Introduction
Myocardial infarction (MI) can lead to ventricular
arrhythmias, heart dysfunction and sudden death (1). Sympathetic neural remodeling,
characterized by cardiac nerve sprouting and sympathetic
hyperinnervation, serves an important role in these outcomes
(2–4). The inflammatory response is a
critical aspect of sympathetic neural remodeling post-MI (5), and anti-inflammatory treatments can
attenuate sympathetic neural remodeling post-MI (6–8).
Thus, attenuating the post-MI inflammatory response may provide an
important strategy to delay sympathetic neural remodeling
post-MI.
Activin A, a transforming growth factor-β
superfamily member, is important in inflammation, by exerting its
function through type II (ActR IIA or ActR II–IIB) and type I (ActR
IB, ALK4) activin receptors (9).
In patients and animals exhibiting heart failure post-MI, activin A
levels are increased and correlate with the degree of cardiac
dysfunction, and its actions are thought to be
inflammatory-mediated (10). In
addition, activin A receptor inhibition impairs expression of
pro-inflammatory factors and increases expression of
anti-inflammatory factors during monocyte differentiation (11). Activin A activates the nuclear
factor (NF)-κB pathway in osteoclast precursors (12), and NF-κB also promotes activin A
production in bone marrow stromal cells (13). Moreover, inhibition of NF-κB
reverses inflammatory-mediated left ventricular remodeling and
cardiac dysfunction post-MI (14).
Therefore, the authors hypothesized that activin A inhibition can
attenuate the inflammatory response post-MI via NF-κB pathway
inactivation.
Nerve growth factor (NGF) is a neurotrophin that
serves an important role in growth, differentiation and survival of
sympathetic adrenergic neurons (15,16).
It is synthesized by inflammatory cells (macrophage and
myofibroblasts) found within the cardiac peri-infarct zone post-MI,
and its synthesis and release are thought to initiate sympathetic
nerve sprouting (5). Transgenic
overexpression of NGF in mice causes cardiac sympathetic
hyperinnervation (17). Activin A
regulates macrophage function in vitro and in vivo
(18,19). Moreover, activin A stimulates the
production of inflammatory mediators [e.g., interleukin (IL)-1β,
tumor necrosis factor (TNF)-α, IL-6, nitric oxide and prostanoids]
in cultured monocyte/macrophage cell lines (20) and promotes differentiation of
fibroblasts into myofibroblasts in human lung fibroblasts, primary
renal interstitial fibroblasts and NRK-49F cells (21,22).
Therefore, the authors also hypothesized that activin A inhibition
could attenuate NGF upregulation post-MI via suppression of the
inflammatory response. The paper investigated whether inhibition of
activin A could reduce post-MI sympathetic neural remodeling via
inhibition of the inflammatory response.
Materials and methods
Animals
The study was approved by the Ethics Committee of
Wuhan University (Wuhan, China). All animal procedures followed the
Guidelines for the Care and Use of Laboratory Animals of the
Institutional Animal Care and Use Committee of Wuhan University
(Wuhan, China), which conform to Guidelines for the Care and Use of
Laboratory Animals published by the US National Institutes of
Health (NIH Publication no. 85-23, revised 1996; Bethesda, MD,
USA). Healthy male Sprague-Dawley rats (weight 200–250 g; 12 h
light/dark cycle) were housed under standard conditions with chow
and water available ad libitum. Animals were acclimated for
1 week prior to the start of experiments.
Myocardial infarction model and
treatment protocol
Animals were anesthetized by intraperitoneal
injection of sodium pentobarbital (40 mg/kg, Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) and subsequently intubated and ventilated
using a small animal ventilator. Left thoracotomy was performed,
and the left anterior descending artery was ligated as previously
described (23). Sham-operated
rats underwent the same protocol, but the coronary arteries were
not tied. Rats surviving 24 h post-operation were divided into
sham, MI, and MI+follistatin-300 (FS) groups (n=6 per group) and
subsequently underwent treatment. FS, a natural activin A inhibitor
(R&D Systems, Inc., Minneapolis, MN, USA), was dissolved in
phosphate-buffered saline (PBS). Rats in the sham and MI groups
were given PBS, whereas rats in the MI+FS group were given FS (1
µg) by intraperitoneal injection once a day for 28 days, as
previously described (24).
Cardiac function
Cardiac function was estimated using transthoracic
echocardiography at 28 days post-MI and treatment. Images of the
left ventricle were acquired at the level of the papillary muscle
(Vevo 770; VisualSonics, Toronto, ON, Canada). Parameters measured
included: Left ventricle ejection fraction (LVEF), left ventricle
fractional shortening (LVFS), left ventricle end-diastolic
dimension (LVEDD), left ventricle end-systolic dimension (LVESD)
and heart rate.
Western blot analysis
Protein was extracted using radioimmunoprecipitation
assay lysate (Beyotime Institute of Biotechnology, Haimen, China)
and phenylmethanesulfonyl fluoride (Abcam, Cambridge, MA, USA) from
the peri-infarct zone of rat hearts at four weeks post-MI in the MI
and MI+FS groups or from the same zone in the sham group. Protein
concentrations were determined using bicinchoninic acid method.
Briefly, 30 µg protein was loaded on a 5 and 10% SDS-PAGE prior to
transfer to polyvinylidene difluoride membranes. Blots were blocked
with 3% bovine serum albumin (Beyotime Institute of Biotechnology)
for detection with phosphoprotein-specific antibodies) or 5%
non-fat milk (for all other antibodies) in Tris-buffered saline
containing 0.1% Tween-20 for 1 h at room temperature, and incubated
with primary antibodies against activin A (cat. no. AF338;
1:10,000; R&D Systems, Inc.), NGF (cat. no. ab52918; 1:400;
Abcam), growth associated protein 43 (GAP43; cat. no. ab75810;
1:100,000; Abcam), tyrosine hydroxylase (TH; cat. no. ab112; 1:200;
Abcam), IL-1β (cat. no. ab9722; 1:1,000, Abcam), TNF-α (cat. no.
ab6671; 1:500; Abcam), phosphorylated IκBα (cat. no. 2859; p-IκBα;
1:1,000; Cell Signaling Technology, Danvers, MA, USA), and
phosphorylated p65 (cat. no. ab86299; p-p65; 1:2,000; Abcam).
Anti-rabbit and rabbit anti-goat horseradish peroxidase
(HRP)-coupled secondary antibodies were used to detect proteins of
interest (cat. no. 7074; 1:2,000; Cell Signaling Technology; and
cat. no. BA1060; 1:1,000; Wuhan Boster Biological Technology, Ltd.,
Wuhan, China; respectively). Blots were developed using an
chemiluminescence reagent (Thermo Fisher Scientific, Inc., Waltham,
MA, USA) and imaged on a Bio-Rad imaging system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Relative intensities of the
protein of interest were normalized to those in the sham group,
which were designated as 100%.
Immunohistochemical analyses
Rats were euthanized and hearts were collected and
embedded in paraffin. Peri-infarct zones were analyzed in cardiac
tissues from the MI and MI+FS groups or the same zone in the sham
group, as previously described (25). Sections were incubated with primary
antibodies to: Activin A; (cat. no. AF338; 1:100; R&D Systems,
Inc.), ED-1 (macrophage-specific marker; cat. no. ab201340; 1:200;
Abcam), α-smooth muscle actin (α-SMA; cat. no. ab5694; 1:100;
Abcam), TH (cat. no. ab112; 1:750; Abcam), and GAP43 (cat. no.
ab75810; 1:500; Abcam). Anti-rabbit/mouse HRP-conjugated secondary
antibodies (cat. nos. 8114 and 8125, respectively; both 1:1,000;
Cell Signaling Technology) were used for immunohistochemical
staining. Imaging was conducted using an Olympus BX51 microscope
(Olympus Corporation, Tokyo, Japan). The number of ED-1-positive
cells per field was measured and expressed as an average number of
cells per mm2. Nerve density
(µm2/mm2) was assessed by calculating TH- and
GAP43-positive nerve areas divided by the total area. All imaging
analysis was done using Image-Pro Plus 6.0 software (Media
Cybernetics, Inc., Rockville, MD, USA).
Activin A ELISA
Blood was collected and serum was analyzed for
activin A using the rat activin A ELISA kit (R&D Systems,
Inc.).
Statistical analysis
All data are presented as mean ± standard deviation.
Differences in mean values between treatment groups were assessed
via one-way analysis of variance using SPSS software (version,
20.0; IBM SPSS, Armonk, NY, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
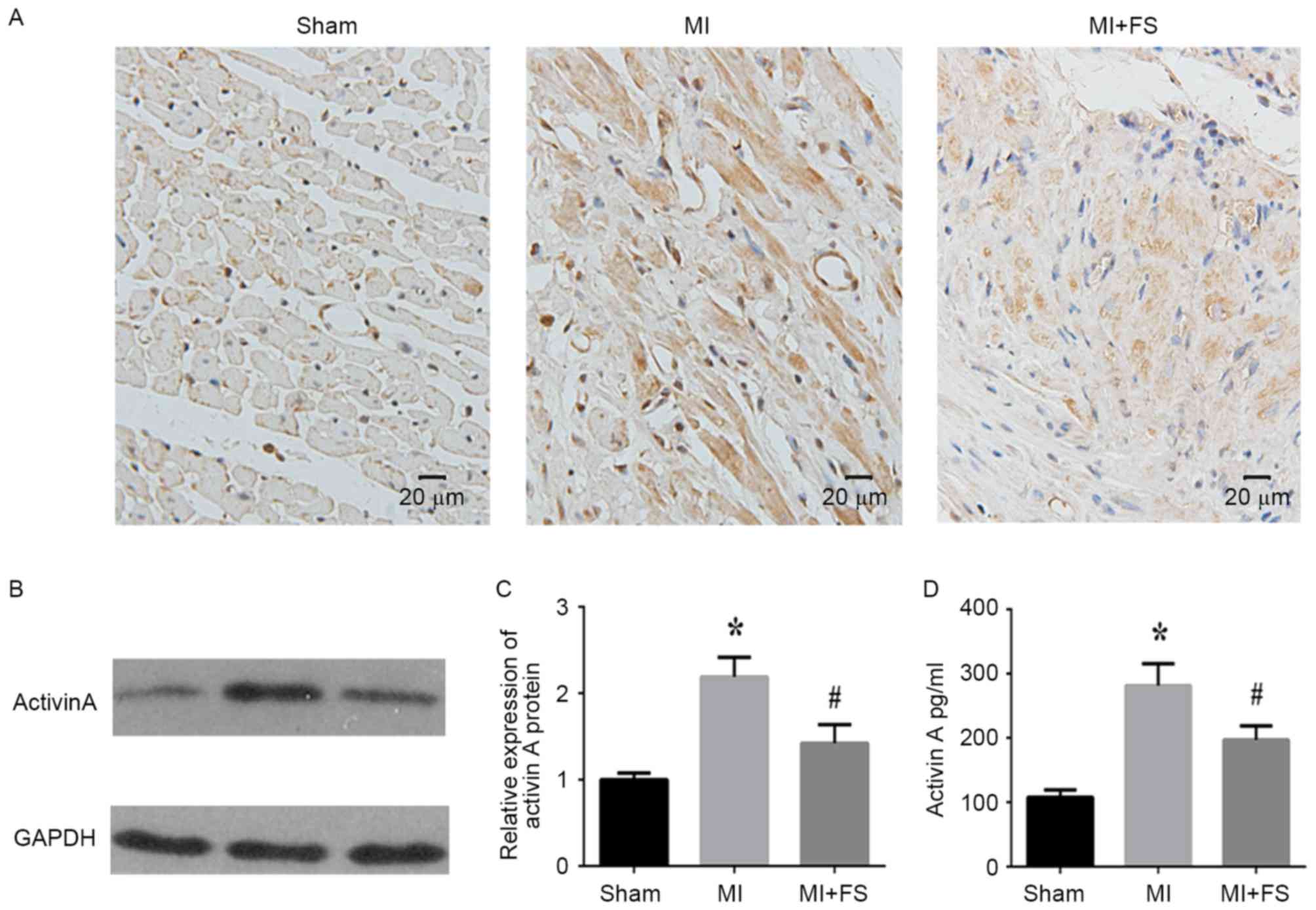
Effect of FS on activin A expression
and production in a rat MI model
To assess the effect of FS, activin A expression was
assessed in cardiac tissues of rats at four weeks post-MI.
Immunohistochemical analyses demonstrated that activin A expression
was higher in the MI when compared with the sham group (Fig. 1A). However, activin A expression
was lower in the MI+FS compared to the MI group (Fig. 1A). Western blotting and ELISA
analyses further validated these findings and showed that activin A
protein expression and serum concentrations were significantly
greater in rats of the MI group compared to the sham group, but
these levels were significantly decreased in rats of the MI+FS vs.
MI group (Fig. 1B-D).
Activin A inhibition downregulated
expression of inflammatory cytokines, NF-κB pathway activation and
inflammatory cell infiltration in rat cardiac tissues post-MI
To determine the effect of activin A inhibition on
the expression of inflammatory cytokines and NF-kB pathway
activation, the authors assessed protein levels of TNF-α, IL-1β,
p-IκBα and p-p65 in the peri-infarct zone of cardiac tissues of
rats at four weeks post-MI. Western blot analyses revealed that
TNF-α, IL1-β1, p-IκBα and p-p65 protein levels were significantly
increased in the MI compared with sham group, and that this
upregulation was significantly attenuated by activin A inhibition
(Fig. 1A-H). Immunohistochemical
analyses demonstrated that the number of infiltrating macrophages
and the proportional area of α-SMA-expressing cells were
significantly increased in the peri-infarct zone in the MI vs. sham
group, and that this upregulation was significantly decreased in
the MI+FS vs. MI group (Fig.
2A-D).
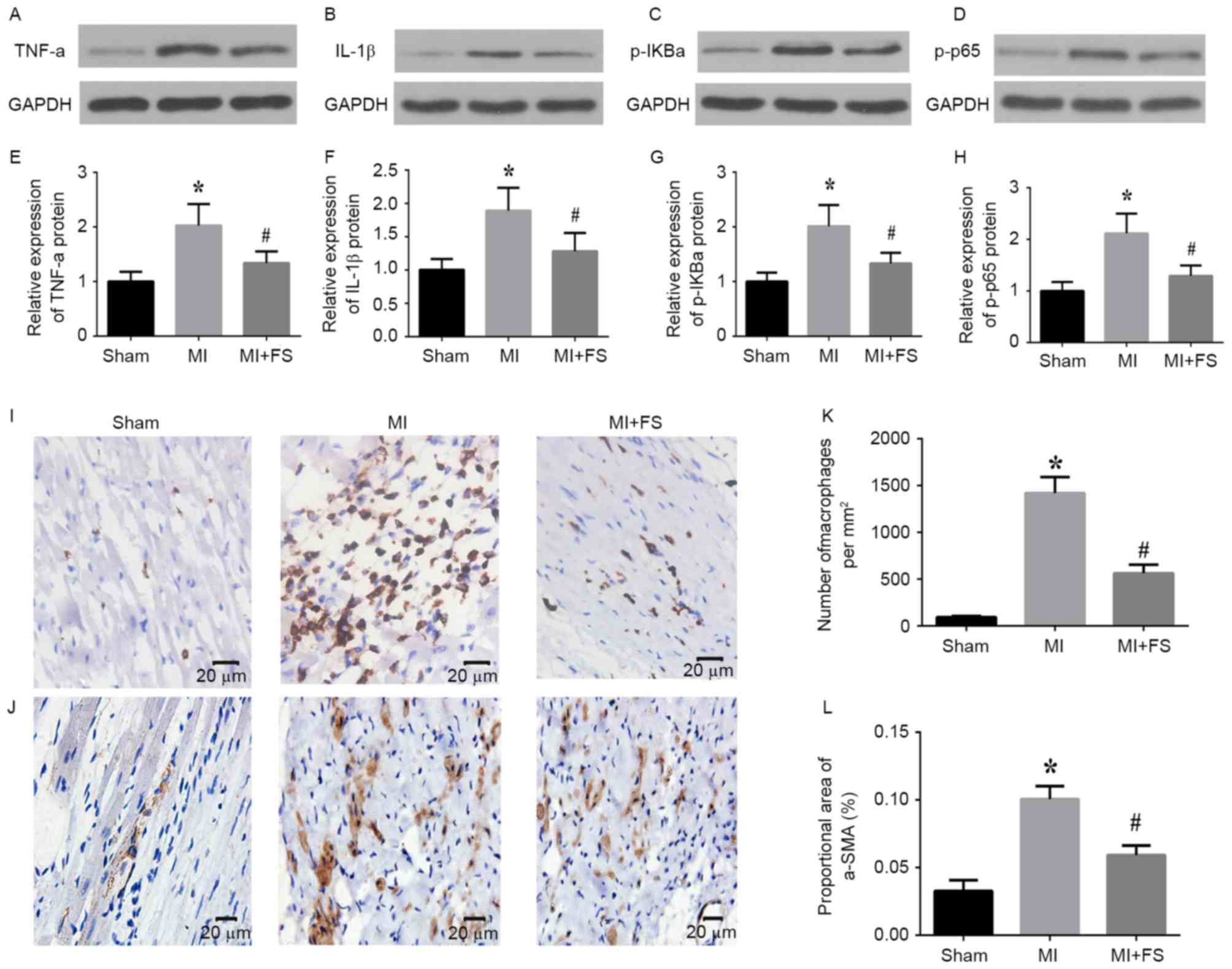 | Figure 2.Effect of activin A inhibition on
expression of inflammatory cytokines, nuclear factor-κB pathway
targets as well as markers of inflammatory cell infiltration in rat
hearts post-MI. (A-D) Representative western blots of TNF-α, IL1-β,
p-IκBα, p-p65 and GAPDH (loading control) protein expression in
peri-infarct zone of rat hearts from sham (left lane), MI (middle
lane), and MI+FS (right lane) groups. (E-H) Quantitative
densitometric analysis of relative TNF-α, IL1-β, p-IκBα and p-p65
protein levels normalized to GAPDH levels in rat heart tissues from
sham, MI, and MI+FS groups. (I and J) Representative
photomicrographs of infiltrating (I) macrophages (ED-1-positive
cells) and (J) myofibroblasts (α-SMA-positive cells) in
peri-infarct zone of rat hearts from sham, MI and MI+FS groups.
Brown color indicates positive expression. Scale bar: 20 µm. (K and
L) Quantitative analysis of the number of macrophages
(ED-1-positive cells) per mm2 and percentage of
myofibroblasts (α-SMA-positive cells; n=6). *P<0.05 vs. sham
group; #P<0.05 vs. MI group. TNF-α; tumor necrosis
factor-α; IL, interleukin; MI, myocardial infarction; FS,
follistatin-300. |
Activin A inhibition downregulated
sympathetic neural remodeling markers in the peri-infarct zone of
rat cardiac tissues post-MI
To determine the effect of activin A inhibition on
sympathetic neural remodeling, the authors assessed GAP43 and TH
expression in the peri-infarct zone of rat cardiac tissues at four
weeks post-MI by immunohistochemical staining. It was observed that
the density of nerve fibers positive for GAP43 and TH was
significantly higher in the MI group than in the sham group,
whereas this increase was reversed via activin A inhibition in the
MI+FS group (Fig. 3A-D). Western
blot analyses validated these findings, indicating that NGF, GAP43
and TH protein levels were considerably increased in the MI group
than in the sham group and that this increase was significantly
decreased in the MI+FS vs. MI group (Fig. 3E-J).
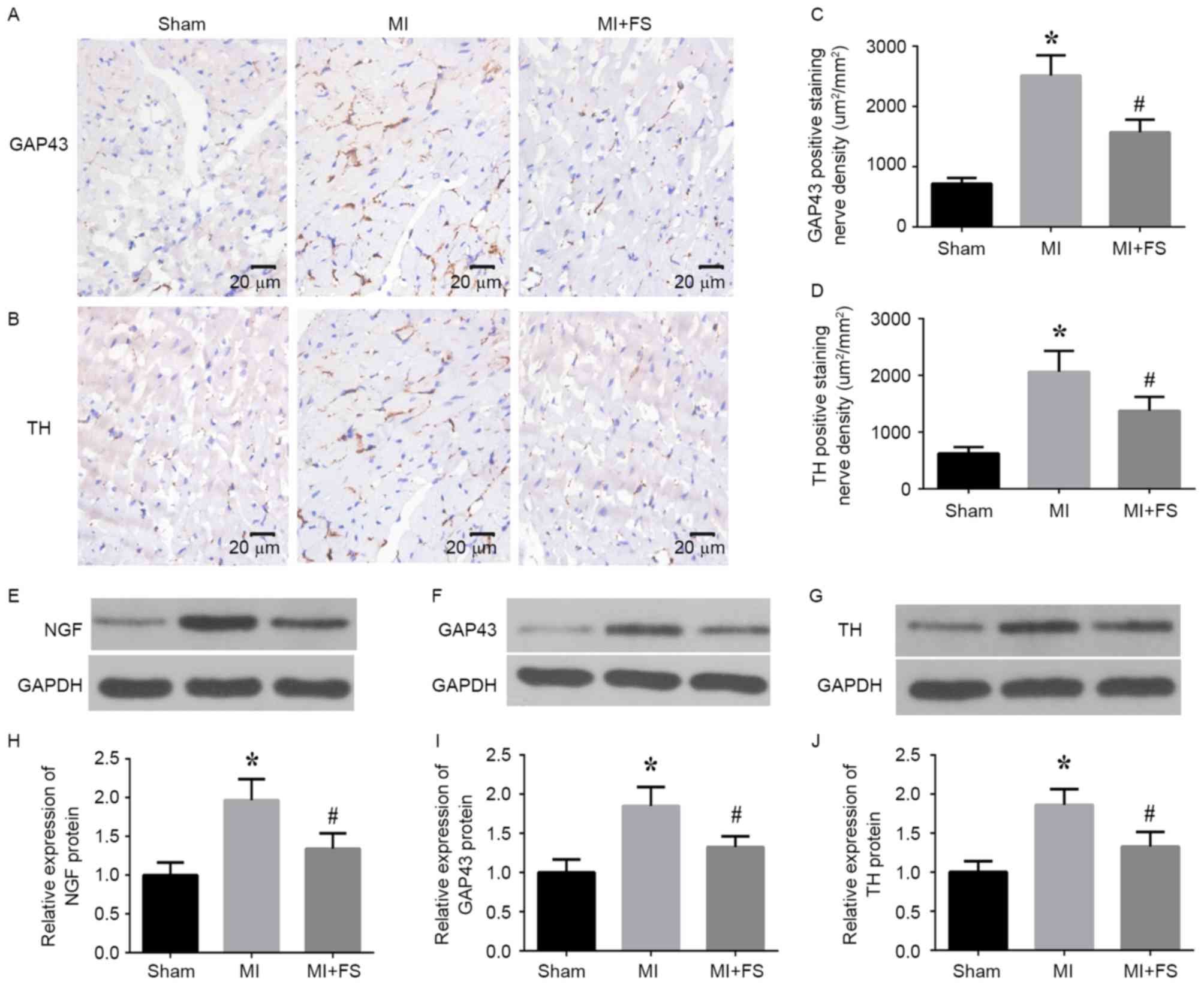 | Figure 3.Effect of FS on NGF, GAP43 and TH
expression in rat hearts post-MI. (A and B) Representative
photomicrographs of GAP43 and TH protein expression at the
peri-infarct border zone in rat heart tissue sections at four weeks
post-MI. Brown color indicates positive expression. Scale bar: 20
µm. (C and D) Quantitative analysis of TH- and GAP43-positive cells
in rat heart tissue sections. (E-G) Representative western blots
for NGF, GAP43, TH and GAPDH (loading control) protein expression
in peri-infarct zone of rat hearts from sham (left lane), MI
(middle lane) and MI+FS (right lane) groups. (H-J) Quantitative
densitometric analysis of relative NGF, GAP43 and TH protein levels
normalized to GAPDH levels in rat heart tissues from sham, MI and
MI+FS groups (n=6). *P<0.05 vs. sham group;
#P<0.05 vs. MI group. Scale bar: 20 µm. MI,
myocardial infarction; FS, follistatin-300; NGF, nerve growth
factor; TH, tyrosine hydroxylase; GAP43, growth associated protein
43. |
Activin A inhibition improved heart
function in rats post-MI
Echocardiography was performed to evaluate cardiac
function in rats four weeks post-MI. The resulting data revealed
that LVEDD and LVESD were significantly increased in the MI group
compared with that sham group, whereas LVEF and LVFS were
significantly decreased in the MI group, when compared with the
sham group. However, inhibition of activin A specifically
ameliorated the changes in these parameters in rats post-MI
(Table I). Notably, heart rate did
not differ among the groups at four weeks post-MI (Table I).
 | Table I.Echocardiographic parameters before
and after FS treatment in a rat MI model. |
Table I.
Echocardiographic parameters before
and after FS treatment in a rat MI model.
| Parameter | Sham | MI | MI+FS |
|---|
| HR (beats/min) |
250±40.27 |
281.4±47.9 |
261±42.24 |
| LVEF (%) |
80±6.69 |
45±4.62a |
54±3.30b |
| LVFS (%) |
44±3.59 |
23±4.19a |
30±2.52b |
| LVEDD (mm) |
6.3±0.44 |
8.0±0.46a |
7.3±0.37b |
| LVESD (mm) |
3.4±0.28 |
5.8±0.58a |
4.0±0.58b |
Discussion
Myocardial necrosis due to MI triggers the
recruitment of inflammatory cells to the site of myocyte loss, and
subsequently promotes secretion and expression of a cascade of
cytokines and chemokines (26).
This inflammation serves an important role in sympathetic neural
remodeling post-MI (5,8). Therefore, blocking the inflammatory
response post-MI may provide a strategy to inhibit sympathetic
neural remodeling. Activin A is a dimeric protein that is
upregulated and associated with inflammation in post-MI heart
failure models (10). FS binds
activin A with high affinity and can regulate endogenous activin A
signaling by inhibiting its interaction with the type II receptor
(27). The authors exploited the
actions of FS as an activin A inhibitor to assess the effect of
activin A inhibition on sympathetic neural remodeling post-MI. The
results revealed that FS can be used as a chemical tool to inhibit
activin A in vivo and inhibition of activin A can
effectively reverse sympathetic neural remodeling by targeting the
inflammatory response in order to improve cardiac function
post-MI.
Activin A is upregulated in the
lipopolysaccharide-induced model of sepsis, and inhibition of
activin A downregulates TNF-α and IL-1β expression as well as
reduces mortality (28). Activin A
also stimulates the production of IL-1β and TNF-α in bone
marrow-derived macrophages (20).
However, whether activin A can impact IL-1β and TNF-α levels
post-MI remains unknown. The present study reported that activin A
inhibition can reduce IL-1β and TNF-α protein expression post-MI,
demonstrating a key role for activin A in targeting inflammatory
mediators during MI. Previous studies have demonstrated that NF-kB
is an important target of inflammatory cytokines induced post-MI
and contributes to the deleterious cardiac remodeling post-MI
(14,29). NF-kB pathways are also direct
regulators of inflammation post-MI (30). Increasing evidence suggests a link
between activin A production and NF-kB activation (12,13,31).
However, whether activin A can impact NF-kB pathway activation
post-MI remains unclear. It was demonstrated that activin A
inhibition can effectively attenuate the activation of NF-kB
targets (p-IκBα and p-p65) in rat hearts post-MI, further
demonstrating a key role for activin A in targeting the
inflammatory response post-MI.
Aberrant sympathetic sprouting is accompanied by
increased NGF expression, which occurs in regions enriched in
inflammatory cells (macrophages and myofibroblasts) within the
peri-infarct zone of the post-MI heart (5). NGF binds to its receptor P75NTR and
activates NF-κB to promote nerve regeneration in Schwan cells
(32), and IL-1 induced by
macrophages also stimulates local NGF production in nerve injury
models (33). Thus, attenuating
inflammatory cell or factors in the peri-infarct zone post-MI may
reduce NGF production, to attenuate aberrant sympathetic nerve
sprouting. Activin A is primarily expressed in
monocytes/macrophages during inflammatory responses (34). It regulates macrophage activation
and function in inflammatory environments (18,19)
and induces inflammatory factors production in monocyte/macrophage
cell lines (20). Additionally,
activin A promotes differentiation of fibroblasts into
myofibroblasts in human lung fibroblasts, primary renal
interstitial fibroblasts and NRK-49F cells (21,22).
The present results indicated that activin A inhibition can reduce
the number of infiltrating macrophages and myofibroblasts as well
as NGF production in the peri-infarct zone. It may be deduced that
activin A inhibition attenuates infiltration of inflammatory cells
and factors post-MI, which may lead to NGF downregulation.
TH- and GAP43-positive nerve fibers are increased
post-MI (35), and upregulation of
both is thought to represent sympathetic nerve remodeling (36). Activin A may directly impact
neuronal cells by inducing neuronal differentiation and survival of
human neuroblastomas (37). The
present study demonstrated that activin A inhibition attenuated the
upregulation of GAP43 and TH expression in rat hearts post-MI.
Possible mechanisms for this reduction may be that inhibition of
activin A suppresses neuronal sprouting, differentiation and
survival to consequently downregulate NGF expression by blocking
the inflammatory response. TH expression can be stimulated by
activin A in combination with basic fibroblast growth factor in
primary neuronal cells and cell lines (38). Activin A can also induce dopamine
beta-hydroxylase gene transcription to promote norepinephrine
secretion (37), suggesting a
direct impact of activin A on sympathetic nerve activity. However,
the specific mechanisms underlying the effects of activin A on
sympathetic neural remodeling remain to be further explored.
Sympathetic neural remodeling plays an important role in heart
dysfunction post-MI, and increased activin A levels correlate with
the degree of heart dysfunction (10). In the current study, activin A
inhibition improved heart function post-MI. The mechanisms for this
improved function may relate to the attenuated sympathetic neural
remodeling response post-MI.
In conclusion, activin A inhibition can attenuate
sympathetic neural remodeling and consequently improve cardiac
function post-MI via inhibition of the inflammatory response. These
findings suggested that activin A is a potential therapeutic target
for sympathetic neural remodeling post-MI.
Acknowledgements
The authors would like to thanks their colleague Rui
Zhao (Wuhan University) for help with data analysis. This work was
funded by the: National Key Basic Research Development Program of
China (The 973 Program, grant no. 2012CB518604), Natural Science
Foundation of China (grant no. 81200139), Fundamental Research
Funds for the Central Universities, China (grant no. 2014302020201)
and Natural Science Foundation of Hubei Province, China (grant no.
2013CFA117). Funders had no role in the study design, data
collection and analysis, decision to publish or preparation of the
manuscript.
References
|
1
|
Solomon SD, Zelenkofske S, McMurray JJ,
Finn PV, Velazquez E, Ertl G, Harsanyi A, Rouleau JL, Maggioni A,
Kober L, et al: Sudden death in patients with myocardial infarction
and left ventricular dysfunction, heart failure, or both. N Engl J
Med. 352:2581–2588. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miyauchi Y, Zhou S, Okuyama Y, Miyauchi M,
Hayashi H, Hamabe A, Fishbein MC, Mandel WJ, Chen LS, Chen PS and
Karagueuzian HS: Altered atrial electrical restitution and
heterogeneous sympathetic hyperinnervation in hearts with chronic
left ventricular myocardial infarction: Implications for atrial
fibrillation. Circulation. 108:360–366. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cao JM, Fishbein MC, Han JB, Lai WW, Lai
AC, Wu TJ, Czer L, Wolf PL, Denton TA, Shintaku IP, et al:
Relationship between regional cardiac hyperinnervation and
ventricular arrhythmia. Circulation. 101:1960–1969. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cao JM, Chen LS, KenKnight BH, Ohara T,
Lee MH, Tsai J, Lai WW, Karagueuzian HS, Wolf PL, Fishbein MC and
Chen PS: Nerve sprouting and sudden cardiac death. Circ Res.
86:816–821. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hasan W, Jama A, Donohue T, Wernli G,
Onyszchuk G, Al-Hafez B, Bilgen M and Smith PG: Sympathetic
hyperinnervation and inflammatory cell NGF synthesis following
myocardial infarction in rats. Brain Res. 1124:142–154. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu T, Zhu W, Gu B, Li S, Wang F, Liu M,
Wei M and Li J: Simvastatin attenuates sympathetic hyperinnervation
to prevent atrial fibrillation during the postmyocardial infarction
remodeling process. J Appl Physiol (1985). 113:1937–1944. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
El-Helou V, Proulx C, Gosselin H, Clement
R, Mimee A, Villeneuve L and Calderone A: Dexamethasone treatment
of post-MI rats attenuates sympathetic innervation of the infarct
region. J Appl Physiol (1985). 104:150–156. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wernli G, Hasan W, Bhattacherjee A, van
Rooijen N and Smith PG: Macrophage depletion suppresses sympathetic
hyperinnervation following myocardial infarction. Basic Res
Cardiol. 104:681–693. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hedger MP and de Kretser DM: The activins
and their binding protein, follistatin-diagnostic and therapeutic
targets in inflammatory disease and fibrosis. Cytokine Growth
Factor Rev. 24:285–295. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yndestad A, Ueland T, Øie E, Florholmen G,
Halvorsen B, Attramadal H, Simonsen S, Frøland SS, Gullestad L,
Christensen G, et al: Elevated levels of activin A in heart
failure: Potential role in myocardial remodeling. Circulation.
109:1379–1385. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
González-Domínguez É, Domínguez-Soto Á,
Nieto C, Flores-Sevilla JL, Pacheco-Blanco M, Campos-Peña V,
Meraz-Ríos MA, Vega MA, Corbí ÁL and Sánchez-Torres C: Atypical
activin A and IL-10 production impairs human CD16+ monocyte
differentiation into anti-inflammatory macrophages. J Immunol.
196:1327–1337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sugatani T, Alvarez UM and Hruska KA:
Activin A stimulates IkappaB-alpha/NFkappaB and RANK expression for
osteoclast differentiation, but not AKT survival pathway in
osteoclast precursors. J Cell Biochem. 90:59–67. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Scicchitano MS, McFarland DC, Tierney LA,
Boyce RW, Frazier KS, Schwartz LW and Thomas HC: Role of p38 in
regulation of hematopoiesis: Effect of p38 inhibition on cytokine
production and transcription factor activity in human bone marrow
stromal cells. Blood Cells Mol Dis. 40:370–380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Onai Y, Suzuki J, Maejima Y, Haraguchi G,
Muto S, Itai A and Isobe M: Inhibition of NF-{kappa}B improves left
ventricular remodeling and cardiac dysfunction after myocardial
infarction. Am J Physiol Heart Circ Physiol. 292:H530–H538. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Levi-Montalcini R: The nerve growth
factor: Its role in growth, differentiation and function of the
sympathetic adrenergic neuron. Prog Brain Res. 45:235–258. 1976.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Glebova NO and Ginty DD: Heterogeneous
requirement of NGF for sympathetic target innervation in vivo. J
Neurosci. 24:743–751. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hassankhani A, Steinhelper ME, Soonpaa MH,
Katz EB, Taylor DA, Andrade-Rozental A, Factor SM, Steinberg JJ,
Field LJ and Federoff HJ: Overexpression of NGF within the heart of
transgenic mice causes hyperinnervation, cardiac enlargement, and
hyperplasia of ectopic cells. Dev Biol. 169:309–321. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Soler Palacios B, Estrada-Capetillo L,
Izquierdo E, Criado G, Nieto C, Municio C, González-Alvaro I,
Sánchez-Mateos P, Pablos JL, Corbí AL and Puig-Kröger A:
Macrophages from the synovium of active rheumatoid arthritis
exhibit an activin A-dependent pro-inflammatory profile. J Pathol.
235:515–526. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Samaniego R, Palacios BS, Domiguez-Soto Á,
Vidal C, Salas A, Matsuyama T, Sánchez-Torres C, de la Torre I,
Miranda-Carús ME, Sánchez-Mateos P and Puig-Kröger A: Macrophage
uptake and accumulation of folates are polarization-dependent in
vitro and in vivo and are regulated by activin A. J Leukoc Biol.
95:797–808. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nüsing RM and Barsig J: Induction of
prostanoid, nitric oxide, and cytokine formation in rat bone marrow
derived macrophages by activin A. Br J Pharmacol. 127:919–926.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yamashita S, Maeshima A, Kojima I and
Nojima Y: Activin A is a potent activator of renal interstitial
fibroblasts. J Am Soc Nephrol. 15:91–101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ohga E, Matsuse T, Teramoto S, Katayama H,
Nagase T, Fukuchi Y and Ouchi Y: Effects of activin A on
proliferation and differentiation of human lung fibroblasts.
Biochem Biophys Res Commun. 228:391–396. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, Wang S, Wier WG, Zhang Q, Jiang H,
Li Q, Chen S, Tian Z, Li Y, Yu X, et al: Exercise improves the
dilatation function of mesenteric arteries in postmyocardial
infarction rats via a PI3K/Akt/eNOS pathway-mediated mechanism. Am
J Physiol Heart Circ Physiol. 299:H2097–H2106. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maeshima A, Mishima K, Yamashita S,
Nakasatomi M, Miya M, Sakurai N, Sakairi T, Ikeuchi H, Hiromura K,
Hasegawa Y, et al: Follistatin, an activin antagonist, ameliorates
renal interstitial fibrosis in a rat model of unilateral ureteral
obstruction. Biomed Res Int. 2014:3761912014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mabe AM, Hoard JL, Duffourc MM and Hoover
DB: Localization of cholinergic innervation and neurturin receptors
in adult mouse heart and expression of the neurturin gene. Cell
Tissue Res. 326:57–67. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Frangogiannis NG: Regulation of the
inflammatory response in cardiac repair. Circ Res. 110:159–173.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Harrison CA, Gray PC, Vale WW and
Robertson DM: Antagonists of activin signaling: Mechanisms and
potential biological applications. Trends Endocrinol Metab.
16:73–78. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jones KL, Mansell A, Patella S, Scott BJ,
Hedger MP, de Kretser DM and Phillips DJ: Activin A is a critical
component of the inflammatory response, and its binding protein,
follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci
USA. 104:pp. 16239–16244. 2007; View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kumar R, Yong QC and Thomas CM: Do
multiple nuclear factor kappa B activation mechanisms explain its
varied effects in the heart? Ochsner J. 13:157–165. 2013.PubMed/NCBI
|
|
30
|
Brown MA and Jones WK: NF-kappaB action in
sepsis: The innate immune system and the heart. Front Biosci.
9:1201–1217. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fukushima N, Matsuura K, Akazawa H, Honda
A, Nagai T, Takahashi T, Seki A, Murasaki KM, Shimizu T, Okano T,
et al: A crucial role of activin A-mediated growth hormone
suppression in mouse and human heart failure. PLoS One.
6:e279012011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Carter BD, Kaltschmidt C, Kaltschmidt B,
Offenhäuser N, Böhm-Matthaei R, Baeuerle PA and Barde YA: Selective
activation of NF-kappa B by nerve growth factor through the
neurotrophin receptor p75. Science. 272:542–545. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lindholm D, Heumann R, Meyer M and Thoenen
H: Interleukin-1 regulates synthesis of nerve growth factor in
non-neuronal cells of rat sciatic nerve. Nature. 330:658–659. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Phillips DJ, Jones KL, Scheerlinck JY,
Hedger MP and de Kretser DM: Evidence for activin A and follistatin
involvement in the systemic inflammatory response. Mol Cell
Endocrinol. 180:155–162. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou S, Chen LS, Miyauchi Y, Miyauchi M,
Kar S, Kangavari S, Fishbein MC, Sharifi B and Chen PS: Mechanisms
of cardiac nerve sprouting after myocardial infarction in dogs.
Circ Res. 95:76–83. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu X, Jiang H, Yu L, Hu X and Liu W:
Desipramine pretreatment improves sympathetic remodeling and
ventricular fibrillation threshold after myocardial ischemia. J
Biomed Biotechnol. 2012:7329092012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Suzuki K, Kobayashi T, Funatsu O, Morita A
and Ikekita M: Activin A induces neuronal differentiation and
survival via ALK4 in a SMAD-independent manner in a subpopulation
of human neuroblastomas. Biochem Biophys Res Commun. 394:639–645.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bao YL, Tsuchida K, Liu B, Kurisaki A,
Matsuzaki T and Sugino H: Synergistic activity of activin A and
basic fibroblast growth factor on tyrosine hydroxylase expression
through Smad3 and ERK1/ERK2 MAPK signaling pathways. J Endocrinol.
184:493–504. 2005. View Article : Google Scholar : PubMed/NCBI
|