Introduction
Nasopharyngeal carcinoma (NPC) is a rare malignancy
among most populations worldwide; based on previous epidemiological
data, global morbidity and mortality of the disease are relatively
low, both <2 per 100,000 persons (1). However, NPC is an endemic disease
that is prevalent in certain regions, such as southern China,
Southeast Asia, Middle East and North Africa (2). For example, in several regions of
southern China, the prevalence is as high as 8–27 per 100,000
persons (3). Etiology of NPC is
complicated, and family history, age and Epstein-Barr virus (EBV)
infection are risk factors for disease development (4); plasma EBV DNA has been recognized as
a biomarker for the prognosis of NPC (5). Genetic factors also contribute to NPC
progression, such as overexpression of CD109 (6) and far upstream element-binding
protein 1 (7). MicroRNAs (miRNAs)
regulate genes at the transcriptional level and serve important
roles during NPC progression. miRNA (miR)-9 was previously
predicted to be a prognostic factor for NPC, and decreased
expression has been associated with advanced tumor-node-metastasis
stage (8).
However, these findings are insufficient to
comprehensively reveal the pathogenesis of NPC. Epigenetic
alterations also serve important roles in disease development. DNA
hypermethylation is the most common epigenetic alteration; it
involves the binding of a methyl group to CpG dinucleotides within
a gene promoter, and is considered a hallmark in various cancer
types (9). The silencing of a
tumor suppressor gene may be caused by abnormal hypermethylation at
the promoter. In NPC, numerous tumor suppressor genes were
previously identified to be methylated (9); for example, expression of the tumor
suppressor gene DLC1 Rho GTPase-activating protein was revealed to
be repressed by promoter hypermethylation in NPC (10). Previous in vitro experiments
indicated that overexpression of cytochrome b5 reductase 2
(CYB5R2) had an inhibitory effect on NPC; however,
CYB5R2 is often inactive in human NPC owing to promoter
hypermethylation (11). Other
tumor suppressor genes, such as Ras association domain family
member 1, cyclin dependent kinase inhibitor 2A, deleted in lung and
esophageal cancer 1, death associated protein kinase 1 and in
particular, ubiquitin C-terminal hydrolase L1, are also methylated
in NPC patients with different incidences (9). These data suggested that there is a
requirement for identifying methylation status as a prognostic
biomarker in NPC.
A previous study using whole-genome methylation
analysis comprised a panel of six hypermethylated genes that were
identified as being correlated with poor survival in patients with
NPC (12). Another study compared
methylome data from methylation chip array analysis with data from
The Cancer Genome Atlas Database and demonstrated that
hypermethylation was a predominant pattern in NPC, and the
methylation was associated with CpG islands in tumor tissues
(13). Nevertheless, there is an
abundance of useful data that remains unexploited in the
aforementioned methylated data sets, which may contribute to our
comprehensive understanding of the correlations between methylation
and NPC etiology.
Therefore, the present study used the two
methylation data sets to find the overlapping methylated regions in
NPC. The identified genes were further investigated, as well as the
potential correlations with each other or with miRNAs. Based on
these comprehensive bioinformatics analyses, the results from the
present study were expected to provide novel insights into the
epigenetic alteration mechanisms in NPC and to identify additional
biomarkers for disease prognosis.
Materials and methods
Data resource
Two data sets (GSE52068 and GSE62336) relating to
NPC methylation were downloaded from the Gene Expression Omnibus
database (http://www.ncbi.nlm.nih.gov/geo); both data sets were
obtained from the Illumina HumanMethylation450 BeadChip platform
(Illumina, Inc., San Diego, CA, USA). There were a total of 48
samples in the GSE52068 data set, which included 24 NPC cases and
24 controls (12), and 50 samples
in the GSE62336 data set, which included 25 NPC cases and 25
controls (13).
Pretreatment of raw data
Signal intensity files within the two data sets were
downloaded, which provided methylated and unmethylated signal
values of the CpG site probes in each sample. Subsequently, the
β-value that indicated the methylation status of a gene was
calculated using the following formula:
β=methylated signalmethylated
signal+unmethylated signal+100
The calculated value was pretreated with the
β-mixture quantile normalization method as previously described
(14).
Prediction of differentially
methylated regions (DMRs)
The COHCAP package, which is a part of the R and
Bioconductor online software suite (http://www.bioconductor.org/packages/3.0/bioc), was
used to predict DMRs between NPC cases and controls in the two data
sets (15). Hypermethylation and
hypomethylation status were selected based on the recommendation of
Sproul et al (16), which
stated that if the CpG site β-value is greater than a certain
value, it is defined as hypermethylation, and if the CpG site
β-value is less than a certain value, it is defined as
hypomethylation (16). Using
COHCAP to compare NPC and control, the Δβ-values, P-values and
false discovery rates (FDRs) were obtained according to the files
containing β values of the CpG site probe. DMRs between the two
samples were identified using the criteria of FDR <0.05 and |Δβ|
>0.1 (Δβ value >0.1 indicated hypermethylation, and Δβ
<-0.1 indicated hypomethylation). Subsequently, the
corresponding gene symbols of the DMRs were established based on
the annotation files in the methylation microarray data sets.
Conversely, DEMs that did not correspond to a gene symbol and DMRs
that corresponded to >1 gene symbol were filtered out.
Differentially methylated CpG islands
(DMCs)
In addition to predicting the methylated site,
COHCAP was also used to predict CpG island methylation, which was
established by calculating the abnormal differentially methylated
site counts in each gene and the average β-values. The
‘COHCAP.avg.by.island’ function implemented in COHCAP package was
used to identify DMCs between NPC and normal samples in each data
set, with the parameters: ‘num.sites=4, which reflected that the
minimal number of differentially methylated sites was 4; |Δβ|
>0.1; and FDR <0.05.
Common DMRs and DMCs in two data
sets
Comparing the DMR and DMC information from the two
databases, overlapping DMRs and DMCs were identified and extracted.
If a DMR appeared in both data sets, it suggested that the
corresponding genes may be highly associated with NPC. From these
resulting data the corresponding genes of the overlapping DMRs were
obtained.
Enrichment analysis of the overlapped
genes
The Database for Annotation, Visualization and
Integration Discovery (version 6.8; http://david.abcc.Ncifcrf.gov) software was used to
perform Gene Ontology (GO; http://www.geneontology.org) term analysis and Kyoto
Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/pathway.html) pathway
enrichment analysis, under the conditions of gene count ≥2 and
P<0.05 based on hypergeometric test, as previously described
(17).
Construction of protein-protein
interaction (PPI) network of overlapping DMR genes
The identified DMR genes were mapped using the
Search Tool for the Retrieval of Interacting Genes (http://string-db.org) database, version 10.0 (18), to explore their potential
interactions at protein level. A PPI network for these genes was
constructed using the parameters: species, Homo sapiens
(hsa); and PPI score=0.4. In addition, it was required that these
PPIs were derived from validated experiments. The PPI network was
visualized with Cytoscape (http://cytoscape.org) software, and the topological
property was analyzed based on the degree of a node. For nodes in
the PPI network, the random walk algorithm was used to analyze them
to obtain marker genes of NPC, as previously described (19).
Identification of miRNAs associated
with DMRs and their targets
Overlapping DMR genes in the two data sets were
further examined to determine whether they encoded miRNAs. The
putative miRNA-coding genes were combined with target gene
information in the miRWalk database to predict their target genes
using the ‘validated target module’ (20). The target genes were compared with
DMRs to select the target genes connecting with other DMR
genes.
NPC related gene analysis
The Comparative Toxicogenomics Database (CTD) is a
comprehensive database that records disease-related genes that have
been previously reported (21).
Based on information in this database, NPC related genes were
downloaded to determine whether they were methylated.
Results
DMRs between NPC samples and controls
in two data sets
Using the thresholds of FDR <0.05 and |Δβ|
>0.1, a total of 4,218 hypermethylated and 2,178 hypomethylated
CpG sites were identified in the GSE52068 data set, which
corresponded to 2,243 and 2,013 genes, respectively. In the
GSE62336 data set, 11,347 hypermethylated and 39 hypomethylated CpG
sites were identified, which corresponded to 4,372 and 37 genes,
respectively.
Overlapped DMR related genes and their
enriched functions and pathways
The identified genes from the two data sets were
compared, and the overlapping DMRs were selected. A total of 3,306
shared CpG sites were identified as hypermethylated, and
corresponded to 1,854 genes; and 18 shared CpG sites were
identified as hypomethylated, corresponding to 18 genes.
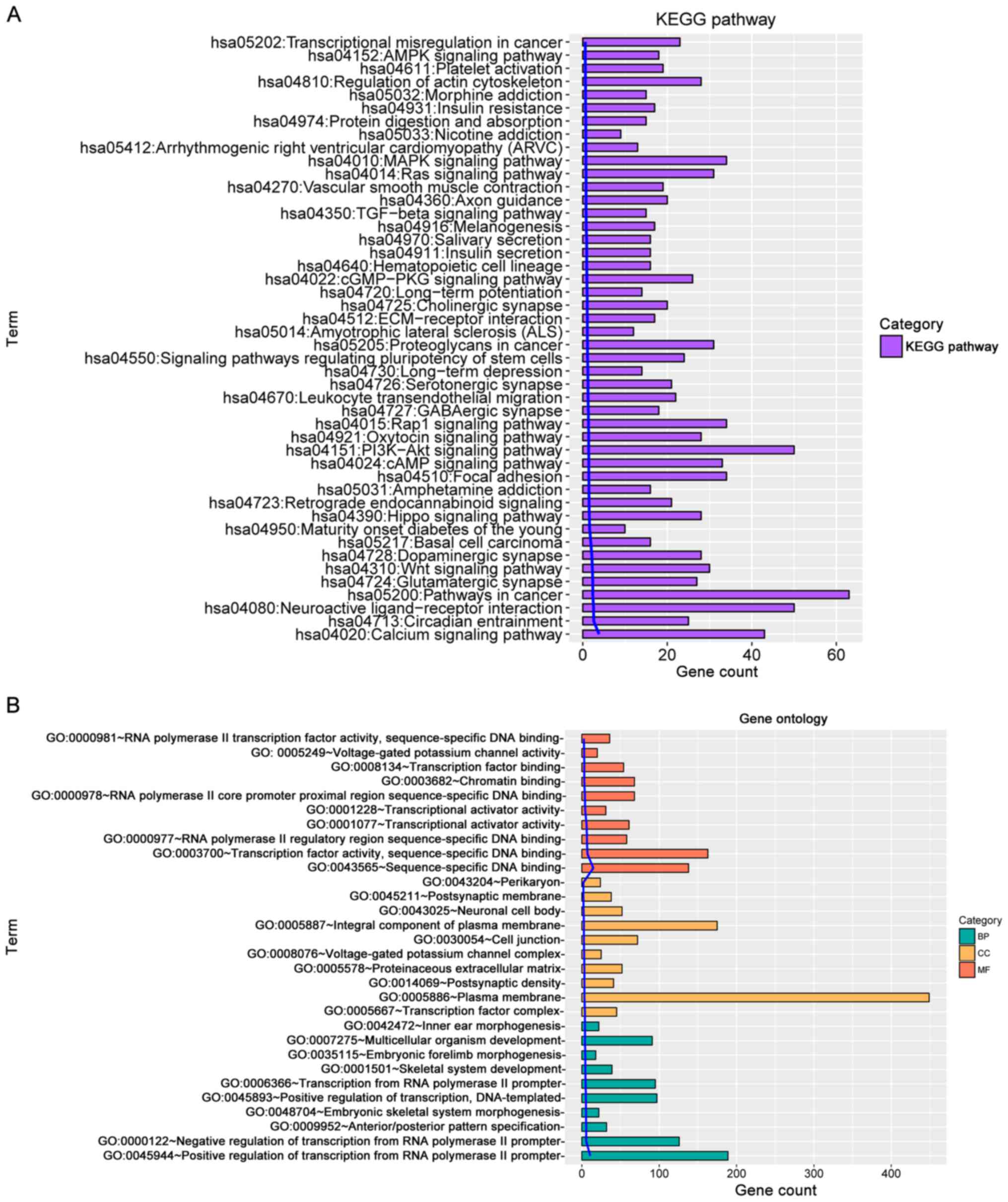
According to KEGG pathway enrichment and GO term
analyses (Fig. 1A and B,
respectively), the selected genes were revealed to be highly
associated with pathways including mitogen-activated protein kinase
(MAPK) signaling pathway, phosphatidylinositol 3-kinase (PI3K)/AKT
signaling pathway, calcium signaling pathway, pathways in cancer
and neuroactive ligand receptor interaction. With regards to the
functions, these genes were significantly enriched in biological
process including positive regulation of transcription from RNA
polymerase II promoter, transcription from RNA polymerase II
promoter and multicellular organism development. Enriched cellular
component annotations included integral component of plasma
membrane, plasma membrane and cell junction. Molecular function
annotations included the following: transcription factor activity,
sequence-specific DNA binding;, transcription factor binding;
sequence-specific DNA binding.
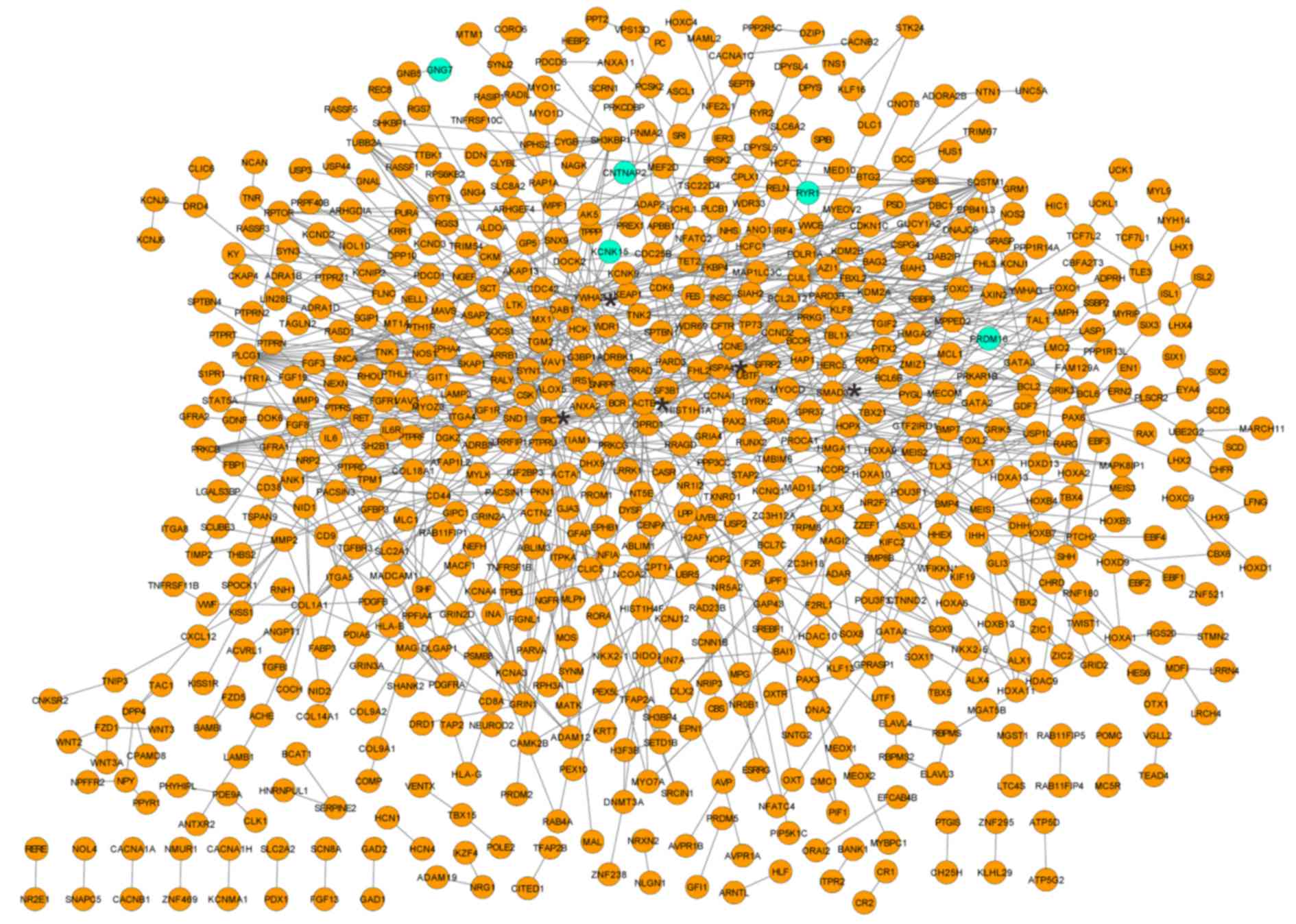
PPI network of the overlapped DMR
corresponding genes
Based on the aforementioned criteria, including PPI
score=0.4, a PPI network was established that contained 677 nodes
and 991 interactions. The random walk algorithm identified 20 nodes
that were predominant with high degree (Fig. 2), such as proto-oncogene
tyrosine-protein kinase SRC (degree=50), SMAD family member 3
(SMAD3; degree=31), tyrosine 3-monooxygenase/tryptophan
5-monooxygenase activation protein ζ (YWHAZ; degree=28), β-actin
(ACTB; degree=27) and Heat shock protein family A member 4 (HSPA4;
degree=24). Notably, these genes were all hypomethylated.
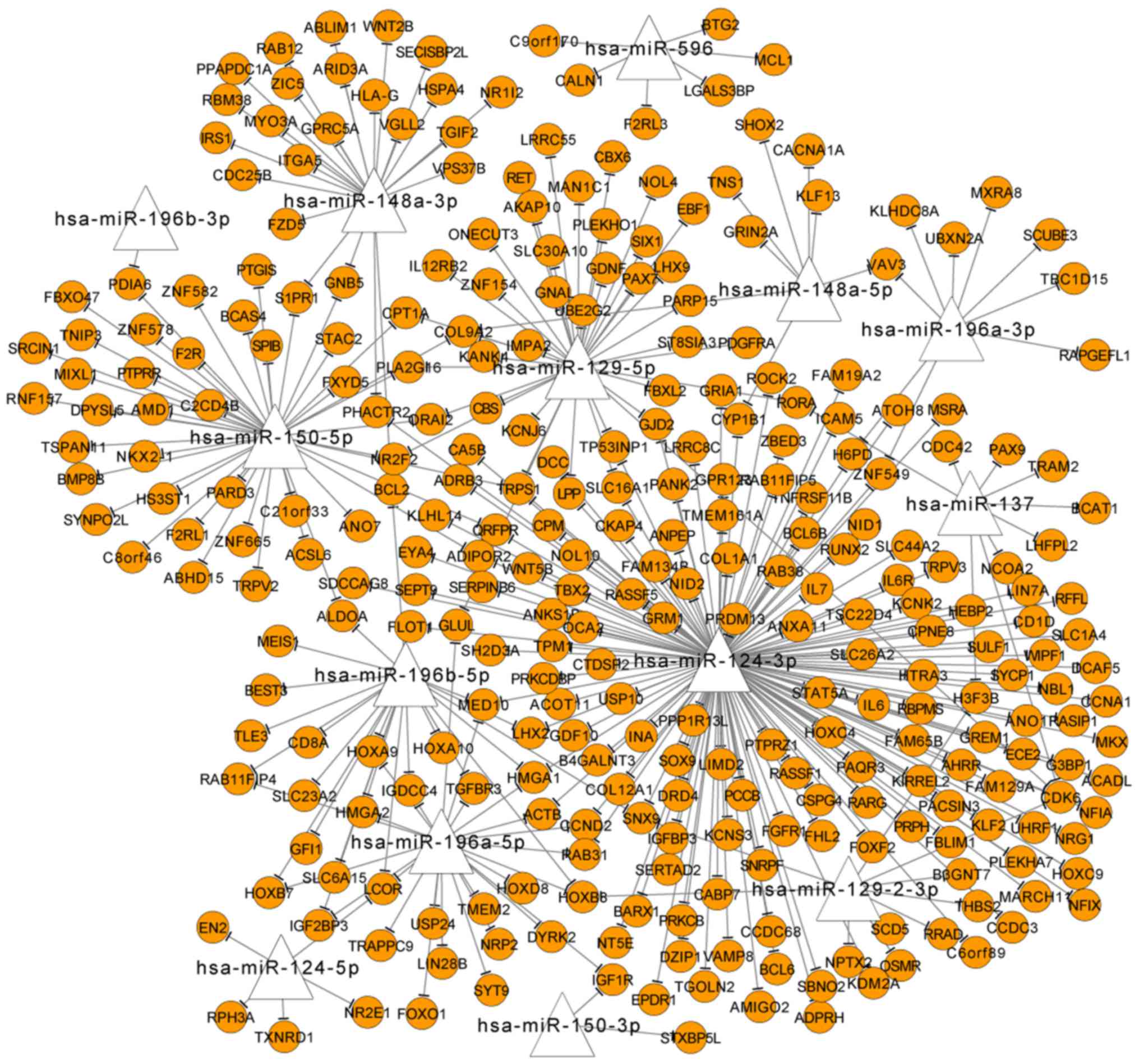
Abnormally methylated miRNAs among the
overlapped DMR genes
Among the overlapped DMR genes, 14 were identified
as miRNA-encoding genes, which regulated 306 other DMR genes, such
as hsa-miR-148a-3p, hsa-miR-129-5p, hsa-miR-150-5p, hsa-miR-124-3p
and hsa-miR-196a-5p (Fig. 3).
Based on the prediction of miRNA-target relationships, HSPA4
was predicted as a target of hsa-miR-148a-3p, and ACTB was
the target of hsa-miR-124-3p and hsa-miR-196a-5p.
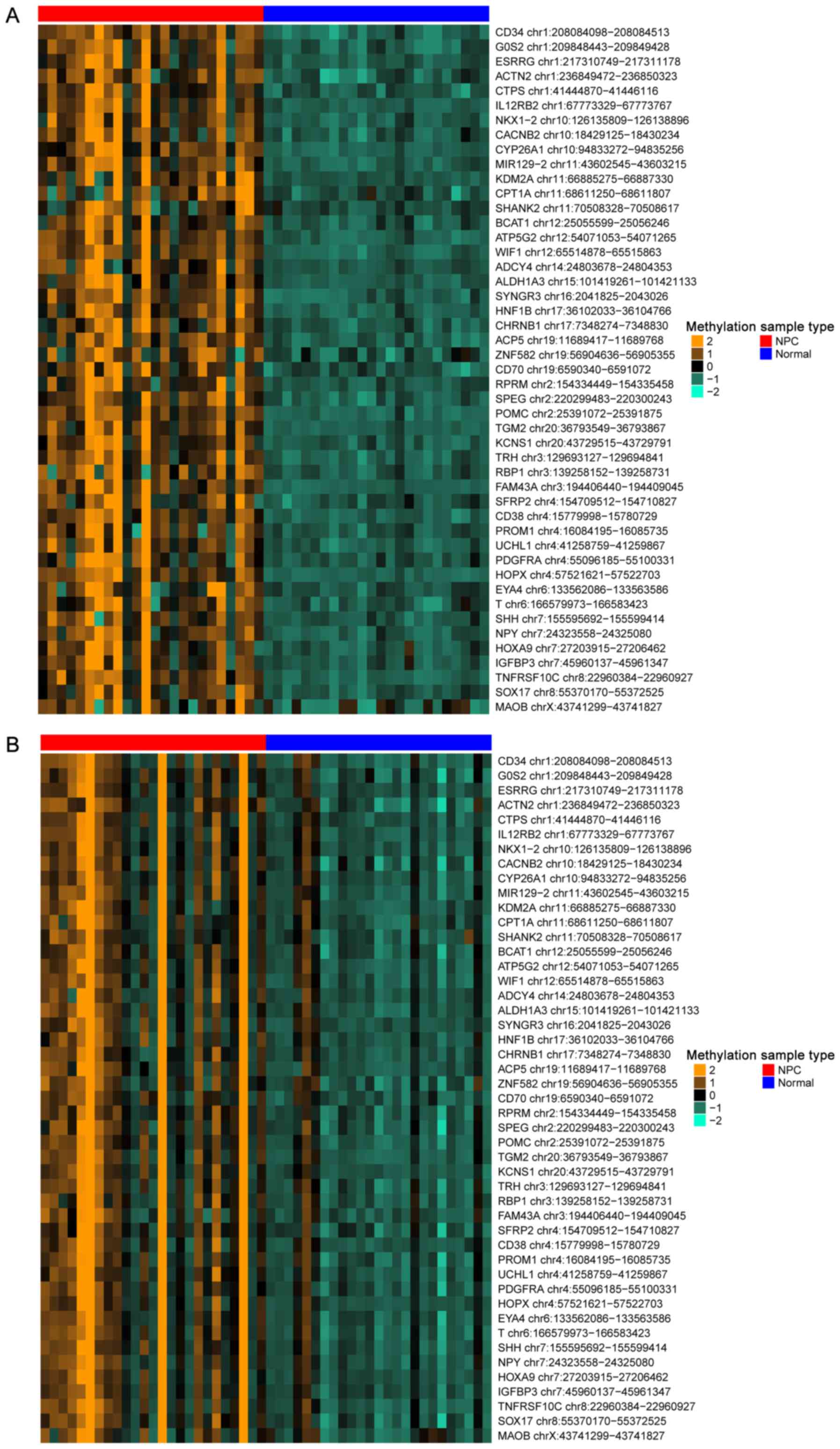
DMC analysis between two different
samples
Using the aforementioned parameters of num.sites=4,
|Δβ| >0.1 and FDR <0.05, a total of 195 DMCs were identified
in the GSE52068 data set, and 798 DMCs were identified in the
GSE62336 data set. Between them, 150 were overlapped and
hypermethylated, and 47 of the overlapped genes were related to
NPC, which were defined as NPC genes; a heat map of the 47
identified NPC genes is presented in Fig. 4. Notably, an miRNA-encoding gene
was also identified, miR129-2 chr11:43602545-43603215.
Identification of NPC related
genes
Based on information obtained from the CTD database,
it was revealed that 583 NPC genes had been previously reported to
associate with hypermethylated DMRs and three NPC genes were
related to hypomethylated DMRs. Notably, 17 of them were
highlighted in the PPI network, such as cell division cycle 42
(CDC42), α1 actin (ACTA1), paired box 6
(PAX6), SMAD3, matrix metallopeptidase 2
(MMP2), collagen type I α1 (COL1A1), protein kinase
Cγ (PRKCG), SRC, phospholipase Cγ (PLCG1),
ubiquitin-conjugating enzyme E2 G2 (UBE2G2), synuclein α
(SNCA), HSPA4, sequestosome 1 (SQSTM1),
ACTB, tyrosine 3-monooxygenase/tryptophan 5-monooxygenase
activation protein γ (YWHAG), fibroblast growth factor
receptor 1 (FGFR1) and YWHAZ. Among them, MMP2
was determined to be an NPC marker gene.
Discussion
The present study identified a number of crucial
genes that were indicted to be hypomethylated, instead of
hypermethylated, in the NPC samples, including SRC, SMAD3,
YWHAZ and HSPA4. These genes were predominant in the PPI
network. Among them, HSPA4 was predicted as a target of
hsa-miR-148a-3p. SRC and SMAD3 were correlated with
NPC based on the CTD database. Most of the identified DMCs were
revealed to be hypermethylated, and 47 were associated with NPC. A
miRNA-encoding gene was also highlighted, miR129-2
chr11:43602545-43603215.
Promoter methylation is implicated in the regulation
of gene expression, and hypomethylation is commonly exhibited in
several cancer types, such as melanoma and squamous cell carcinoma
(SCC) (22). Hypomethylation of
deleted in split hand/split foot 1 was demonstrated to result in
the overexpression of this gene, which was associated with poor
prognosis in patients with SCC (22). SRC encodes a
tyrosine-protein kinase protein; it is an oncogene that has a
similar function as the v-src gene of the Rous sarcoma
virus. Activation of cellular SRC (c-SRC) was previously reported
to promote metastasis of NPC by regulating the PI3K/AKT signaling
pathway (23). The long non-coding
RNA actin filament associated protein 1-antisense RNA 1 (AFAP1-AS1)
was demonstrated to act as an adaptor that connects other proteins
like SRC in the mediation of actin filament integrity (24). In NPC, upregulation of AFAP1-AS1
expression may lead to tumor metastasis and has been associated
with poor prognosis (24). These
data suggested that overexpression of SRC may be associated
with metastasis and poor prognosis of NPC. In the present study,
SRC was demonstrated to be hypomethylated and was implicated
in NPC. On this basis, it was inferred that hypomethylation of SRC
may result in the upregulation of this gene, which may contributes
to the NPC metastasis and poor prognosis.
SMAD3 is a member of SMAD family of signal
transducers and transcriptional regulators. Activated by
transforming growth factor-β (TGF-β), SMAD3 serves a crucial
role in promoting carcinogenesis (25). A previous study on TGF-β/Smad
signaling in NPC reported that the expression of SMAD3 was
significantly increased in the CNE2 NPC cell line (26). In addition, downregulation of
SMAD3 expression by miR-145 was demonstrated to inhibit the
invasion and metastasis of NPC (27), which suggested that the elevated
SMAD3 expression level may account for metastasis of NPC.
Other previous studies have reported the relationship between
SMAD3 expression and methylation, and it was demonstrated
that different SMAD3 expression levels had similar promoter
methylation patterns in the maintenance of self-tolerance (28). However, unlike these
autoimmune-associated lesions, results from the present study
indicated that SMAD3 was hypomethylated in NPC, which
suggested that SMAD3 hypomethylation may result in the
upregulated expression of this gene and may account for NPC
metastasis. These results indicated that the upregulation of
SMAD3 by hypomethylation may be linked to poor prognosis of
NPC.
HSPA4 belongs to the HSP70 protein family. Induced
by Nibrin, an important DNA repair protein that maintains genomic
integrity in the advanced stages NPC, HSPA4 was reported to
facilitate the metastatic activity in tumor cells (29). In addition, polymorphism of another
HSP70 family member, HSP70-2, was demonstrated to be involved with
susceptibility to NPC (30). These
data suggested that abnormal expression of HSP70-related genes may
be highly associated with NPC progression. However, a connection
between HSPA4 expression and its methylation status have not
been reported. In the present study, HSPA4 was indicated to
be hypomethylated in NPC samples, which may account for an
upregulation in expression. These results indicated that HSPA4 may
be important to NPC metastasis and hypomethylation may be a
predictive factor for disease prognosis.
In terms of miRNAs, miR-148a was previously reported
to be downregulated in NPC (31),
and it may be involved in epithelial-mesenchymal transition (EMT)
and metastasis in hepatocellular carcinoma (32); however, this function has not been
reported in NPC. The present study predicted that HSPA4 was
a target of hsa-miR-148a-3p. Combining this with the
hypomethylation of HSPA4, it may be inferred that
hsa-miR-148a-3p binds to the gene promoter region that controls
methylation of this gene during NPC metastasis. However, this
hypothesis requires further analysis through dual-luciferase
reporter system.
By binding to the phosphoserine-containing proteins,
the YWHAZ protein serves an important role in the regulation of
signal transduction (33). In NPC
cells, EBV-miR-BART10-3p was reported to induce the expression of
β-catenin by inhibiting the expression of β-transducin
repeat-containing E3 ubiquitin protein ligase (BTRC), which
facilitates EMT and promotes metastasis (34). Formation of the β-catenin/YWHAZ
complex was reported to inhibit the binding of β-catenin to BTRC,
which ensures the stability of β-catenin and thus promotes EMT and
metastasis in lung cancer (35).
Neither the function nor the methylation status of YWHAZ in NPC has
been reported; however, hypermethylation in the gene promoter was
previously demonstrated to suppress the expression of this gene.
The hypomethylation of YWHAZ reported in the present study
may indicate an increase in gene expression. Therefore, it was
speculated that, similar to lung cancer, the increase in
YWHAZ expression may also lead to the EMT in NPC and promote
metastasis. This hypothesis, however, needs to be validated in
future studies.
miR129-2 was previously demonstrated to act as a
tumor suppressor, and the methylation of miR129-2 was reported to
be a frequent event in lymphoid malignancies (36). Differential methylation of this
miRNA has been indicated in rectal cancer and colorectal cancer
(37,38). In the present study, miR129-2 was
revealed to be hypermethylated in NPC, which suggested that
expression may be inhibited by hypermethylation in NPC
development.
It should be noted that the inferred effects of the
identified methylated genes and miRNAs, as well as their predictive
correlations in NPC, require further experimental validation.
Despite this limitation, the present study may be of great value
for providing novel methylation biomarkers for NPC detection and
prognosis.
In conclusion, several novel methylated genes and
miRNA were identified that may serve as potential biomarkers for
NPC prognosis. Hypomethylation of SRC, SMAD3, YWHAZ and
HSPA4, and the hypermethylation of miR129-2, may be linked
with poor prognosis of NPC. Nevertheless, all the predicted results
need to be further validated by experiments.
Glossary
Abbreviations
Abbreviations:
|
NPC
|
nasopharyngeal carcinoma
|
|
DMR
|
differentially methylated region
|
|
PPI
|
protein-protein interaction
|
|
EBV
|
Epstein-Barr virus
|
|
FDR
|
false discovery rate
|
|
CTD
|
Comparative Toxicogenomics
Database
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
Tang LL, Chen WQ, Xue WQ, He YQ, Zheng RS,
Zeng YX and Jia WH: Global trends in incidence and mortality of
nasopharyngeal carcinoma. Cancer Lett. 374:22–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chang ET and Adami HO: The enigmatic
epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol
Biomarkers Prev. 15:1765–1777. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tay JK, Chan SH, Lim CM, Siow CH, Goh HL
and Loh KS: The role of Epstein-Barr virus DNA load and serology as
screening tools for nasopharyngeal carcinoma. Otolaryngol Head Neck
Surg. 155:274–280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dai W, Zheng H, Cheung AK, Tang CS, Ko JM,
Wong BW, Leong MM, Sham PC, Cheung F, Kwong DL, et al: Whole-exome
sequencing identifies MST1R as a genetic susceptibility gene in
nasopharyngeal carcinoma. Proc Natl Acad Sci USA. 113:pp.
3317–3322. 2016; View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chan KC: Plasma Epstein-Barr virus DNA as
a biomarker for nasopharyngeal carcinoma. Chin J Cancer.
33:598–603. 2014.PubMed/NCBI
|
|
6
|
Jia W, Ren C, Wang L, Zhu B, Jia W, Gao M,
Zeng F, Zeng L, Xia X, Zhang X, et al: CD109 is identified as a
potential nasopharyngeal carcinoma biomarker using aptamer selected
by cell-SELEX. Oncotarget. 7P:1–55342. 2016.
|
|
7
|
Liu ZH, Hu JL, Liang JZ, Zhou AJ, Li MZ,
Yan SM, Zhang X, Gao S, Chen L, Zhong Q and Zeng MS: Far upstream
element-binding protein 1 is a prognostic biomarker and promotes
nasopharyngeal carcinoma progression. Cell Death Dis. 6:e19202015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu J, Xu X, Liu X, Peng Y, Zhang B, Wang
L, Luo H, Peng X, Li G, Tian W, et al: Predictive value of miR-9 as
a potential biomarker for nasopharyngeal carcinoma metastasis. Br J
Cancer. 110:392–398. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tian F, Yip SP, Kwong DL, Lin Z, Yang Z
and Wu VW: Promoter hypermethylation of tumor suppressor genes in
serum as potential biomarker for the diagnosis of nasopharyngeal
carcinoma. Cancer Epidemiol. 37:708–713. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feng X, Ren C, Zhou W, Liu W, Zeng L, Li
G, Wang L, Li M, Zhu B, Yao K and Jiang X: Promoter
hypermethylation along with LOH, but not mutation, contributes to
inactivation of DLC-1 in nasopharyngeal carcinoma. Mol Carcinog.
53:858–870. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xiao X, Zhao W, Tian F, Zhou X, Zhang J,
Huang T, Hou B, Du C, Wang S, Mo Y, et al: Cytochrome b5 reductase
2 is a novel candidate tumor suppressor gene frequently inactivated
by promoter hypermethylation in human nasopharyngeal carcinoma.
Tumor Biol. 35:3755–3763. 2014. View Article : Google Scholar
|
|
12
|
Jiang W, Liu N, Chen XZ, Sun Y, Li B, Ren
XY, Qin WF, Jiang N, Xu YF, Li YQ, et al: Genome-wide
identification of a methylation gene panel as a prognostic
biomarker in nasopharyngeal carcinoma. Mol Cancer Ther.
14:2864–2873. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dai W, Cheung AK, Ko JM, Cheng Y, Zheng H,
Ngan RK, Ng WT, Lee AW, Yau CC, Lee VH and Lung ML: Comparative
methylome analysis in solid tumors reveals aberrant methylation at
chromosome 6p in nasopharyngeal carcinoma. Cancer Med. 4:1079–1090.
2015. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Teschendorff AE, Marabita F, Lechner M,
Bartlett T, Tegner J, Gomez-Cabrero D and Beck S: A beta-mixture
quantile normalization method for correcting probe design bias in
Illumina Infinium 450 k DNA methylation data. Bioinformatics.
29:189–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Warden CD, Lee H, Tompkins JD, Li X, Wang
C, Riggs AD, Yu H, Jove R and Yuan YC: COHCAP: An integrative
genomic pipeline for single-nucleotide resolution DNA methylation
analysis. Nucleic Acids Res. 41:e1172013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sproul D, Nestor C, Culley J, Dickson JH,
Dixon JM, Harrison DJ, Meehan RR, Sims AH and Ramsahoye BH:
Transcriptionally repressed genes become aberrantly methylated and
distinguish tumors of different lineages in breast cancer. Proc
Natl Acad Sci USA. 108:pp. 4364–4369. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang DW, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43(Database issue): D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Köhler S, Bauer S, Horn D and Robinson PN:
Walking the interactome for prioritization of candidate disease
genes. Am J Hum Genet. 82:949–958. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dweep H and Gretz N: miRWalk2.0: A
comprehensive atlas of microRNA-target interactions. Nat Methods.
12:6972015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Davis AP, Murphy CG, Johnson R, Lay JM,
Lennon-Hopkins K, Saraceni-Richards C, Sciaky D, King BL,
Rosenstein MC, Wiegers TC and Mattingly CJ: The comparative
toxicogenomics database: Update 2013. Nucleic Acids Res.
41(Database issue): D1104–D1114. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Venza M, Visalli M, Catalano T, Beninati
C, Teti D and Venza I: DSS1 promoter hypomethylation and
overexpression predict poor prognosis in melanoma and squamous cell
carcinoma patients. Hum Pathol. 60:137–146. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ke L, Xiang Y, Guo X, Lu J, Xia W, Yu Y,
Peng Y, Wang L, Wang G, Ye Y, et al: c-Src activation promotes
nasopharyngeal carcinoma metastasis by inducing the
epithelial-mesenchymal transition via PI3K/Akt signaling pathway: A
new and promising target for NPC. Oncotarget. 7:28340–28355. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bo H, Gong Z, Zhang W, Li X, Zeng Y, Liao
Q, Chen P, Shi L, Lian Y, Jing Y, et al: Upregulated long
non-coding RNA AFAP1-AS1 expression is associated with progression
and poor prognosis of nasopharyngeal carcinoma. Oncotarget.
6:20404–20418. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Javelaud D and Mauviel A: Crosstalk
mechanisms between the mitogen-activated protein kinase pathways
and Smad signaling downstream of TGF-beta: implications for
carcinogenesis. Oncogene. 24:5742–5750. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiao J, Xiang Q, Xiao YC, Su ZJ, Huang ZF,
Zhang QH, Tan Y, Li XK and Huang YD: The effect of transforming
growth factor-beta1 on nasopharyngeal carcinoma cells: Insensitive
to cell growth but functional to TGF-beta/Smad pathway. J Exp Clin
Cancer Res. 29:352010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang H, Sun P, Lei Z, Li M, Wang Y, Zhang
HT and Liu J: miR-145 inhibits invasion and metastasis by directly
targeting Smad3 in nasopharyngeal cancer. Tumour Biol.
36:4123–4131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Busque L, Belisle C, Provost S, Giroux M
and Perreault C: Differential expression of SMAD3 transcripts is
not regulated by cis-acting genetic elements but has a gender
specificity. Genes Immun. 10:192–196. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Silva J, Teixeira AL, Lobo F, Maurício J
and Medeiros R: DNA repair system and prostate cancer progression:
The role of NBS1 polymorphism (rs1805794). DNA Cell Biol.
31:1182–1186. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ghorbani MJ, Salehi Z, Sabet EE and
Ejtehadi F: Anatlysis of HSPA1B A1267G gene polymorphism in peptic
ulcer. Mol Biol (Mosk). 48:728–732. 2014.(In Russian). View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen HC, Chen GH, Chen YH, Liao WL, Liu
CY, Chang KP, Chang YS and Chen SJ: MicroRNA deregulation and
pathway alterations in nasopharyngeal carcinoma. Br J Cancer.
100:1002–1011. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang JP, Zeng C, Xu L, Gong J, Fang JH
and Zhuang SM: MicroRNA-148a suppresses the epithelial-mesenchymal
transition and metastasis of hepatoma cells by targeting Met/Snail
signaling. Oncogene. 33:4069–4076. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jarczak J, Kaba J and Bagnicka E: The
validation of housekeeping genes as a reference in quantitative
real time PCR analysis: Application in the milk somatic cells and
frozen whole blood of goats infected with caprine arthritis
encephalitis virus. Gene. 549:280–285. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yan Q, Zeng Z, Gong Z, Zhang W, Li X, He
B, Song Y, Li Q, Zeng Y, Liao Q, et al: EBV-miR-BART10-3p
facilitates epithelial-mesenchymal transition and promotes
metastasis of nasopharyngeal carcinoma by targeting BTRC.
Oncotarget. 6:41766–41782. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen CH, Chuang SM, Yang MF, Liao JW, Yu
SL and Chen JJ: A novel function of YWHAZ/β-catenin axis in
promoting epithelial-mesenchymal transition and lung cancer
metastasis. Mol Cancer Res. 10:1319–1331. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Maia BM, Rocha RM and Calin GA: Clinical
significance of the interaction between non-coding RNAs and the
epigenetics machinery: Challenges and opportunities in oncology.
Epigenetics. 9:75–80. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vymetalkova V, Vodicka P, Pardini B, Rosa
F, Levy M, Schneiderova M, Liska V, Vodickova L, Nilsson TK and
Farkas SA: Epigenome-wide analysis of DNA methylation reveals a
rectal cancer-specific epigenomic signature. Epigenomics.
8:1193–1207. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bandres E, Agirre X, Bitarte N, Ramirez N,
Zarate R, Roman-Gomez J, Prosper F and Garcia-Foncillas J:
Epigenetic regulation of microRNA expression in colorectal cancer.
Int J Cancer. 125:2737–2743. 2009. View Article : Google Scholar : PubMed/NCBI
|