Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common malignancy and the third leading cause of cancer-related
mortality worldwide (1). Despite
major advancements in the diagnosis and treatment of HCC that have
been made over the past few decades, the prognosis of patients with
HCC remains unsatisfactory owing to its high recurrence rate,
metastasis and poor therapeutic response (2,3).
Hepatocarcinogenesis is a complex, multi-step process that involves
the accumulation of a number of genetic and epigenetic alterations
(4). However, the molecular
mechanisms of HCC pathogenesis are not yet fully understood.
Therefore, investigations are urgently needed to explore new
potential therapeutic targets and to elucidate the relevant
molecular pathways involved in HCC progression and metastasis.
As a member of the high mobility group AT-hook
(HMGA) protein family, HMGA1 is involved in a number of biological
processes, such as stem cell self-renewal, cell proliferation,
differentiation and neoplastic transformation (5,6).
Moreover, knockdown of HMGA1 expression may interfere with
tumorigenic growth and may reduce cell invasion and migration
capabilities in certain cancer cell lines (7,8). A
previous study revealed that HMGA1 is overexpressed in HCCs, and
patients with a detectable level of HMGA1 mRNA in tumor sections
have an increased risk of recurrence/metastasis and a shorter
survival time (9). However, little
is known about the role of HMGA1 in the development of HCC.
Integrin-linked kinase (ILK) is an intracellular
serine/threonine protein kinase and adaptor protein that interacts
with the cytoplasmic domains of β1 and β3 integrins (10). ILK regulates several cellular
processes that are necessary for cancer progression, including cell
proliferation, survival, migration and invasion, and angiogenesis
(11–13). As a component of the
phosphatidylinositol 3-kinase pathway, activated ILK is able to
directly phosphorylate protein kinase B (PKB/Akt) on Ser473 and
glycogen synthase kinase 3β (GSK3β) on Ser9, resulting in the
activation of PKB/Akt and the inhibition of GSK3β, respectively
(14). Furthermore, ILK expression
and activity are often elevated in human malignancies (15).
In a previous study, gene expression profile
analysis of a MCF-7 breast epithelial cell line stably transfected
with HMGA1 identified integrins and their signaling pathways as
significantly upregulated genes (16). The present study focused on the ILK
gene, as previous reports indicated its dysregulation in HCC and
its involvement in promoting HCC cell growth, motility and invasion
(17,18). However, the association between
HMGA1 and ILK in cancer has not yet been characterized. The present
study hypothesized that there is an HMGA1/ILK axis in HCC and
demonstrated that the knockdown of HMGA1 expression suppressed cell
growth, migration and invasion, and induced apoptosis; these
effects were reversed by overexpression of ILK in MHCC97H cell
cultures. The antagonistic effects of ILK were attenuated in the
presence of the Akt inhibitor MK2206. In addition, matrix
metalloproteinase (MMP)2, MMP9, CyclinD1 and c-Myc were identified
as possible downstream effectors of the HMGA1/ILK/Akt/GSK3β
signaling pathway.
Materials and methods
Cell culture
The human HCC cell line MHCC97H was obtained from
The Liver Cancer Institute of Fudan University (Shanghai, China)
and cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% fetal bovine serum (FBS; HyClone; GE Healthcare Life Sciences,
Logan, UT, USA), 100 U/ml penicillin and 100 µg/ml streptomycin.
Cells were maintained at 37°C and 5% CO2 in a humidified
incubator.
Plasmids and lentiviral vector
The lentiviral short hairpin (sh)RNA vector against
HMGA1 (shHMGA1,
5′-GATCCAGCGAAGTGCCAACACCTATTCAAG-AGATAGGTGTTGGCACTTCGCTTTTTTTA-3′)
and the scrambled control shRNA vector (shControl) were obtained
from GeneChem Co., Ltd. (Shanghai, China). The ILK expression
plasmid GV144-enhanced green fluorescent protein (EGFP)-ILK and the
negative control plasmid GV144-EGFP were also from GeneChem Co.,
Ltd.
Plasmids and lentiviral
transduction
MHCC97H cells (5×106 cells/ml, 37°C) were
pre-transfected with 4 µg/ml GV144-EGFP-ILK or GV144-EGFP using
FuGENE 6 Transfection Reagent (Roche Diagnostics GmbH, Mannheim,
Germany), according to the manufacturer's protocol. Following 6 h
incubation, the cells were transfected with LV-shHMGA1 or
LV-shControl [multiplicity of infection (MOI)=100] for 2 days,
according to Recombinant Lentivirus Operation Manual (GeneChem Co.,
Ltd.). The infection conditions were initial incubation for 10 min
at room temperature followed by centrifugation at 1,900 × g for 30
min at 32°C. Then the incubation for 24 h at 37°C with the Akt
inhibitor MK2206 (5 µM; Selleck Chemicals, Houston, TX, USA) or
dimethylsulfoxide (DMSO) was carried out. There was a total of
seven groups in these experiments: i) DMSO as Blank control; ii)
shControl; iii) shHMGA1; iv) shHMGA1 + EGFP; v) shHMGA1 + ILK; vi)
shHMGA1 + ILK + DMSO; and vii) shHMGA1 + ILK + MK2206. MOI 20, 50,
80, 100, 150 and 200 were also assessed. The knockdown efficiency
of HMGA1, measured with reverse transcription-quantitative
polymerase chain reaction (RT-qPCR), reached 95% when MOI was at
100, where the cell viability was still at a high level.
RT-qPCR
Total RNA was extracted (2×105 cells/ml)
from cultured MHCC97H cells using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). The ratio of absorbance at 260 and
280 nm was used to access the purity of RNA and
RiboGreen™ used for RNA quantification based on the
RiboGreen RNA quantitation assay. Subsequently, reverse transcribed
into cDNA using the RevertAid First Strand cDNA Synthesis kit
(Fermentas; Thermo Fisher Scientific, Inc.). qPCR was performed
using SYBR-Green PCR Master Mix (Tiangen Biotech Co., Ltd.,
Beijing, China) and an ABI 7900 Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). GAPDH was used as an
internal control. Primer sequences used for qPCR are shown in
Table I. Relative expression
levels (represented as fold change) of the target genes were
calculated using the 2−ΔΔCq method (19). RNA extraction, cDNA synthesis, and
were qPCR performed according to the manufacturer's protocols. The
PCR program was at 98°C for 5 min on the initial cycle then 30 sec
rest followed by 25 cycles, annealing was performed at 72°C for 30
sec, extension temperature was settled at 72°C for 15 sec per kb
and final extension was at 68°C for 5 min. Each experiment was
performed in triplicate.
 | Table I.Primer sequences used in reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences used in reverse
transcription-quantitative polymerase chain reaction.
| Gene | Primer sequence
(5′→3′) |
|---|
| HMGA1 | F:
TCCAAGAAGGCATCCGCATT |
|
| R:
AGGAGCAGGTGGAAGAGTGA |
| ILK | F:
TTTGCAGTGCTTCTGTGGGAA |
|
| R:
CTACTTGTCCTGCATCTTCTC |
| MMP2 | F:
GATGTTGTCTTGTGAGCGTGC |
|
| R:
CTGGGGCAGTCCAAAGAACT |
| MMP9 | F:
TGTACCGCTATGGTTACACTCG |
|
| R:
GGCAGGGACAGTTGCTTCT |
| CyclinD1 | F:
CAGAAGAGCGCGAGGGAG |
|
| R:
TCGTTGAGGAGGTTGGCATC |
| c-Myc | F:
GGACTTGTTGCGGAAACGAC |
|
| R:
CTCAGCCAAGGTTGTGAGGT |
| GAPDH | F:
TGCACCACCAACTGCTTAGC |
|
| R:
GGCATGGACTGTGGTCATGAG |
Western blot analysis
Whole-cell protein was extracted (1×106
cells) using Radioimmunoprecipitation Assay Lysis Buffer (Solarbio
Science & Technology Co., Ltd., Beijing, China). Equal amounts
(50 µg) of proteins, quantified by Bradford assay, were separated
by 8–12% SDS-PAGE and transferred to polyvinylidene difluoride
membranes (Millipore; Merck KGaA, Darmstadt, Germany). The
membranes were blocked for 1 h with 5% bovine serum albumin (Abcam,
Cambridge, UK) in Tris-buffered saline containing 0.05% Tween-20
(TBST) and then incubated at 4°C overnight with the following
rabbit anti-human primary antibodies: anti-HMGA1 (ab205768;
1:5,000), anti-ILK (ab196013; 1:5,000), anti-Akt (ab81283;
1:5,000), anti-GSK3β (ab32391; 1:5,000), anti-MMP2 (ab92536;
1:5,000), anti-MMP9 (ab38898; 1:1,000), anti-cyclinD1 (ab16663;
1:25) and anti-c-Myc (ab3207; 1:10,000) (all from Abcam);
anti-phosphorylated (p)-AKT (Ser473) (4060; 1:2,000) and
anti-p-GSK3β (Ser9) (5558; 1:1,000) (Cell Signaling Technology,
Inc., Danvers, MA, USA); and anti-β-actin (TA-09, 1:1,000)
(Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China).
Following primary antibody incubation, the membranes were washed
with TBST to remove excess antibodies and incubated with
horseradish peroxidase-conjugated goat anti-rabbit (IgG HRP;
PV-6001; 1:10,000) and rabbit anti-mouse (IgG HRP; PV-6002;
1:1,000) secondary antibodies (Zhongshan Golden Bridge
Biotechnology Co., Ltd.) for 1 h at room temperature. The signals
were visualized using SuperSignal West Pico Chemiluminescent
Substrate (Thermo Fisher Scientific, Inc.), and band intensities
were quantified by ImageJ software (National Institutes of Health,
Bethesda, MD, USA). β-actin was used as an internal control.
Cell proliferation assay
MHCC97H cells were seeded at 2×103
cells/well and incubated at 37°C for 12 h in a 96-well plate. Prior
to detection, Cell Counting Kit-8 (CCK-8) Reagent (10 µl/well;
Solarbio Science & Technology Co., Ltd.) was added to each well
and cells were incubated for 2 h at 37°C in a 5% CO2
atmosphere. The absorbance was then measured at 450 nm using a
microplate reader and the OD value of each well was used to
represent cell proliferation.
Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) apoptosis assay
An Annexin V-FITC Apoptosis Detection kit (Beyotime
Institute of Biotechnology, Haimen, China) was used strictly
according to the manufacturer's protocol to detect apoptosis.
Following their respective treatments, aforementioned, MHCC97H
cells (1×105) from each of the seven experimental groups
were collected and resuspended in 500 µl Binding Buffer. Annexin
V-FITC (5 µl) and PI (5 µl) were added to the cell suspensions and
were incubated for 15 min at room temperature in the dark; the
apoptosis rates of these cells were immediately assessed by a
FACScan flow cytometry and analyzed using BD FACStation software
version 6.1 (BD Biosciences, Franklin Lakes, NJ).
Cell invasion assay
The invasive capabilities of the 7 experimental
groups of MHCC97H cells were detected using 24-well Matrigel-coated
chambers (pore size, 8 µm; BD Biosciences). Cells
(1×105) were resuspended in 200 µl serum-free medium and
seeded into the upper chamber, and 600 µl DMEM containing 10% FBS
was added to the lower chamber as a chemoattractant. Following 24 h
incubation at 37°C, the cells remaining in the upper chamber were
removed with a cotton swab and the cells that migrated to the lower
surface of membrane were fixed with 100% methanol, stained with
0.1% crystal violet and observed by light and electron microscopy
using Gen5 software version 2.0 (BioTek Instruments, Inc.,
Winooski, VT, USA).
Wound-healing assay for cell
migration
Cells (1×105 cells) were seeded in 6-well
plates and grown to confluence 100% at 37°C. The monolayer of cells
was scratched with a pipette tip to create wound, and the dead
cells were washed away with PBS prior to culturing in 10% FBS
supplemented DMEM at 37°C. Images were captured by light and
electron microscopy at 0 and 24 h, and cell migration was assessed
by the width of the wound.
Statistical analysis
Results are presented as mean ± standard deviation
of three independent experiments. Statistical significances between
different groups were analyzed one-way analysis of variance
followed by Tukey comparison test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Alteration of HMGA1, ILK, p-Akt and
p-GSK3β expression in MHCC97H cells treated with shRNA, ILK and/or
MK2206
As shown in Fig.
1A, MHCC97H cells transfected with shHMGA1 exhibited a
significant reduction in HMGA1 mRNA expression (P<0.05 vs.
shControl), even when the same cells expressed the ILK vector or
are treated with the Akt-specific inhibitor MK2206. shHMGA1
transfection also resulted in a significant decrease in the level
of ILK mRNA expression in MHCC7H cells (P<0.05 vs. shControl);
however, this decrease in expression was recovered by ILK
overexpression (P<0.05 vs. shHMGA + EGFP); ShHMGA1 transfection
resulted in a great increase in the level of ILK mRNA in MHCC7H
cells compared with HMGA1 mRNA (P<0.05 vs. shHMGA + ILK +
MK2206; Fig. 1B). Western blot
analysis demonstrated similar results: protein expression levels of
HMGA1 and ILK were reduced in cells exposed to shHMGA1, and ILK
expression vector was only able to recover the expression of ILK
and not HMGA1 (Fig. 1C). The
protein expression levels of p-Akt and p-GSK3β were also reduced by
shHMGA1 treatment, but were recovered in cells overexpressing of
ILK. In MHCC97H cells co-treated with shHMGA1, the ILK expression
vector and MK2206, the protein expression levels of HMGA1, p-Akt
and p-GSK3β were similar to those cells treated with shHMGA1
only.
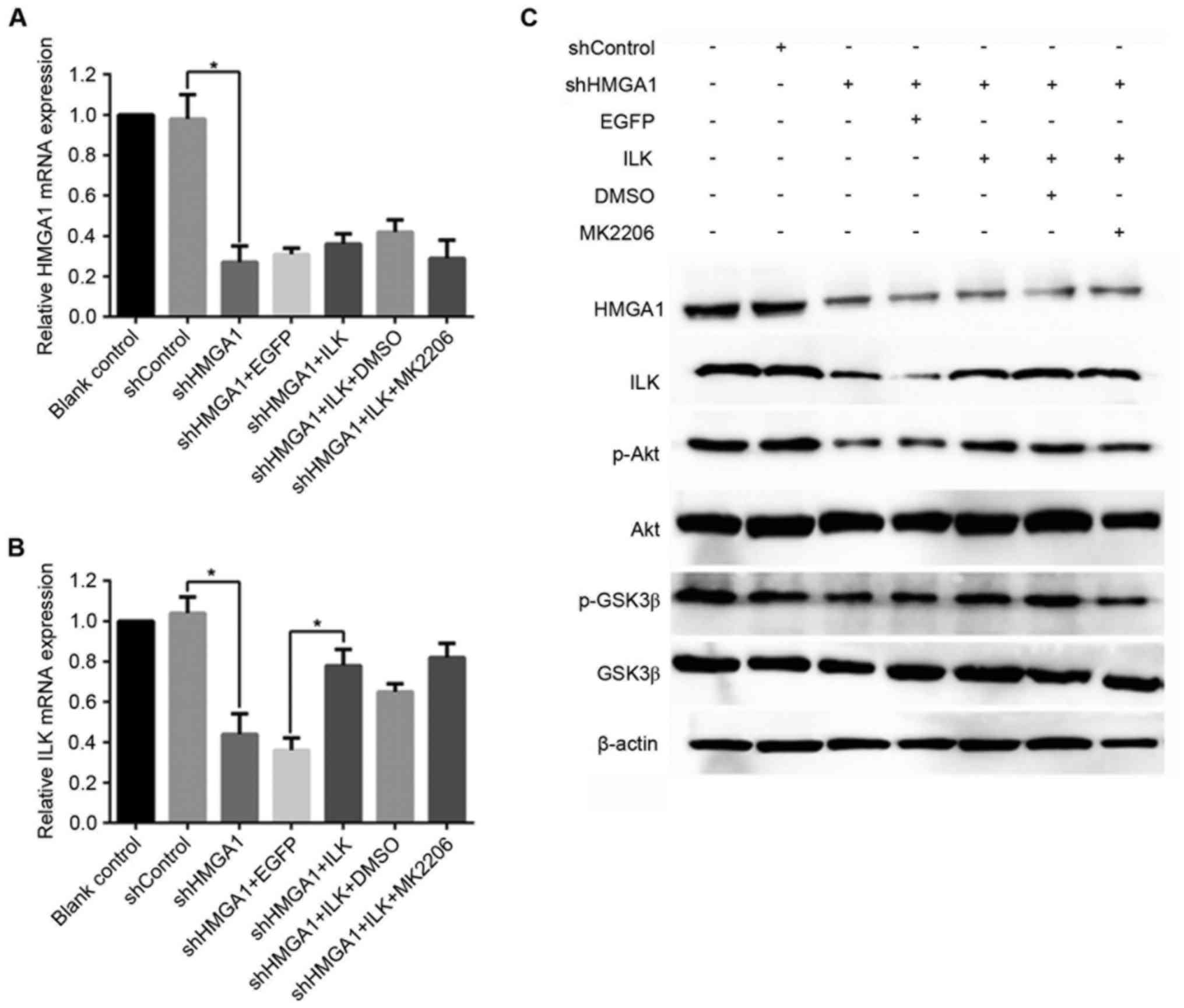 | Figure 1.mRNA and protein expression levels of
HMGA1, ILK, p-Akt and p-GSK3β in MHCC97H hepatocellular carcinoma
cells treated with shHMGA1, ILK expression vector and/or MK2206.
Alterations in mRNA expression levels of (A) HMGA1 and (B) ILK were
evaluated by reverse transcription-quantitative polymerase chain
reaction. *P<0.05. (C) Protein expression levels of HMGA1, ILK,
total Akt, p-Akt, total GSK3β and p-GSK3β were examined by western
blot analysis; GAPDH was used as an internal control. DMSO,
dimethylsulfoxide; EGFP, enhanced green fluorescent protein; GSK3β,
glycogen synthase kinase 3β; HMGA1, high mobility group AT-hook 1;
ILK, integrin-linked kinase; MK2206, an Akt-specific inhibitor; p,
phosphorylated; sh, short hairpin RNA. |
HMGA1 participates in MHCC97H cell
proliferation and inhibits cell apoptosis through the ILK/Akt/GSK3β
signaling pathway
Cell proliferation was detected using the CCK-8
reagent, and the OD450 value of each sample represented the cell
viability. As shown in Table II,
the OD450 of the Blank control and shControl groups were similar.
The OD450 of shHMGA1-treated MHCC97H cells was obviously less than
cells treated with shControl (P<0.05), but was similar to that
of cells co-treated with shHMGA1 + EGFP, shHMGA1 + ILK + DMSO and
shHMGA1 + ILK + MK2206. Similarly, the differences were identified
between the shHMGA1 + EGFP group and the shHMGA1 + ILK group
(P<0.05), as well as between the shHMGA1 + ILK + DMSO group and
the shHMGA1 + ILK + MK2206 group (P<0.05).
| Table II.Cell proliferation and apoptosis assay
results. |
Table II.
Cell proliferation and apoptosis assay
results.
| Group | OD450a | Apoptosis rate
(%)a,b |
|---|
| Blank control | 1.11±0.11 | 3.5±0.8 |
| shControl | 1.17±0.09 | 4.8±1.1 |
| shHMGA1 |
0.36±0.07c |
32.6±2.7c |
| shHMGA1 + EGFP | 0.37±0.02 |
36.3±4.5c |
| shHMGA1 + ILK |
0.58±0.04d |
13.4±1.1d |
| shHMGA1 + ILK +
DMSO | 0.52±0.03 | 14.8±1.2 |
| shHMGA1 + ILK +
MK2206 |
0.39±0.06e |
23.2±1.8e |
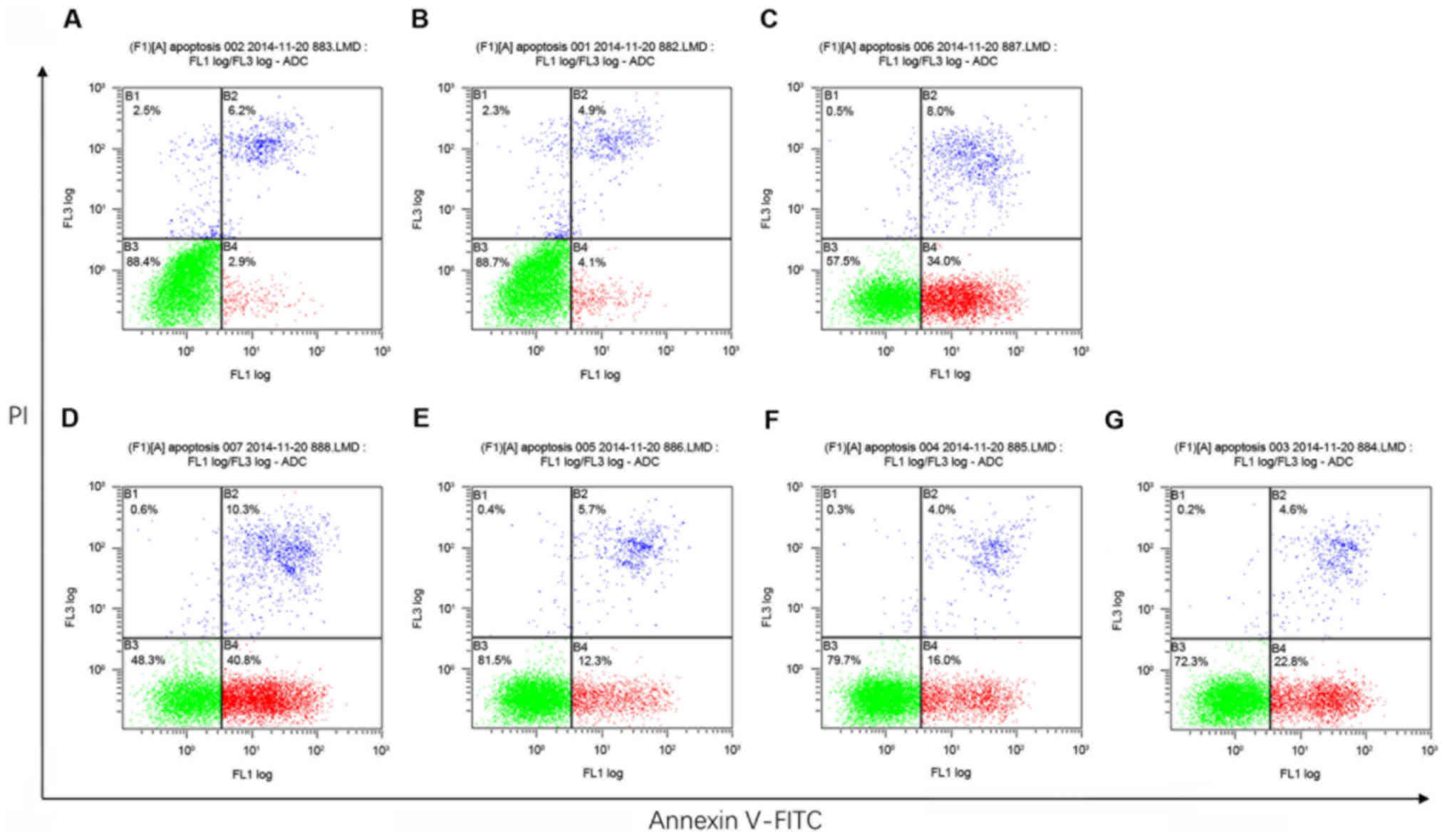
Annexin V-FITC/PI apoptosis detection by flow
cytometry revealed that, compared with the Blank control and the
shControl groups, MHCC97H cells treated with either shHMGA1 or
shHMGA1 + EGFP exhibited a slight increase in the rate of apoptosis
(P<0.05; Fig. 2; Table II). Cells in the shHMGA1 + ILK
group and the shHMGA1 + ILK + DMSO group were moderate lower than
cells in the shHMGA1 group and the shHMGA1 + EGFP group
(P<0.05). The apoptosis rate of cells in the shHMGA1 + ILK +
MK2206 group was substantial higher than cells in the shHMGA1 + ILK
group or the shHMGA1 + ILK + DMSO group (P<0.05).
HMGA1 participates in MHCC97H cell
invasion and migration through the ILK/Akt/GSK3β signaling
pathway
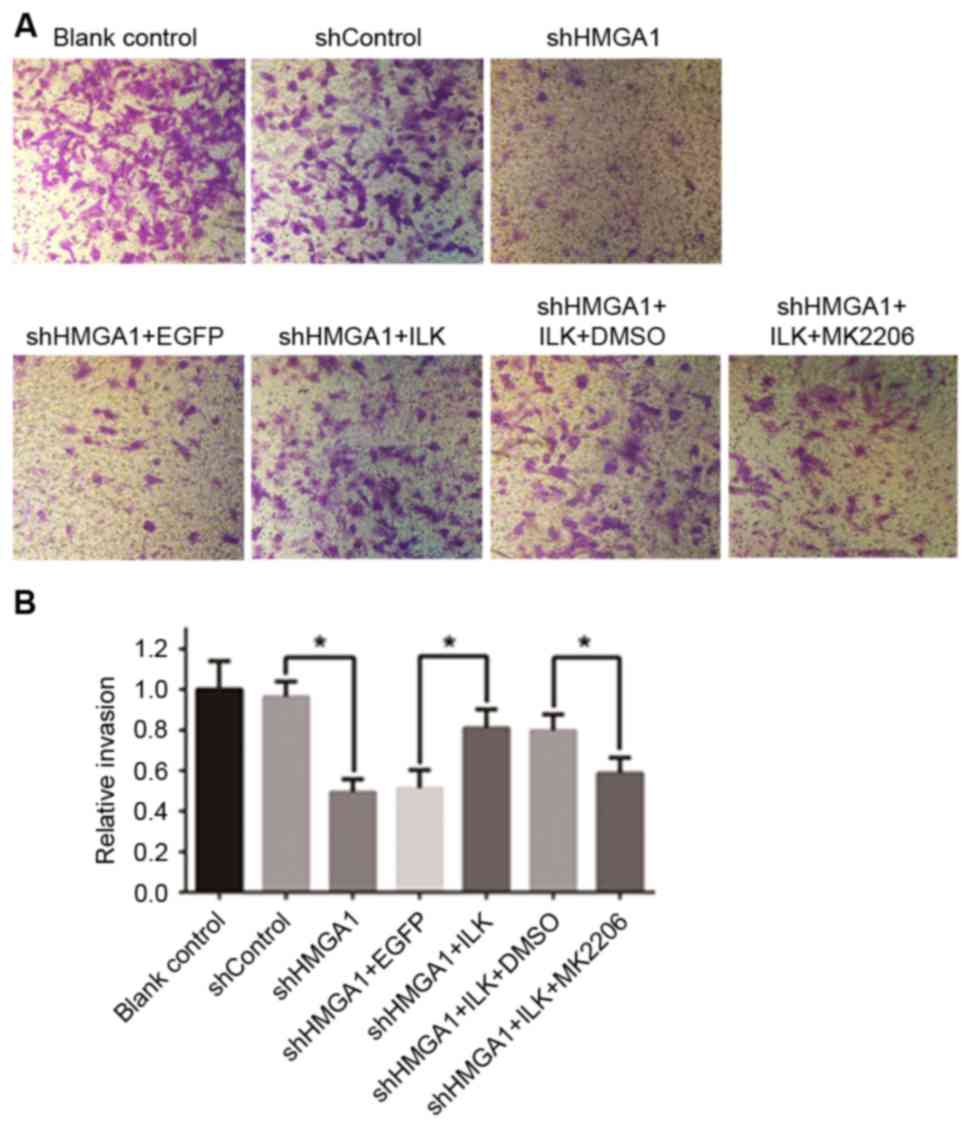
The invasive capabilities of MHCC97H cells cultured
with the various aforementioned treatments were detected with
24-well Matrigel-coated chambers (Fig.
3), and a wound-healing assay was used to examine cell
migration (Fig. 4). The invasive
and migratory capabilities of MHCC97H cells were suppressed by
knockdown of HMGA1 by shRNA transfection; however, cells
co-transfected with the ILK expression vector, which increased of
ILK protein expression, attenuated this decreased cell migration
and invasion. Notably, cells co-cultured with shHMGA1 + ILK +
MK2206 exhibited a decrease in invasion and migration, which
indicated that MK2206 was able to cancel the effects induced by the
upregulation of ILK protein expression. Significant differences
were identified in the invasion assay in relation to shHMGA1 vs.
shControl, shHMGA1 + ILK vs. shHMGA1 + EGFP, and shHMGA1 + ILK +
MK2206 vs. shHMGA1 + ILK + DMSO (P<0.05; Fig. 3B).
HMGA1 serves a role in regulating
MMP2, MMP9, CyclinD1, c-Myc expression in MHCC97H cells
As the results of RT-qPCR analysis demonstrate
(Fig. 5A-D), the shRNA knockdown
of HMGA1 expression in MHCC97H cells led to a significant decrease
in mRNA expression levels of MMP2, MMP9, CyclinD1 and C-Myc with
the (P<0.05 vs. shControl). These shHMGA1-induced decreases in
expression were reversed in MHCC97H cells co-treated with the ILK
expression vector (P<0.05 vs. shHMGA1 + EGFP). MK2206 exposure
was able to inhibit the effects of ILK expression in shHMGA1
co-treated cells, and this led to the decreased expression of MMP2,
MMP9, CyclinD1 and C-Myc (P<0.05 vs. shHMGA1 + ILK + DMSO). The
protein expression levels, as detected by western blot analysis, of
MMP2, MMP9, CyclinD1 and C-Myc in MHCC97H cells were similar to
their mRNA expression levels in the various treatments, and were
influenced by shHMGA1 and ILK expression (Fig. 5E).
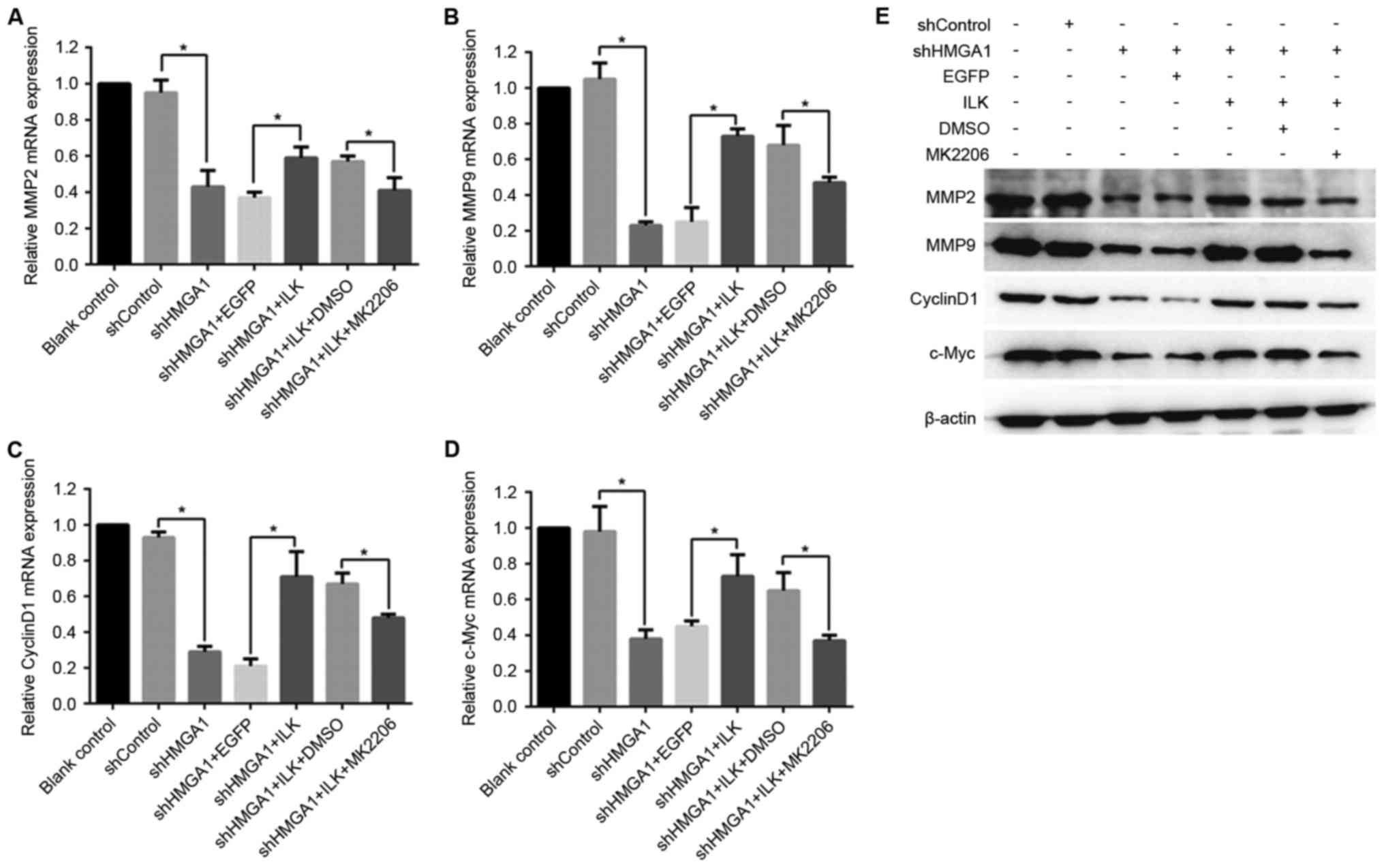 | Figure 5.mRNA and protein expression of MMP2,
MMP9, CyclinD1 and c-Myc in MHCC97H hepatocellular carcinoma cells
treated with shHMGA1, ILK expression vector and/or MK2206. Changes
in mRNA expression levels were determined by reverse
transcription-quantitative polymerase chain reaction for (A) MMP2,
(B) MMP9, (C) CyclinD1 and (D) c-Myc. *P<0.05. (E) Protein
expression levels of MMP2, MMP9, CyclinD1 and c-Myc were detected
by western blot analysis; β-actin was used as an internal control.
DMSO, dimethylsulfoxide; EGFP, enhanced green fluorescent protein;
HMGA1, high mobility group AT-hook 1; ILK, integrin-linked kinase;
MK2206, an Akt-specific inhibitor; MMP, matrix metalloproteinase;
sh, short hairpin RNA. |
Discussion
An increasing number of studies have indicated that
HMGA1 is overexpressed in almost all aggressive cancers, including
HCC, and regulates many processes that are characteristically
implicated in tumorigenesis (20,21).
However, the roles and mechanisms of HMGA1 in HCC pathogenesis
remain elusive. The present study provided, to the best of our
knowledge, the first evidence that the effects of HMGA1 expression
on MHCC97H cell malignant phenotypes are achieved through the
ILK/Akt/GSK3β signaling pathway.
Numerous additional studies have demonstrated that
ILK also has significant effects on the development and progression
of human carcinoma (11–13,15,18,22).
ILK serves a crucial role in diverse cellular functions that are
associated with cell survival, proliferation, motility,
epithelial-mesenchymal transition and angiogenesis (11–13).
Akt and GSK3β are two major downstream substrates of the
intracellular serine/threonine kinase ILK in cancer cells (14). ILK/Akt/GSK3β signaling has been
demonstrated to be a major pathway involved in various cancers
(22). It has been reported that
ILK was overexpressed during liver oncogenesis and cirrhosis, and
this overexpression was strongly associated with Akt activation
(18). One study revealed that ILK
knockdown significantly suppressed HCC cell growth, motility and
invasion in vitro and inhibited tumorigenesis in vivo
with reduced p-Akt (Ser473) expression (17). Another study demonstrated that HCC
cell lines exhibited increased sensitivity to epidermal growth
factor receptor inhibitors and decreased AKT activation when cells
were transformed with a kinase-inactive ILK (23). Taken together, these data suggested
that the regulatory function of ILK in HCC pathogenesis may involve
the Akt pathway.
HMGA1 and ILK serve important roles in tumor
development and both are highly expressed in HCC (15,22,24,25).
The present study aimed to determine whether HMGA1 regulated the
ILK/Akt/GSK3β signaling pathway in MHCC97H cells. The results
demonstrated that ILK expression levels were decreased by
shRNA-mediated knockdown of HMGA1 expression, but were not affected
by treatment with the Akt inhibitor MK2206. In addition, HMGA1
knockdown suppressed Akt and GSK3β phosphorylation, which was
recovered in MHCC97H cells transfected with an ILK expression
vector. Co-treatment with MK2206 was able to disrupt the effects of
ILK overexpression, and reduced HMGA1 expression levels caused by
shHMGA1 were unaffected by ILK/MK2206 treatment. These data
indicated that HMGA1 may be an upstream element in the
ILK/Akt/GSK3β pathway, which carries out the regulatory functions
in MHCC97H cells.
Based on the well-defined role of ILK in human
malignancy, the present study hypothesized that the promoting
effects of HMGA1 on cell malignant phenotypes may at least
partially be mediated by ILK. This theory was supported by the
results demonstrating that the knockdown of HMGA1 expression was
able to inhibit the proliferative and invasive ability of MHCC97H
cells and to induce apoptosis, whereas the effects of shHMGA1 were
reversed by ILK overexpression, which itself was subverted when
cells were co-treated with MK2206.
To further define the downstream molecular
mechanisms underlying HMGA1/ILK/Akt/GSK3β-axis-mediated cancer cell
progression, the present study analyzed the mRNA and protein
expressions of MMP2, MMP9, CyclinD1 and c-Myc, which have been
identified as targets of the Akt/GSK3β pathway (26–28).
MMPs, particularly MMP2 and MMP9, are well-known extracellular
matrix-degrading enzymes that serve important roles in tumor
invasion and metastasis (29).
CyclinD1 has been revealed to be overexpresses in various tumors
and promotes the G1/S shift that leads to cell proliferation
(30,31). The multifunctional, nuclear
phosphoprotein c-Myc is involved in cell cycle progression,
apoptosis and cellular transformation (32). Results from the present study
demonstrated that the alterations in MMP2, MMP9, CyclinD1 and c-Myc
expression correlated with the shHMGA1-induced changes to MHCC97H
cells malignant phenotype, which suggested that HMGA1 may promote
HCC growth and metastasis via activation of ILK/Akt/GSK3β signaling
and subsequent upregulation of MMP2, MMP9, CyclinD1 and c-Myc
expression in MHCC97H cells.
In conclusion, the results from the present study
suggested that HMGA1 participates in the survival and metastasis of
HCC cells through ILK/Akt/GSK3β signaling, and that MMP2, MMP9,
CyclinD1 and c-Myc are part of the downstream effectors of this
pathway. These data provide the basis for exploring the application
of HMGA1 inhibition as a therapy for patients with HCC and a new
treatment strategy to prevent HCC development.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maluccio M and Covey A: Recent progress in
understanding, diagnosing, and treating hepatocellular carcinoma.
CA Cancer J Clin. 62:394–399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Teufel A, Staib F, Kanzler S, Weinmann A,
Schulze-Bergkamen H and Galle PR: Genetics of hepatocellular
carcinoma. World J Gastroenterol. 13:2271–2282. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shah SN and Resar LM: High mobility group
A1 and cancer: Potential biomarker and therapeutic target. Histol
Histopathol. 27:567–579. 2012.PubMed/NCBI
|
|
5
|
Jung KY, Chen K, Kretzler M and Wu C:
TGF-beta1 regulates the PINCH-1-integrin-linked kinase-alph-aparvin
complex in glomerular cells. J Am Soc Nephrol. 18:66–73. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Attwell S, Mills J, Troussard A, Wu C and
Dedhar S: Integration of cell attachment, cytoskeletal
localization, and signaling by integrin-linked kinase (ILK),
CH-ILKBP, and the tumor suppressor PTEN. Mol Biol Cell.
14:4813–4825. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Z, Nie F, Wang S and Li L: Histone H4
Lys 20 monomethylation by histone methylase SET8 mediates Wnt
target gene activation. Proc Natl Acad Sci USA. 108:pp. 3116–3123.
2011; View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Curtin JC and Lorenzi MV: Drug discovery
approaches to target Wnt signaling in cancer stem cells.
Oncotarget. 1:552–566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chang ZG, Yang LY, Wang W, Peng JX, Huang
GW, Tao YM and Ding X: Determination of high mobility group A1
(HMGA1) expression in hepatocellular carcinoma: A potential
prognostic marker. Dig Dis Sci. 50:1764–1770. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hannigan GE, Leung-Hagesteijn C,
Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC and Dedhar
S: Regulation of cell adhesion and anchorage-dependent growth by a
new beta 1-integrin-linked protein kinase. Nature. 379:91–96. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Novak A, Hsu SC, Leung-Hagesteijn C,
Radeva G, Papkoff J, Montesano R, Roskelley C, Grosschedl R and
Dedhar S: Cell adhesion and the integrin-linked kinase regulate the
LEF-1 and beta-catenin signaling pathways. Proc Natl Acad Sci USA.
95:pp. 4374–4379. 1998; View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Attwell S, Roskelley C and Dedhar S: The
integrin-linked kinase (ILK) suppresses anoikis. Oncogene.
19:3811–3815. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Troussard AA, Costello P, Yoganathan TN,
Kumagai S, Roskelley CD and Dedhar S: The integrin linked kinase
(ILK) induces an invasive phenotype via AP-1 transcription
factor-dependent upregulation of matrix metalloproteinase 9
(MMP-9). Oncogene. 19:5444–5452. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Delcommenne M, Tan C, Gray V, Rue L,
Woodgett J and Dedhar S: Phosphoinositide-3-OH kinase-dependent
regulation of glycogen synthase kinase 3 and protein kinase B/AKT
by the integrin-linked kinase. Proc Natl Acad Sci USA. 95:pp.
11211–11216. 1998; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tan C, Cruet-Hennequart S, Troussard A,
Fazli L, Costello P, Sutton K, Wheeler J, Gleave M, Sanghera J and
Dedhar S: Regulation of tumor angiogenesis by integrin-linked
kinase (ILK). Cancer Cell. 5:79–90. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reeves R, Edberg DD and Li Y:
Architectural transcription factor HMGI(Y) promotes tumor
progression and mesenchymal transition of human epithelial cells.
Mol Cell Biol. 21:575–594. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chan J, Ko FC, Yeung YS, Ng IO and Yam JW:
Integrin-linked kinase overexpression and its oncogenic role in
promoting tumorigenicity of hepatocellular carcinoma. PLoS One.
6:e169842011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peroukides S, Bravou V, Varakis J,
Alexopoulos A, Kalofonos H and Papadaki H: ILK overexpression in
human hepatocellular arcinoma and liver cirrhosis correlates with
activation of Akt. Oncol Rep. 20:1337–1344. 2008.PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fusco A and Fedele M: Roles of HMGA
proteins in cancer. Nat Rev Cancer. 7:899–910. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fedele M and Fusco A: HMGA and cancer.
Biochim Biophys Acta. 1799:48–54. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Persad S and Dedhar S: The role of
integrin-linked kinase (ILK) in cancer progression. Cancer
Metastasis Rev. 22:375–384. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fuchs BC, Fujii T, Dorfman JD, Goodwin JM,
Zhu AX, Lanuti M and Tanabe KK: Epithelial-to-mesenchymal
transition and integrin-linked kinase mediate sensitivity to
epidermal growth factor receptor inhibition in human hepatoma
cells. Cancer Res. 68:2391–2399. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
McDonald PC, Oloumi A, Mills J, Dobreva I,
Maidan M, Gray V, Wederell ED, Bally MB, Foster LJ and Dedhar S:
Rictor and integrin-linked kinase interact and regulate Akt
phosphorylation and cancer cell survival. Cancer Res. 68:1618–1624.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zheng Y, Ritzenthaler JD, Sun X, Roman J
and Han S: Prostaglandin E2 stimulates human lung carcinoma cell
growth through induction of integrin-linked kinase: The involvement
of EP4 and Sp1. Cancer Res. 69:896–904. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liang J and Slingerland JM: Multiple roles
of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell
Cycle. 2:339–345. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang S and Basson MD: Integrin-linked
kinase: A multi-functional regulator modulating extracellular
pressure-stimulated cancer cell adhesion through focal adhesion
kinase and AKT. Cell Oncol. 31:273–289. 2009.PubMed/NCBI
|
|
28
|
Pontier SM, Huck L, White DE, Rayment J,
Sanguin-Gendreau V, Hennessy B, Zuo D, St-Arnaud R, Mills GB,
Dedhar S, et al: Integrin-linked kinase has a critical role in
ErbB2 mammary tumor progression: Implications for human breast
cancer. Oncogene. 29:3374–3385. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weaver AM: Invadopodia: Specialized cell
structures for cancer invasion. Clin Exp Metastasis. 23:97–105.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Attwooll C, Denchi E Lazzerini and Helin
K: The E2F family: Specific functions and overlapping interests.
EMBO J. 23:4709–4716. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hulit J, Lee RJ, Russell RG and Pestell
RG: ErbB-2-induced mammary tumor growth: The role of cyclinDl and
p27kipl. Biochem Pharmacol. 64:827–836. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
McMahon SB: MYC and the control of
apoptosis. Cold Spring Harb Perspect Med. 4:a0144072014. View Article : Google Scholar : PubMed/NCBI
|