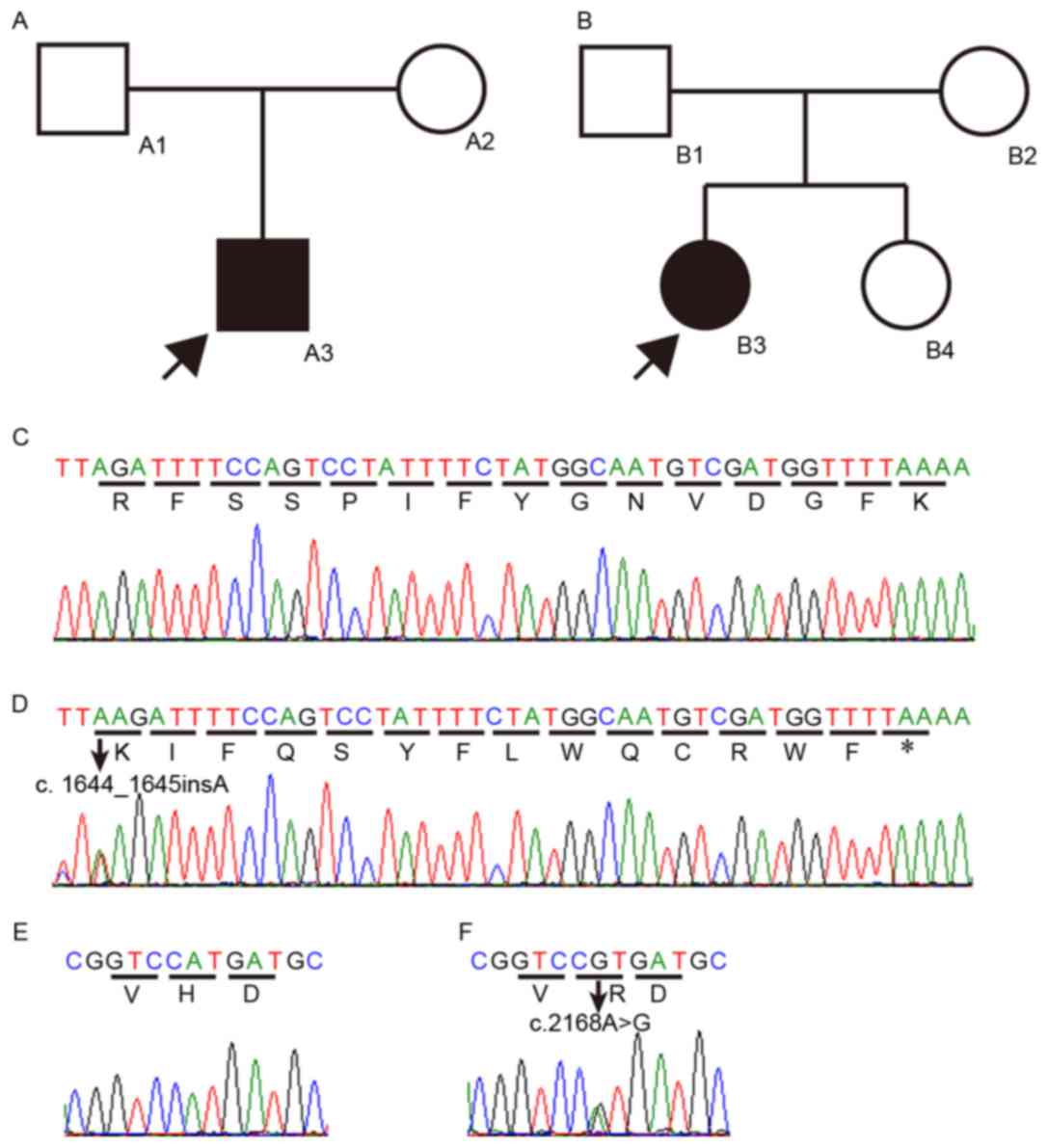
The novel compound heterozygous mutation,
p.R549Kfs*15 and p.H723R, in SLC26A4 was detected in a
4-year-old boy (A3) from family A and a 5-year-old girl (B3) from
family B in the present study (Fig.
1A-F). The two families were from different cities of the
Jiangsu province of China, and there was no relationship between
the families. There was no history of hearing loss in the families
of the two the two individuals or any clinical evidence showing any
syndrome or disease, for example hypothyroidism. There was no
history of previous illness, including meningitis, or trauma, and
pregnancies were normal. There were no occurrences of congenital
NSHL or syndromic hearing infection, including PS, in either
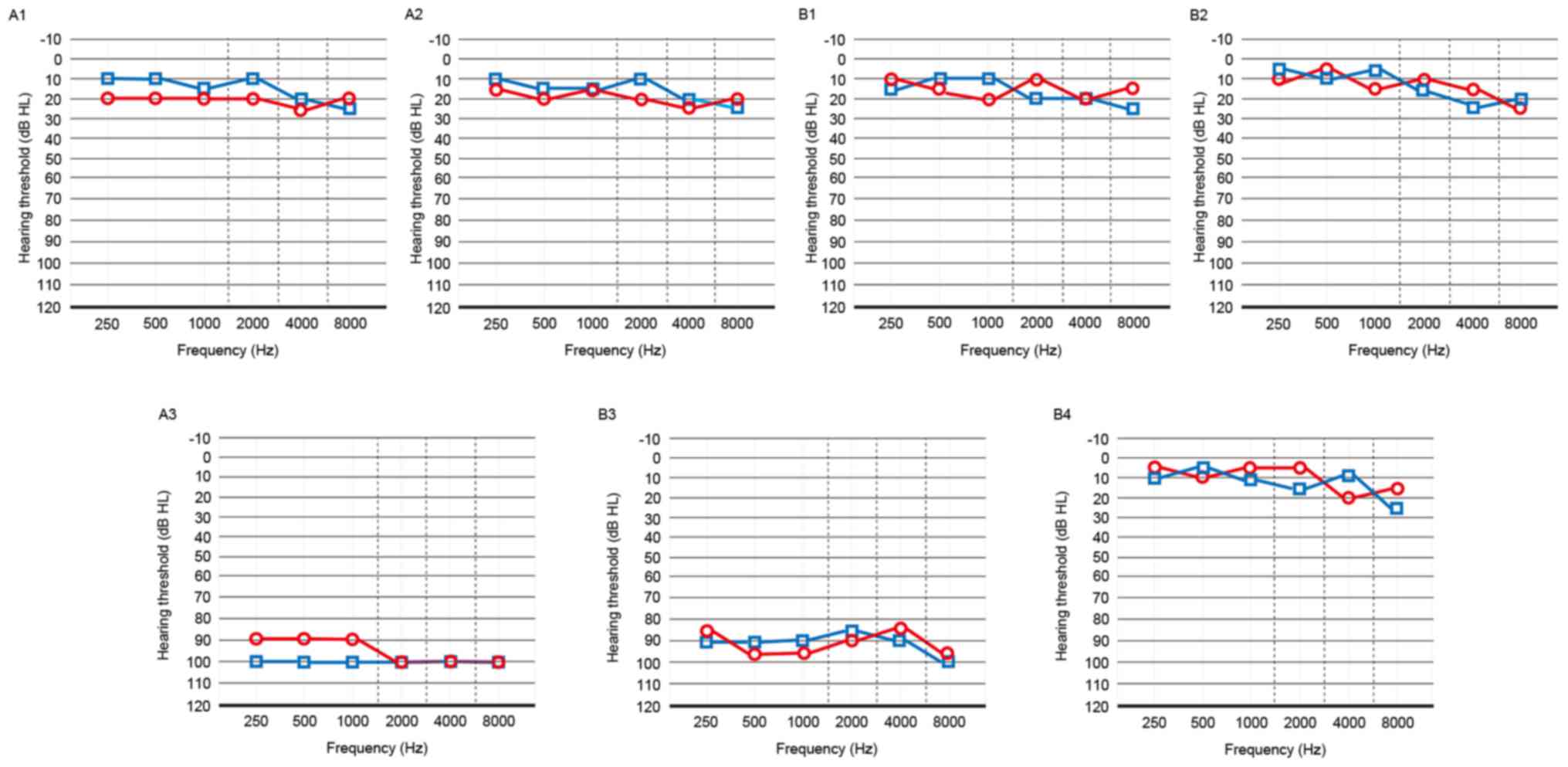
family. Audiological examinations were performed and the results of
PTA test showed that the two probands (A3 and B3) exhibited
bilateral profound SHL (Fig. 2).
This type of hearing cannot be improved with ear hearing aids and
they presented with deficiency in language development. The results
of the ABR showed that no representative wave was initiated under
the highest intensity (105 dB), and the DPOAE showed no
representative response in the frequency range of 1–4 kHz in the
two probands. The results of the otoscopic examinations revealed a
normal external auditory canal and tympanic membranes, and
tympanometric results were normal. The two probands showed no
vestibular symptoms and had a walking age of 12 months. Their
thyroids were of normal size, and the results of the laboratory
investigations showed normal blood parameters and thyroid function.
The other members of the two families had normal hearing (Fig. 2).
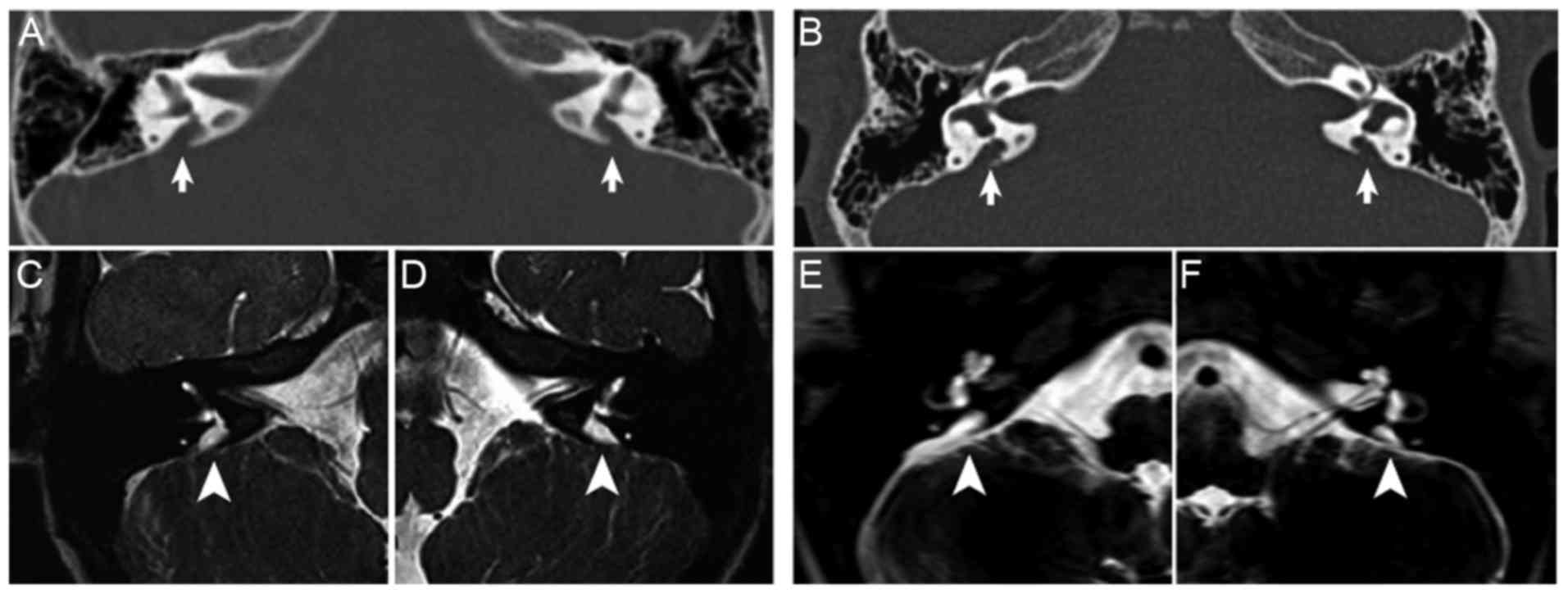
Radiological examination
EVA was defined as a diameter at the midpoint
between the common crus and the external aperture of >1.5 mm
(10). The temporal bone CT
examinations of the two probands showed no cochlear malformations,
however, bilateral EVA was present and the width of the vestibular
aqueduct was >1.5 mm (Fig. 3A and
B). On MRI examination, enlargement of the endolymphatic sac
and duct was observed in the left and right ears of the two
probands (Fig. 3C-F).
Mutation analysis
Sequencing of the 20 SLC26A4 coding exons in
the 52 patients with EVA revealed the same compound heterozygous
mutation in the two probands, the 4-year-old boy and a 5-year-old
girl, from the two separate families. The two variants comprised an
insertion of an adenine (c.1644_1645insA) in exon 15 (Fig. 1C) and an adenine to guanine
substitution (c.2168A>G) in exon 19 (Fig. 1E). The first insertion was
predicted to cause a frameshift and produce a truncated protein by
a premature stop (p.R549Kfs*15). This variation has not been
reported previously. Individuals A1 in family A, and B1 and B4 in
family B were heterozygous for this variation. The second mutation
led to the substitution of histidine by arginine (p.H723R). This
variation was present in the NCBI dbSNP (rs121908362) and Deafness
Variation Databases of the University of Uiowa (http://www.healthcare.uiowa.edu/labs/pendredandbor/slcMutations.htm).
The missense mutation identified in SLC26A4
has been previously reported to cause PS or NSHL with EVA (11,12).
Patient A2 in family A and B2 in family B were heterozygous for
this variation. The affected members (A3 and B3) of the two
families were heterozygous for the p.R549Kfs*15 and p.H723R
mutations. None of the compound heterozygous mutations were
detected in the 60 ethnically-matched normal control subjects or in
the 50 remaining NSHL patients with EVA. This finding indicated
that the p.R549Kfs*15 variation was the NSHL-causing mutation in
the two families, as a compound heterozygous mutation with
p.H723R.
Discussion
In the present study, a novel compound heterozygous
mutation of SLC26A4 (c.1644_1645insA, c.2168A>G) was
identified in two patients with profound bilateral SHL and EVA. The
parents of the two patients and an unaffected member in family B
(B4) carried a mutant allele separately with a normal allele and
had normal hearing. The compound heterozygous mutation was
inherited from the mutant alleles of the father and mother in the
two probands. Therefore, it was concluded that the two affected
families reported were consistent with an autosomal recessive
disorder caused by bi-allelic function loss of the pendrin protein.
Compound heterozygous patients with two SLC26A4 mutant
alleles have been reported in up to 50% of Chinese patients with HL
and exhibit significant genetic heterogeneity in Asian populations
(13–15). The mutant allele of c.2168A>G
(p. H723R) has been reported to be a common pathogenic mutation
among Chinese, Korean and Japanese patients with HL (14–17).
The incidence of this mutation in patients with NSHL and EVA in the
present study was 9/52 (17.31%; 95% CI, 8.23–30.33%). It has been
reported that the mutant pendrin comprised p.H723R is localized
predominantly in endoplasmic reticulum and was found to lack the
activity of anion exchange (18).
It was also found that patients with two bi-allelic mutations of
p.H723R or a compound heterozygous mutation with the other mutation
often suffered from bilateral SHL (19–21).
The other mutant allele c.1644_1645insA mutation of
SLC26A4 was identified as a novel compound heterozygous
mutation with p.H723R in the present study. The incidence of this
mutation in patients with NSHL and EVA in the present study was
2/52 (3.85%; 95% CI, 0.47–13.21%). The predicted result of the
c.1644_1645insA mutation was a frameshift beginning at codon 549
(Arg to Lys) and a premature translation stop at position 562 in
exon 15. The different conserved amino acid change (p.H723R) in
exon 19 of pendrin caused by mutation of c.2168A>G and the early
translational termination in exon 15 at the coding region of
carboxyl terminus caused by c.1644_1645insA led to a deleterious
effect on protein function, eventually resulting in a pathologic
phenotype with profound HL and EVA.
Pendrin, as with other proteins in the SLC26 family,
carries a sulfate transporter and anti-sigma factor antagonist
(STAS) domain in its carboxy-terminus. Aravind and Koonin (22) reported that the STAS domain
encompassed amino acids 535–573 and 654–729 of pendrin. The
p.R549Kfs*15 and p.H723R mutations identified in the two probands
in the present study were involved in the STAS domain. The STAS
domain in proteins of the SLC26 family is reported to be involved
in nucleotide binding and interactions with other proteins
(23,24). The function of the STAS domain can
be affected or lost by mutations reported in a number of patients
with PS or EVA, which indicates that the STAS domain may be an
important intracellular component of pendrin in the function of
plasma membrane targeting and retention. The majority of the
mutations in the STAS domain lead to its reduced or complete loss
of function as an anion transporter, including iodide efflux
(18,25–27).
It has also been confirmed that the c.2168A> G
(p.H723R) mutant is a hot-spot region of the SLC26A4
mutation in Chinese patients with PS and EVA (12,16),
also affecting the putative protein kinase A (PKA) binding site.
The PKA and STAS domains located in the carboxyl terminus region of
pendrin are considered to be important components in the targeting
of proteins to the plasma membrane (28).
In addition, the c.1644_1645insA (p.R549Kfs*15)
mutation identified in the present study can cause an early stop
codon and truncated protein. The early stop codon causes
nonsense-mediated mRNA decay and these mRNAs are rapidly decayed
(29). As the c.2168A>G
(p.H723R) mutant is on another allele, the patients reported had no
pendrin proteins with a normal function and exhibited NSHL with
EVA. A number of reasons indicate the novel compound heterozygous
mutation of the SLC26A4 gene, including p.R549Kfs*15 and
p.H723R, as the pathologic mutation. The 20 coding exon sequencing
analysis of SLC26A4 in the 52 NSHL patients with EVA
revealed the compound heterozygous mutation (c.1644_1645insA,
c.2168A>G) in two individuals (2/52; 3.85%). The high prevalence
of this novel compound heterozygous mutation of SLC26A4 was
almost 4% among the patients with NSHL and EVA in the present study
(P=0.03 with Fisher's exact test). Therefore, the novel compound
heterozygous mutation of SLC26A4 identified in the present
study expands on the wide mutational spectrum of SLC26A4.
The results of the present study provide a foundation for future
investigations of the molecular mechanisms of SLC26A4 mutations
associated with HL and EVA.
Acknowledgements
The present study was supported by the General
Program from Natural Science Foundation of China to Dr Gao Xia
(grant nos. 81371090 and 81570921). The authors would like to thank
the Core Medical and Genetics Laboratory of Drum Tower Hospital
Affiliated to Nanjing University Medical School.
Glossary
Abbreviations
Abbreviations:
|
EVA
|
enlarged vestibular aqueduct
|
|
NSHL
|
nonsyndromic hearing loss
|
|
STAS
|
anti-sigma factor antagonist
|
|
SHL
|
sensorineural hearing loss
|
|
PS
|
Pendred syndrome
|
|
MD
|
Mondini dysplasia
|
|
PTA
|
pure-tone audiometry
|
|
CT
|
computed tomography
|
|
MRI
|
magnetic resonance imaging
|
|
ABR
|
auditory brainstem response
|
|
DPOAE
|
distortion product otoacoustic
emissions
|
|
PCR
|
polymerase chain reaction
|
|
NCBI
|
National Center for Biotechnology
Information
|
|
PKA
|
putative protein kinase A
|
References
|
1
|
Usami S, Abe S, Weston MD, Shinkawa H, Van
Camp G and Kimberling WJ: Non-syndromic hearing loss associated
with enlarged vestibular aqueduct is caused by PDS mutations. Hum
Genet. 104:188–192. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pique LM, Brennan ML, Davidson CJ,
Schaefer F, Greinwald J Jr and Schrijver I: Mutation analysis of
the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital
hearing loss. Peer J. 2:e3842014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reardon W, OMahoney CF, Trembath R, Jan H
and Phelps PD: Enlarged vestibular aqueduct: A radiological marker
of pendred syndrome, and mutation of the PDS gene. QJM. 93:99–104.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Campbell C, Cucci RA, Prasad S, Green GE,
Edeal JB, Galer CE, Karniski LP, Sheffield VC and Smith RJ: Pendred
syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel
mutations and possible genotype-phenotype correlations. Hum Mutat.
17:403–411. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pryor SP, Madeo AC, Reynolds JC, Sarlis
NJ, Arnos KS, Nance WE, Yang Y, Zalewski CK, Brewer CC, Butman JA
and Griffith AJ: SLC26A4/PDS genotype-phenotype correlation in
hearing loss with enlargement of the vestibular aqueduct (EVA):
Evidence that Pendred syndrome and non-syndromic EVA are distinct
clinical and genetic entities. J Med Genet. 42:159–165. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Everett LA, Glaser B, Beck JC, Idol JR,
Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, et
al: Pendred syndrome is caused by mutations in a putative sulphate
transporter gene (PDS). Nat Genet. 17:411–422. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kopp P, Pesce L and Solis-S JC: Pendred
syndrome and iodide transport in the thyroid. Trends Endocrinol
Metab. 19:260–268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chai Y, Huang Z, Tao Z, Li X, Li L, Li Y,
Wu H and Yang T: Molecular etiology of hearing impairment
associated with nonsyndromic enlarged vestibular aqueduct in East
China. Am J Med Genet A. 161A:1–2233. 2013.PubMed/NCBI
|
|
9
|
Van Nierop JW, Huinck WJ, Pennings RJ,
Admiraal RJ, Mylanus EA and Kunst HP: Patients with pendred
syndrome: Is cochlear implantation beneficial? Clin Otolaryngol.
41:386–394. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cremers CW, Admiraal RJ, Huygen PL, Bolder
C, Everett LA, Joosten FB, Green ED, van Camp G and Otten BJ:
Progressive hearing loss, hypoplasia of the cochlea and widened
vestibular aqueducts are very common features in Pendred's
syndrome. Int J Pediatr Otorhinolaryngol. 45:113–123. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Coyle B, Reardon W, Herbrick JA, Tsui LC,
Gausden E, Lee J, Coffey R, Grueters A, Grossman4 A, Phelps PD, et
al: Molecular analysis of the PDS gene in Pendred syndrome. Hum Mol
Genet. 7:1105–1112. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reyes S, Wang G, Ouyang X, Han B, Du LL,
Yuan HJ, Yan D, Dai P and Liu XZ: Mutation analysis of SLC26A4 in
mainland Chinese patients with enlarged vestibular aqueduct.
Otolaryngol Head Neck Surg. 141:502–508. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peng Q, Huang S, Liang Y, Ma K, Li S, Yang
L, Li W, Ma Q, Liu Q, Zhong B and Lu X: Concurrent genetic and
standard screening for hearing impairment in 9317 southern chinese
newborns. Genet Test Mol Biomarkers. 20:603–608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsukamoto K, Suzuki H, Harada D, Namba A,
Abe S and Usami S: Distribution and frequencies of PDS (SLC26A4)
mutations in Pendred syndrome and nonsyndromic hearing loss
associated with enlarged vestibular aqueduct: A unique spectrum of
mutations in Japanese. Eur J Hum Genet. 11:916–922. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shin JW, Lee SC, Lee HK and Park HJ:
Genetic screening of GJB2 and SLC26A4 in korean cochlear
implantees: Experience of soree ear clinic. Clin Exp
Otorhinolaryngol. 5 Suppl 1:S10–S13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang QJ, Zhao YL, Rao SQ, Guo YF, Yuan H,
Zong L, Guan J, Xu BC, Wang DY, Han MK, et al: A distinct spectrum
of SLC26A4 mutations in patients with enlarged vestibular aqueduct
in China. Clin Genet. 72:245–254. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu CC, Yeh TH, Chen PJ and Hsu CJ:
Prevalent SLC26A4 mutations in patients with enlarged vestibular
aqueduct and/or Mondini dysplasia: A unique spectrum of mutations
in Taiwan, including a frequent founder mutation. Laryngoscope.
115:1060–1064. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yoon JS, Park HJ, Yoo SY, Namkung W, Jo
MJ, Koo SK, Park HY, Lee WS, Kim KH and Lee MG: Heterogeneity in
the processing defect of SLC26A4 mutants. J Med Genet. 45:411–419.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pang X, Chai Y, Chen P, He L, Wang X, Wu H
and Yang T: Mono-allelic mutations of SLC26A4 is over-presented in
deaf patients with non-syndromic enlarged vestibular aqueduct. Int
J Pediatr Otorhinolaryngol. 79:1351–1353. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sagong B, Baek JI, Lee KY and Kim UK: A
novel frameshift mutation of SLC26A4 in a korean family with
nonsyndromic hearing loss and enlarged vestibular aqueduct. Clin
Exp Otorhinolaryngol. 10:50–55. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yao J, Qian X, Bao J, Wei Q, Lu Y, Zheng
H, Cao X and Xing G: Probing the effect of two heterozygous
mutations in codon 723 of SLC26A4 on deafness phenotype based on
molecular dynamics simulations. Sci Rep. 5:108312015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aravind L and Koonin EV: The STAS domain-a
link between anion transporters and antisigma-factor antagonists.
Curr Biol. 10:R53–R55. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sharma AK, Ye L, Baer CE, Shanmugasundaram
K, Alber T, Alper SL and Rigby AC: Solution structure of the
guanine nucleotide-binding STAS domain of SLC26-related SulP
protein Rv1739c from Mycobacterium tuberculosis. J Biol Chem.
286:8534–8544. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH,
Millen L, Goto H, Naruse S, Soyombo A, Thomas PJ and Muallem S:
Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell
Biol. 6:343–350. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Taylor JP, Metcalfe RA, Watson PF, Weetman
AP and Trembath RC: Mutations of the PDS gene, encoding pendrin,
are associated with protein mislocalization and loss of iodide
efflux: Implications for thyroid dysfunction in Pendred syndrome. J
Clin Endocrinol Metab. 87:1778–1784. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pera A, Dossena S, Rodighiero S, Gandía M,
Bottà G, Meyer G, Moreno F, Nofziger C, Hernández-Chico C and
Paulmichl M: Functional assessment of allelic variants in the
SLC26A4 gene involved in Pendred syndrome and nonsyndromic EVA.
Proc Natl Acad Sci USA. 105:pp. 18608–18613. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Scott DA, Wang R, Kreman TM, Andrews M,
McDonald JM, Bishop JR, Smith RJ, Karniski LP and Sheffield VC:
Functional differences of the PDS gene product are associated with
phenotypic variation in patients with Pendred syndrome and
non-syndromic hearing loss (DFNB4). Hum Mol Genet. 9:1709–1715.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bizhanova A, Chew TL, Khuon S and Kopp P:
Analysis of cellular localization and function of carboxy-terminal
mutants of pendrin. Cell Physiol Biochem. 28:423–434. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Frischmeyer PA and Dietz HC:
Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet.
8:1893–1900. 1999. View Article : Google Scholar : PubMed/NCBI
|