Introduction
Epilepsy is a disease caused by the synchronous
abnormal discharge of intracranial neurons, it is predominantly
characterized by Convulsive status epilepticus (1). The pathology of epilepsy is a complex
process, including genetic factors, brain diseases including
intracranial tumor, intracranial infection, craniocerebral injury,
cerebrovascular disease, and systemic diseases, such as anoxia,
metabolic disease and endocrine disease (2). Epilepsy is a life-threatening
neurological disease that affects many people (3,4).
Surgery can sometimes result in positive outcomes for patients with
drug-resistant seizures (5);
however, sometimes completely removing lesions is a problem
(6).
A previous study observed that mutational hot spots
in the transmembrane domains of calcium voltage-gated channel
subunit alpha1 and ATPase/Na+/K+ transporting subunit alpha 2
proteins were highly associated with epilepsy in hemiplegic
migraine (7). Brain inflammation
is considered serve an important role in epilepsy (8). The pro-inflammatory C5a receptor,
C5ar1, represents a novel target for improved anti-epileptic drug
development, which may be beneficial for pharmaco-resistant
patients (9).
Inflammatory processes in the central nervous system
(CNS) have been reported to serve a vital role in human epilepsy
and in experimental models of seizures (10). CNS inflammation is characterized by
a disturbance of glial cell functions: A previous study has
demonstrated the direct effects of several antiepileptic drugs
(AEDs) on glial viability through detecting the gap junctional
network, microglial activation and cytokine expression in an in
vitro astroglia/microglia co-culture model (11). Anti-inflammatory therapies both in
clinical and in experimental settings obtained a good therapeutic
result. All the above highlight the important role of brain
inflammation in the aetiopathogenesis of seizures. Certain
components of the inflammatory gene network might contribute to the
process (12).
Glia serve a pivotal role in the initiation and
maintenance of the CNS immune response, and it involves in defining
central nervous system architecture, brain metabolism, and the
survival of neurons, development and modulation of synaptic
transmission, propagation of nerve impulses, and many other
physiological functions (13). In
the recently years, the contribution of glial cells, mainly
astrocytes and microglia, to the pathophysiology of epilepsy is
increasingly appreciated (11,14).
Microglia are the major immune cells in the brain,
and serve an immune monitoring function. They have been
traditionally studied in various contexts of disease, and their
activation has been assumed to induce mostly detrimental effects
(15). Microglia-driven epilepsy
may be a primary pathogenic process; experimental and clinical
studies support a pathogenic role of microglial activation and
proliferation in epileptogenesis (16). Previous studies have demonstrated
that microglia activated by recurrent seizures continuously secrete
a variety of immune effector molecules, such as interleukin (IL)-1,
IL-6, IL-8, tumor necrosis factor (TNF)-α, transforming growth
factor (TGF)-β, and oxygen free radicals, such as reactive oxygen
species, which can damage other neurons, glial cells and the blood
brain barrier, causing local or extensive injury of central nervous
system, aggravate epilepsy and cause seizures (16,17).
High-mobility group box 1 (HMGB1) is a nuclear
protein with cytokine-type functions upon its extracellular
release. HMGB1 activates inflammatory pathways by stimulating
multiple receptors, chiefly toll-like receptor 4 (TLR4) and
receptor for advanced glycation end products (RAGE) (18). A study on a model of renal ischemic
reperfusion injury (IRI) demonstrated that the HMGB1-TLR4
inflammatory signaling pathway was inhibited by dexamethasone
treatment via the attenuated translocation of HMGB1 from the
nucleus to the cytoplasm, and the down-regulation of TLR4
expression (19). Quercetin has
hepatoprotective and anti-fibrotic effects to liver fibrosis
through modulating the HMGB1-TLR2/4-nuclear factor (NF)-κB
signaling pathways (20).
Experimental models of seizures and temporal lobe epilepsy verified
that hyperexcitability of HMGB1 and TLR4 signaling leads to the
development and perpetuation of seizures (21). IL-1 type 1 receptor/TLR, which can
be activated by proinflammatory cytokines such as HMGB1, serves a
key role in seizures (22). HMGB1
contributes to the overexpression of P-glycoprotein in mouse
epileptic brain tissues via activation of TLR4/RAGE receptors and
the downstream transcription factor NF-κB in brain microvascular
endothelial cells (23).
The present study aimed to explore a novel way for
the treatment of intractable epilepsy by targeting microglia and
blocking the associated inflammatory signaling pathways, receiving
the effect of reducing nerve injury and cytotoxic substance
release.
Materials and methods
Specimen collection
The present study was ethically approved by The
First Affiliated Hospital of Zhengzhou University (Zhengzhou,
China) and written informed consent was obtained from every
participant. Brain tissue was collected post-surgery from both
patients with intractable epilepsy (EP group) after resection, and
the control brain tissue of patients with craniocerebral trauma
induced intracranial hypertension. All brain tissue was frozen in
liquid nitrogen.
Immunohistochemical staining
(IHC)
IHC was performed as previous described (5). Brain tissue sections (5 µm) for
immunohistochemical analysis were fixed in 4% paraformaldehyde
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and
paraffin-embedded. Sections of the brain regions were selected for
the IHC study following fixing of the sections with acetone.
Briefly, slides were incubated with 0.03%
H2O2 in PBS for 10 min at room temperature,
then incubated in 5% normal goat serum in PBS for 15 min. Sections
were then incubated with primary antibodies: Anti-HMGB1 (cat. no.
ab18256, 1:100; Abcam, Cambridge, UK), anti-NF-κB p65 (cat. no.
ab16502, 1:5,000; Abcam) and anti-OX42 (cat. no. ab1211, 1:500;
Abcam) in 3% bovine albumin serum in PBS at 4°C overnight.
Immunoreactivity was detected using avidin-biotin-peroxidase
technique; sections were incubated with 3, 3′-diaminobenzidine as
the chromogen for 20 min at 37°C. Images were acquired using an
upright optical microscope (DM4000; Leica Microsystems, Mannheim,
Germany). Prior to mounting, the cell nuclei were counterstained by
Hoechst 33342 fluorescent dye (1:1,000; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA USA) for 10 min at 37°C. Samples
without the addition of a primary antibody were used as the
negative control. For each field, the number of positively stained
capillaries were counted under a light microscope and the
percentage of positive cells was calculated from the average value
of five fields.
Western blotting
Total protein was extracted from brain tissue or
cultured cells in different groups as described previously
(24). Protein concentration was
determined using a bicinchoninic acid protein assay kit. Proteins
(30 µg/lane) were separated using 10% SDS-PAGE and transferred onto
polyvinylidene difluoride membranes. The membranes were incubated
overnight with the following primary antibodies: Anti-HMGB1 (cat.
no. ab18256; 1:100; Abcam), anti-TLR4 (cat. no. 293072; 1:1,000;
Santa Cruz, Biotechnology, Inc., Dallas, TX, USA), anti-RAGE (cat.
no. AB15323; 1:1,000; Santa Cruz Biotechnology, Inc.), anti-NF-κB
p65 (cat. no. ab16502; 1:5,000; Abcam), anti-inducible nitric oxide
synthase (cat. no. 49055; iNOS; 1:200; Abcam) and anti-β-actin
(cat. no. A1978; 1:50,000; Sigma-Aldrich; Merck KGaA) at 4°C.
Subsequently, the membranes were incubated with horseradish
peroxidase (HRP)-conjugated anti-rabbit IgG secondary antibody
(cat. no. 6721; 1:5,000; Abcam) at 37°C for 30 min and exposed to
X-ray film using an enhanced chemiluminescence system (Thermo
Fisher Scientific, Inc.). The intensity of the bands was measured
using Lab-works (version 4.0; Ultra-Violet Products Ltd.,
Cambridge, UK).
Reagents and cell lines
Human microglia (HM) cells were obtained from Dr.
Dai Antibody's laboratory (Case Western Reserve University,
Cleveland, OH, USA) and were cultured as described previously
(25). HM cells were cultured in
Dulbecco's modified Eagle's medium supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin-G, 100 µg/ml streptomycin and 0.01 M Hepes buffer
(Thermo Fisher Scientific, Inc.) in a humidified 5% CO2
atmosphere at 37°C.
HM microglia were stimulated with epileptic agent
coriaria lactone (CL), and the expression levels of HMGB1, TLR4,
RAGE and p-NF-κB p65 were detected by western blotting after
collecting cells.
Plasmid transfection
Full-length human HMGB1 cDNA was amplified by
polymerase chain reaction from pCMV4-RelA plasmid (Shanghai
GenePharma Co., Ltd., Shanghai, China) using the forward primer
5′-GGTCGGTACCATGGACGAACTGTTCCCCCT-3′ and the reverse primer
5′-CCATCTCGAGTTAGGAGCTGATCTGACTCA-3′, inserted into a pcDNA3.1
vector (Shanghai GenePharma Co., Ltd.) tagged with FLAG. HMGB1
small interfering (si)RNA (HMGB1-KD) and its control siRNA were
purchased from Santa Cruz Biotechnology, Inc. Transient
transfection of HM cells with pcDNA3.1/HMGB1 cDNA or control
pcDNA3.1, and its control siRNA was carried out using Lipofectamine
2000 reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol.
Enzyme-linked immunosorbent assay
(ELISA)
The expression levels of cytokines IL-1 (cat. no.
68616), IL-6 (cat. no. 68834), TNF-α (cat. no. 55845), TGF-β (cat.
no. 31366) and IL-10 (cat. no. 68540) in the cell culture
supernatant were detected by ELISA (MLBIO; Shanghai Enzyme-linked
Biotechnology Co., Ltd., Shanghai, China). A 96-well plate was
coated with the primary monoclonal antibody of IL-1, IL-6, TNF-α,
TGF-β or IL-10. Standards and supernatant samples were added to the
wells. Following washing with 5% non-fat dry milk in PBS with 0.05%
Tween-20 (Sigma-Aldrich; Merck KGaA), the wells were incubated with
HRP-conjugated anti-rabbit IgG antibody (cat. no. 6721; 1:1,000;
Abcam) for 10 min at 37°C. The plate was washed again with the
aforementioned solution, followed by incubation with
tetramethylbenzidene solution for 15 min at 37°C. This reaction was
stopped by adding hydrochloride solution. Optical density was
measured by using a microplate reader (Bio-Rad 550; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) set to 450 nm.
Cytotoxicity/cell viability assay
The potential toxic effect of HMGB1 on HM cells was
analyzed using the 3-(4,
5-dimethylthylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
method. Cells were seeded into 96-well plates at a concentration of
8×103 cells per well. After being grown for 24 h, cells
were incubated at 37°C with either HMGB1 (10, 100 and 500 ng/ml) or
untreated control medium (PBS), respectively, for different periods
of time (4, 8, 16 and 24 h). Subsequently, 50 µl MTT tetrazolium
salt (Sigma-Aldrich; Merck KGaA) was diluted with PBS to a
concentration of 5 mg/ml, then the mixture was added to each well
and incubated for another 2 h at 37°C. The medium was then
aspirated from each well and 200 µl dimethyl sulfoxide was added to
dissolve the formazan crystals. The resulting solution (150 µl) was
transferred to a 96-well plate and the absorbance of each well was
determined using a Tecan infinite M200 Pro plate reader at a
wavelength of 570 nm. Each experiment was replicated three times.
The potential toxic effect of CL on HM cells was also analyzed with
an MTT assay.
5-ethynyl-2-deoxyuridine (EdU)
staining
The EdU assay is a method of detecting proliferating
cells with the advantage of being simple and convenient, avoiding
DNA denaturation and maintaining the shape of the sample. It has
replaced the traditional method of bromodeoxyuridine staining. It
is a method of detection of DNA synthesis in proliferating cells
and relies on the incorporation of labeled DNA precursors into
cellular DNA during the S phase of the cell cycle (1). EdU staining was conducted using
Cell-Light™ EdU kit (Guangzhou RiboBio Co., Ltd.,
Guangzhou, China), according to the manufacturer's protocol.
Paraffin sections were de-paraffinized in xylene for 10 min twice,
rinsed in an alcohol gradient (100, 95, 85%) for 10 min, and rinsed
in deionized water for 5 min. After washing with 2 mg/ml glycine
solution diluted in double distilled water for 10 min, the sections
were permeabilized with 0.5% Triton X-100 in PBS for 20 min, and
then washed twice with PBS for 10 min. The Apollo reaction buffer
liquid, catalyst, fluorescent dyes and buffer additives (Guangzhou
RiboBio Co., Ltd.) were dissolved in deionized water, and shaken to
make the Apollo 567 staining reaction solution (Guangzhou RiboBio
Co., Ltd.). The sections were then incubated for 30 min without
light. The sections were washed twice with PBS for 10 min at 37°C.
For subsequent DNA staining, sections were counterstained with
Hoechst 33342 for 30 min away from light at 37°C. The slides were
then washed twice with PBS for 3 min, and observed immediately
under a fluorescent microscope (magnification, ×400). All the
procedures were conducted at room temperature. EdU-positive cells
were detected by an Olympus BX51 microscope (Olympus Corporation,
Tokyo, Japan). Images of the ApolloW 567 were captured with a ‘red’
filter set: Excitation, 550 nm; emission, 565 nm; filter, 555±15
nm. Images of the Hoechst 33342 were captured with a ‘blue’ filter
set: Excitation, 350 nm; emission, 461 nm; filter, 405±15 nm.
Statistical analysis
All data are expressed as the mean ± standard
deviation. The statistical analyses were performed using GraphPad
Prism 6.0 software (GraphPad Prism Inc., USA). Comparison between
two groups was performed by unpaired Student's t-test. Comparison
among multiple groups was performed by one-way analysis of variance
with a least significant difference post-hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
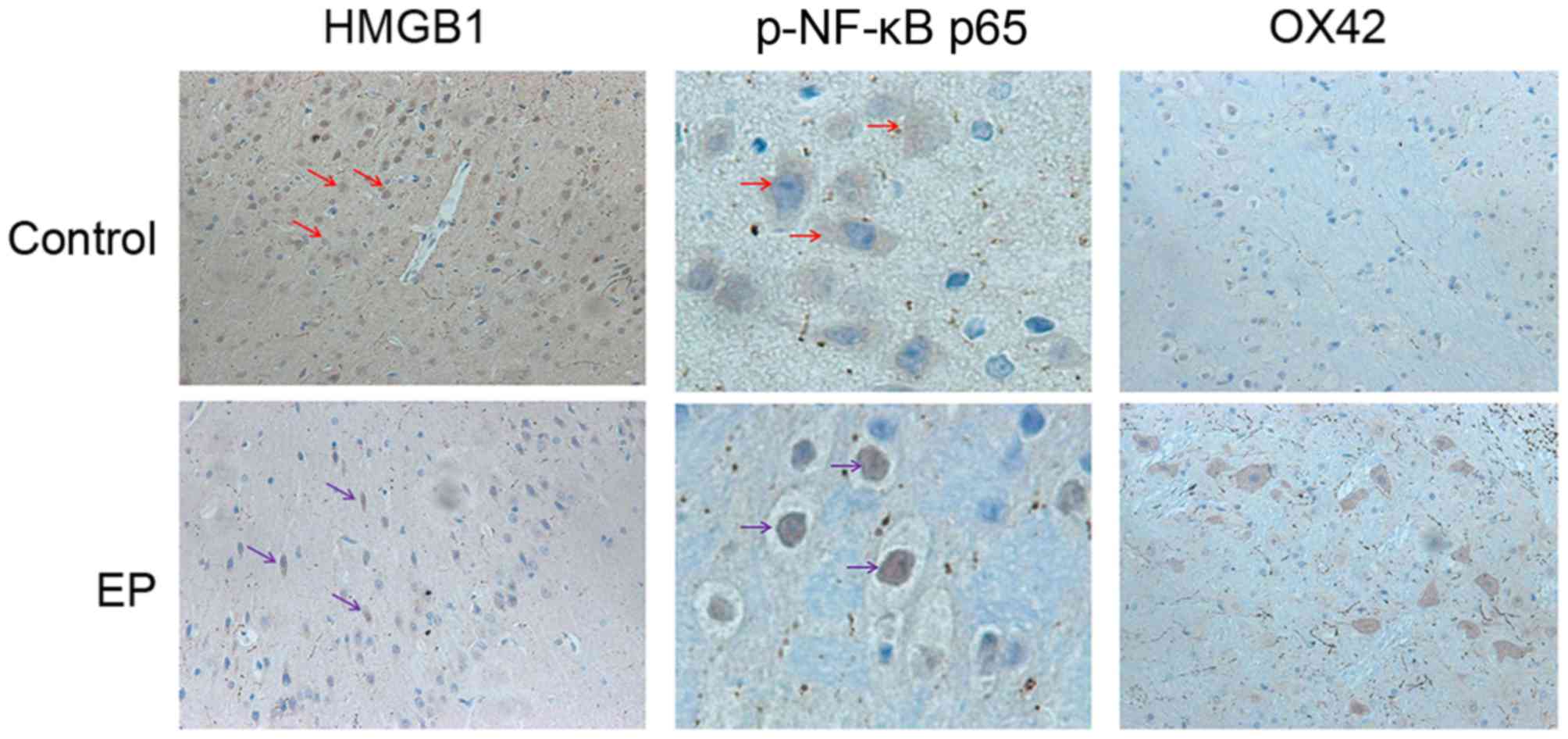
Expression and distribution of HMGB1,
NF-κB p65 and OX42 in the epileptic brain tissues of patients with
refractory epilepsy brain tissue
To explore the expression level and changes in the
distribution pattern of HMGB1 in epileptic brain tissues, resection
of brain tissue after surgery was collected. The distribution of
HMGB1, NF-κB p65 and OX42 in the control and EP group was observed
using IHC. In the control group, HMGB1 immunoreactivity was
detected mostly in the nuclei of the glial cells and pyramidal
neurons. In the EP group, cytoplasmic staining of HMGB1 was
significantly increased in the pyramidal neurons (Fig. 1). Next to glial cells with nuclear
staining, glial cells with both nuclear and cytoplasmic staining
were also present. These results indicated the nuclear to
cytoplasmic translocation of HMGB1 in neurons and glial cells, and
predicted its subsequent release into the extracellular space.
Higher expression levels of NF-κB p65 were detected in the EP group
in comparison with the control group. Positive NF-κB p65 staining
was detected mainly in the cell nucleus in the EP group, suggesting
the cytoplasmic to nuclear translocation of NF-κB p65 in neurons
and glial cells. Next, IHC staining of the brain tissues of
patients was conducted, using a marker for reactive microglia
(OX42). The results reflected that the cell density of the two was
similar.
Comparing the level of HMGB1, TLR4,
RAGE, NF-κB p65 and iNOS in the brain tissues of the control and
the EP groups
To investigate the expression level of HMGB1, TLR4,
RAGE, NF-κB p65 and iNOS in the control and the EP brain tissues,
western blotting was performed. From the result, HMGB1 was notably
increased, and the TLR4 and RAGE signaling pathway was markedly
activated; their expression levels were both significantly
increased. Both genetic and biochemical data support that TLR4 and
RAGE signaling pathway could eventually lead to the activation of
NF-κB (23). Thus, in the present
study, upregulation of NF-κB was observed (Fig. 2).
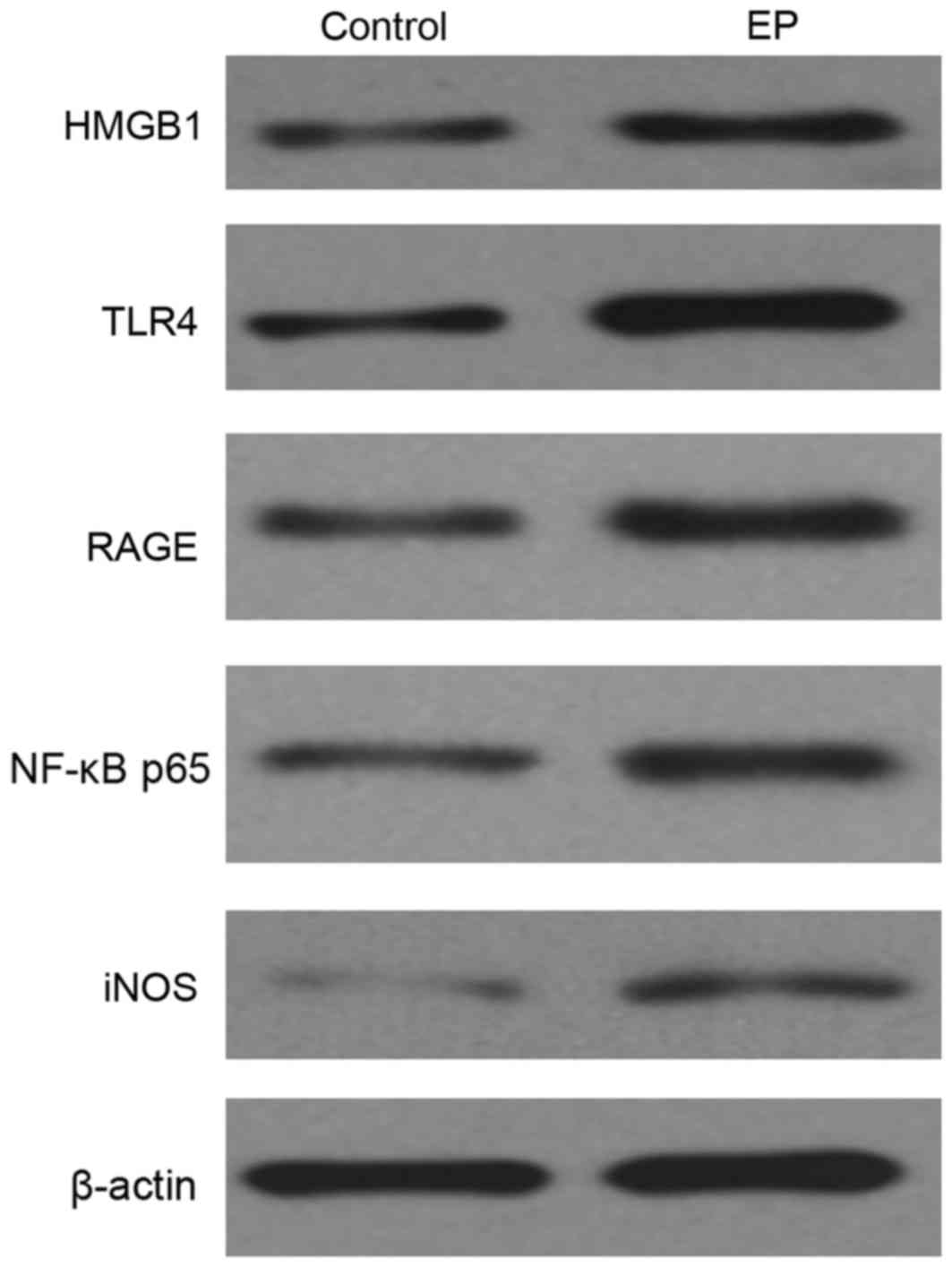 | Figure 2.Expression level of HMGB1, TLR4, RAGE,
NF-κB p65 and iNOS in the brain tissues of the control and the EP
group, as detected by western blotting. HMGB1, high-mobility group
box-1; NF-κB, nuclear factor-κB; EP, refractory epilepsy group;
TLR4, toll-like receptor 4; RAGE, receptor for advanced glycation
end products; iNOS, inducible nitric oxide synthase. |
Comparing the level of cytokines of IL-1, IL-6,
TNF-α, TGF-β and IL-10 in the cell supernatant of the control and
the CL-induced epileptic cell model. To determine the levels of
cytokines of IL-1, IL-6, TNF-α, TGF-β and IL-10 in the cell
supernatant of the control and the CL-induced epileptic cell model,
ELISA was used to determine the concentration of each cytokine.
From the result (Fig. 3), CL
effectively enhanced the level of the all the above cytokines, and
induced overexpression of HMGB1.
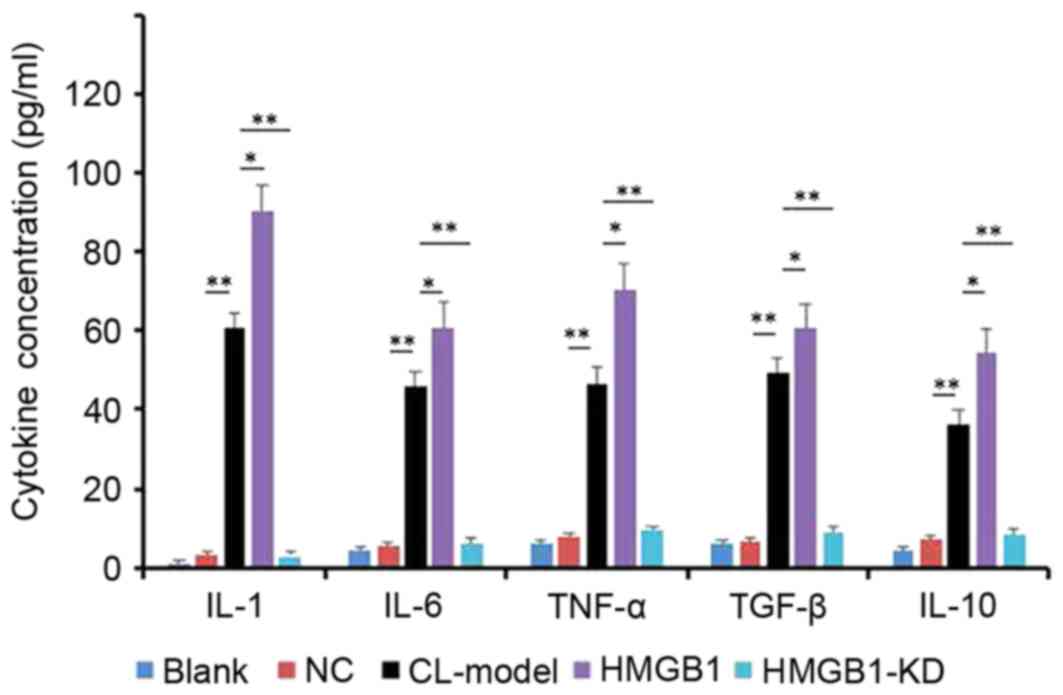 | Figure 3.Levels of cytokines, as detected by
ELISA. HM cells were induced by CL, then transfected with HMGB1
mimic and HMGB1-KD. Data are presented as the mean ± standard
deviation. *P<0.05, **P<0.01. HMGB1, high-mobility group
box-1; NC, negative control; IL, interleukin; TNF, tumor necrosis
factor; TGF, transforming growth factor; CL-Model, cariaria lactone
induced epilepsy cell model; CL-HM, cariaria lactone induced
epilepsy cell model plus HMGB1; CL-HM-KD, cariaria lactone induced
epilepsy cell model plus HMGB1 siRNA; siRNA, small interfering RNA;
HMGB1-KD, HMGB1 siRNA. |
Expression of HMGB1, TLR4, RAGE and
NF-κB p65 in HM cells
Western blotting was used to detect expression of
HMGB1, TLR4, RAGE and NF-κB p65 in HM cells. From the result
(Fig. 4A), CL appeared to increase
the levels of HMGB1, TLR4, RAGE, NF-κB p65 and iNOS in HM cells;
overexpression of HMGB1 enhanced this result (Fig. 4B), and inhibition of HMGB1 with
HMGB1-KD blocked the function of CL to HM cells.
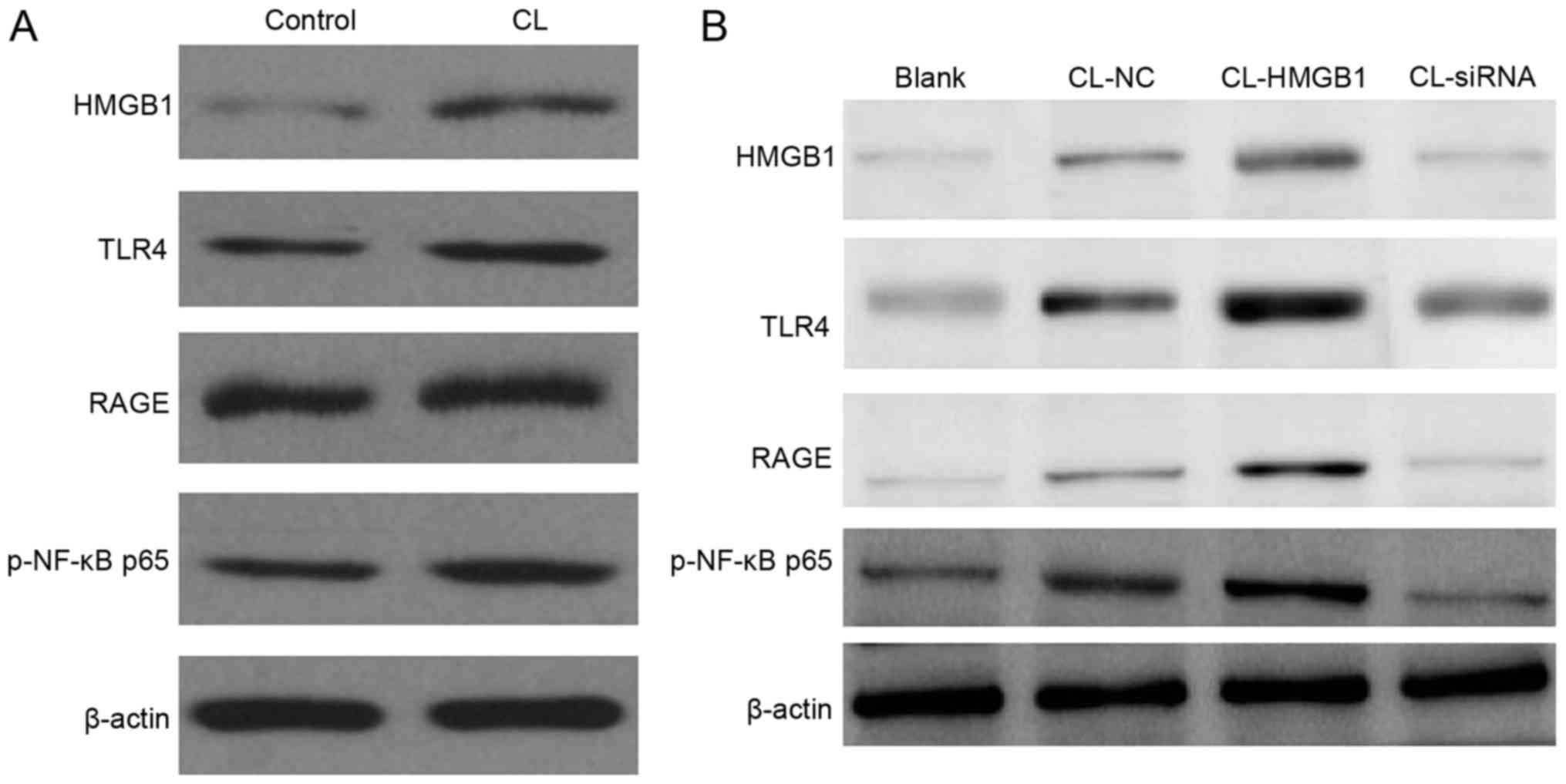 | Figure 4.Western blotting of protein expression
levels in HM cells. (A) CL significantly increases the levels of
HMGB1, TLR4, RAGE, NF-κB p65 and iNOS in HM cells. (B)
Overexpression of HMGB1 this increased further, and inhibition of
HMGB1 by siRNA blocked the function of CL to HM cells. HMGB1,
high-mobility group box-1; NF-κB, nuclear factor-κB; EP, refractory
epilepsy group; TLR4, toll-like receptor 4; RAGE, receptor for
advanced glycation end products; iNOS, inducible nitric oxide
synthase; p-phosphorylated; siRNA, small interfering RNA; NC,
negative control; CL, cariaria lactone. |
HMGB1 had no significant effect on the
viability of HM cells
As presented in Fig.
5, the overexpression of HMGB1 did not significantly decrease
the viability of HM cells at any tested concentrations or time
points, compared with the control cells. Additionally, incubation
with CL followed by overexpression also had no effect on the
viability of HM cells.
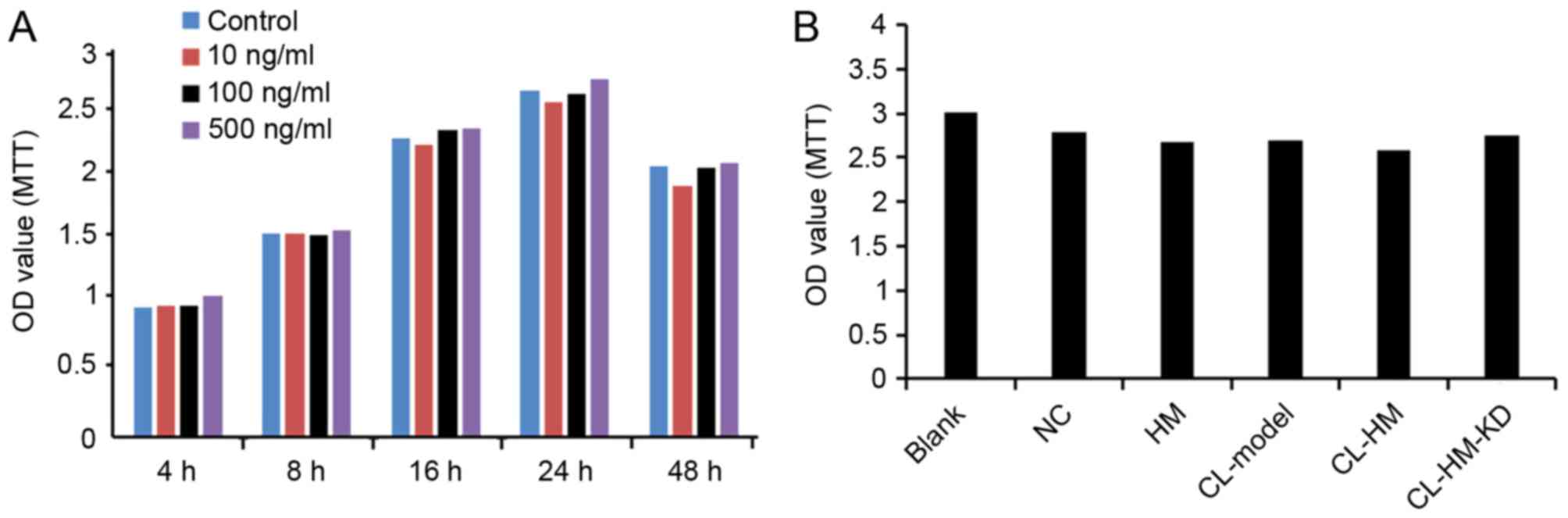 | Figure 5.HMGB1 has no significant effect on
viability of HM cells. (A) Cells were transfected with different
concentrations of HMGB1 for the indicated time periods. Cell
viability was determined using MTT assay. (B) Comparison of cell
viability in each group of Blank, NC, HM, CL-Model, CL-HM and
CL-HM-KD. Data from three independent experiments are presented as
the mean ± standard deviation (n=3). HMGB1, high-mobility group
box-1; Blank, blank group; NC, negative control group; HM, HMGB1;
CL-Model, cariaria lactone induced epilepsy cell model; CL-HM,
cariaria lactone induced epilepsy cell model plus HMGB1; and
CL-HM-KD, cariaria lactone induced epilepsy cell model plus HMGB1
siRNA; OD, optical density. |
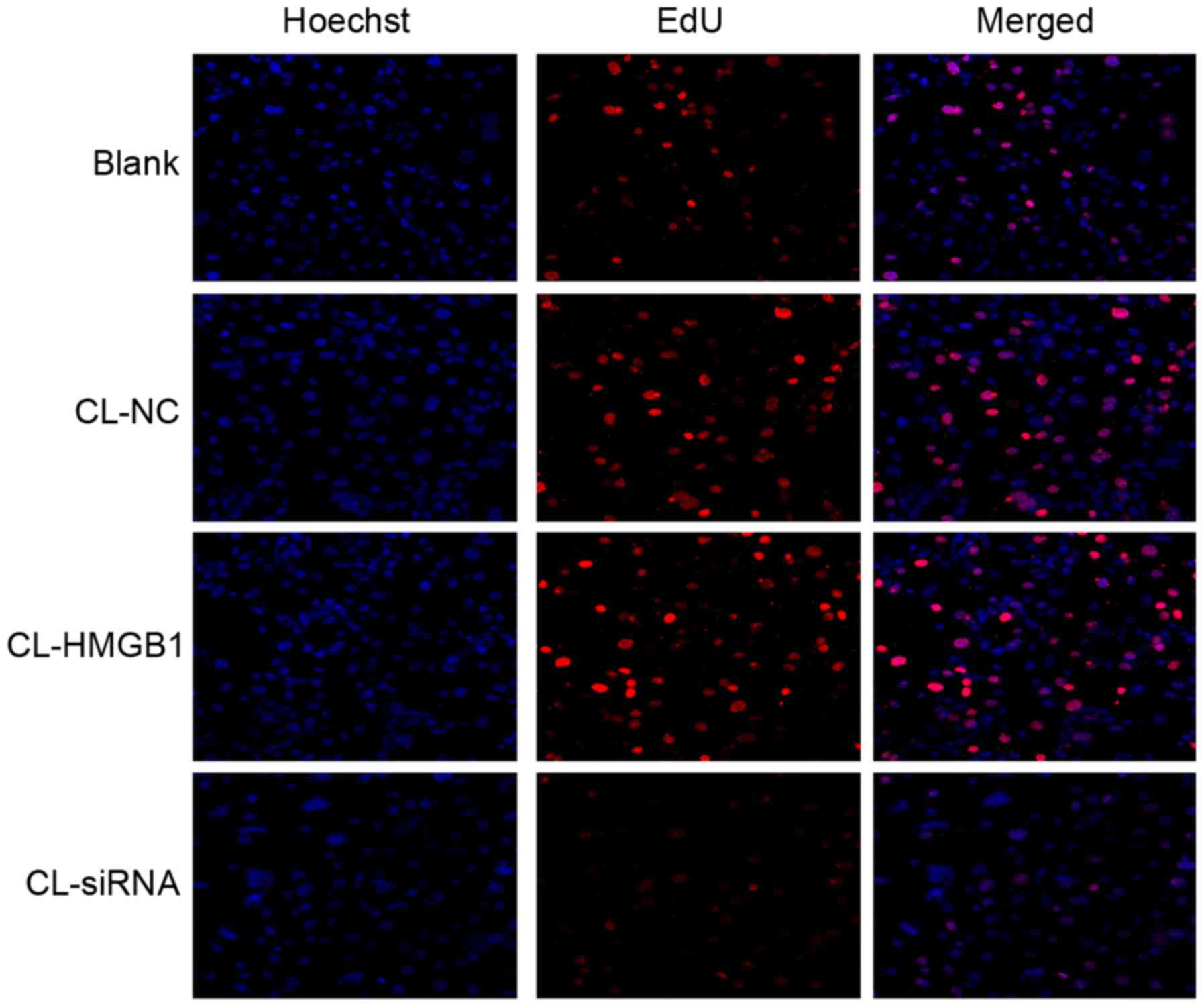
EdU assay
To determine the proliferation of HM cells, an EdU
assay was used. Similar to the MTT results, there were no
significant differences among each group (Fig. 6), which reflects that HMGB1 and CL
had no significant effect on the proliferation of HM cells.
Discussion
Epilepsy is a common disease of the central nervous
system, with common symptoms including frequent repetitive muscle
jerks, usually arrhythmic, which severely threatens the health of
many people (18,26,27).
The treatment methods include chemotherapy conservative treatment
and surgery (28). However,
neither of them can definitely receive encouraging results
(29). The pathology for this
disease is very complex, including genetic factors, CNS
inflammation, and brain diseases and disorders (30). Among them, CNS inflammation has
attracted increasing attention (10).
The role of complement system-mediated inflammation
is of key interest in seizure and epilepsy pathophysiology
(9). Administration of selective
serotonin reuptake inhibitors could lessen their antidepressant
effect over time. as well as elevated seizure outcomes observed in
people with epilepsy (4). A
previous study demonstrated that microglial cells in epileptic rats
feature widespread, activation-associated morphological changes
such as increase in cell number density, massive up-regulation of
CD11b and de-ramification (14).
In gliomas, microglia and macrophages represent a significant
component of the inflammatory response, and the balance of pro- and
anti-inflammatory functions of them dictates their antitumor
activity (31). Recent evidence
implicates glial cells and neuroinflammation in the pathogenesis of
epilepsy, with the promise of targeting these cells to complement
existing strategies (32).
HMGB1 is a central late pro-inflammatory cytokine
that triggers the inflammatory response. Our study verified the
role of HMGB1 in regulating brain injury in the epileptic brain
tissues of patients. A previous study demonstrated that expression
of HMGB1, TLR2, TLR4 and RAGE were up-regulated in rasmussen
encephalitis (33). Another study
also revealed cell loss and neurogenesis, which involve
inflammatory signaling pathways, were upregulated in human epilepsy
(34).
The HMGB1-TLR2/4-NF-κB signaling pathways is a
classic inflammatory signaling pathway (19–22),
usually involved in mediating multiple inflammatory pathways, and
during this process, microglia activation releases cytokines. HMGB1
may signal through TLR4, and results in the activation of NF-κB,
which participates in regulation of inflammation and activation of
immune cells (35). In addition,
knockdown of HMGB1 expression significantly inhibits the expression
levels of NF-κB/p65 and suppresses the nuclear translocation and
DNA-binding activity of NF-κB/p65 (36).
In conclusion, the present study demonstrated that
the epileptogenous agent CL effectively increased HMGB1 and
cytokines levels in HM cells, which had the similar result of brain
tissue of post-surgery patients with intractable epilepsy, while
overexpression of HMGB1 could aggravate this result, and inhibition
of HMGB1 could block that result. These results suggested that
HMGB1 serves a vital role in epileptogenesis through microglial
activation, via TLR4-NF-κB signaling pathway activation. The
present study provided evidence for a clinical treatment method for
the patients with epilepsy. The development of a novel,
non-invasive biomarker for HMGB1 may allow early identification of
patients at high risk of developing epilepsy.
References
|
1
|
Snoeijen-Schouwenaars FM, van Ool JS, Tan
IY, Schelhaas HJ and Majoie MH: Evaluation of perampanel in
patients with intellectual disability and epilepsy. Epilepsy Behav.
66:64–67. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gavvala JR and Schuele SU: New-onset
seizure in adults and adolescents: A review. JAMA. 316:2657–2668.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pohlmann-Eden B, Aldenkamp A, Baker GA,
Brandt C, Cendes F, Coras R, Crocker CE, Helmstaedter C,
Jones-Gotman M, Kanner AM, et al: The relevance of neuropsychiatric
symptoms and cognitive problems in new-onset epilepsy-Current
knowledge and understanding. Epilepsy Behav. 51:199–209. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singh T and Goel RK: Managing
epilepsy-associated depression: Serotonin enhancers or serotonin
producers? Epilepsy Behav. 66:93–99. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jehi LE, Palmini A, Aryal U, Coras R and
Paglioli E: Cerebral cavernous malformations in the setting of
focal epilepsies: Pathological findings, clinical characteristics
and surgical treatment principles. Acta Neuropathol. 128:55–65.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zubkov S and Friedman D: Epilepsy
treatment and creativity. Epilepsy Behav. 57:230–233. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Prontera P, Sarchielli P, Caproni S,
Bedetti C, Cupini LM, Calabresi P and Costa C: Epilepsy in
hemiplegic migraine: Genetic mutations and clinical implications.
Cephalalgia. 2017.(Epub ahead of print). PubMed/NCBI
|
|
8
|
Wang C, Ding H, Tang X, Li Z and Gan L:
Liuweibuqi capsules suppress inflammation by affecting T cell
polarization and survival in chronic obstructive pulmonary disease.
Med Chem Res. 26:2816–2823. 2017. View Article : Google Scholar
|
|
9
|
Okuneva O, Li Z, Körber I, Tegelberg S,
Joensuu T, Tian L and Lehesjoki AE: Brain inflammation is
accompanied by peripheral inflammation in Cstb-/-mice, a model for
progressive myoclonus epilepsy. J Neuroinflammation. 13:2982016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Benson MJ, Thomas NK, Talwar S, Hodson MP,
Lynch JW, Woodruff TM and Borges K: A novel anticonvulsant
mechanism via inhibition of complement receptor C5ar1 in murine
epilepsy models. Neurobiol Dis. 76:87–97. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Alyu F and Dikmen M: Inflammatory aspects
of epileptogenesis: Contribution of molecular inflammatory
mechanisms. Acta Neuropsychiatr. 29:1–16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dambach H, Hinkerohe D, Prochnow N,
Stienen MN, Moinfar Z, Haase CG, Hufnagel A and Faustmann PM: Glia
and epilepsy: Experimental investigation of antiepileptic drugs in
an astroglia/microglia co-culture model of inflammation. Epilepsia.
55:184–192. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Patterson KP, Brennan GP, Curran M,
Kinney-Lang E, Dubé C, Rashid F, Ly C, Obenaus A and Baram TZ:
Rapid, coordinate inflammatory responses after experimental febrile
status epilepticus: Implications for epileptogenesis. eNeuro.
2:2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
von Bernhardi R, Eugenín-von Bernhardi J,
Flores B and Eugenín León J: Glial cells and integrity of the
nervous system. Adv Exp Med Biol. 949:1–24. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Papageorgiou IE, Fetani AF, Lewen A,
Heinemann U and Kann O: Widespread activation of microglial cells
in the hippocampus of chronic epileptic rats correlates only
partially with neurodegeneration. Brain Struct Funct.
220:2423–2439. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luo C, Ikegaya Y and Koyama R: Microglia
and neurogenesis in the epileptic dentate gyrus. Neurogenesis
(Austin). 3:e12355252016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Najjar S, Pearlman D, Miller DC and
Devinsky O: Refractory epilepsy associated with microglial
activation. Neurologist. 17:249–254. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mazarati A, Maroso M, Iori V, Vezzani A
and Carli M: High-mobility group box-1 impairs memory in mice
through both toll-like receptor 4 and receptor for advanced
glycation end products. Exp Neurol. 232:143–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Xia J, Zhang Y, Xiao F, Wang J,
Gao H, Liu Y, Rong S, Yao Y, Xu G and Li J: HMGB1-TLR4 signaling
participates in renal ischemia reperfusion injury and could be
attenuated by dexamethasone-mediated inhibition of the ERK/NF-κB
pathway. Am J Transl Res. 8:4054–4067. 2016.PubMed/NCBI
|
|
20
|
Li X, Jin Q, Yao Q, Xu B, Li Z and Tu C:
Quercetin attenuates the activation of hepatic stellate cells and
liver fibrosis in mice through modulation of HMGB1-TLR2/4-NF-κB
signaling pathways. Toxicol Lett. 261:1–12. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zurolo E, Iyer A, Maroso M, Carbonell C,
Anink JJ, Ravizza T, Fluiter K, Spliet WG, van Rijen PC, Vezzani A
and Aronica E: Activation of Toll-like receptor, RAGE and HMGB1
signalling in malformations of cortical development. Brain.
134:1015–1032. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maroso M, Balosso S, Ravizza T, Liu J,
Bianchi ME and Vezzani A: Interleukin-1 type 1 receptor/Toll-like
receptor signalling in epilepsy: The importance of IL-1beta and
high-mobility group box 1. J Intern Med. 270:319–326. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen Y, Huang XJ, Yu N, Xie Y, Zhang K,
Wen F, Liu H and Di Q: HMGB1 contributes to the expression of
P-glycoprotein in mouse epileptic brain through toll-like receptor
4 and receptor for advanced glycation end products. PLoS One.
10:e01409182015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ratovitski T, Chaerkady R, Kammers K,
Stewart JC, Zavala A, Pletnikova O, Troncoso JC, Rudnicki DD,
Margolis RL, Cole RN and Ross CA: Quantitative proteomic analysis
reveals similarities between huntington's disease (HD) and
huntington's disease-like 2 (HDL2) human brains. J Proteome Res.
15:3266–3283. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smith AM, Gibbons HM, Oldfield RL, Bergin
PM, Mee EW, Curtis MA, Faull RL and Dragunow M: M-CSF increases
proliferation and phagocytosis while modulating receptor and
transcription factor expression in adult human microglia. J
Neuroinflammation. 10:852013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Delahaye-Duriez A, Srivastava P, Shkura K,
Langley SR, Laaniste L, Moreno-Moral A, Danis B, Mazzuferi M,
Foerch P, Gazina EV, et al: Rare and common epilepsies converge on
a shared gene regulatory network providing opportunities for novel
antiepileptic drug discovery. Genome Biol. 17:2452016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mameniskiene R and Wolf P: Epilepsia
partialis continua: A review. Seizure. 44:74–80. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ertürk Çetin Ö, İşler C, Uzan M and Özkara
Ç: Epilepsy-related brain tumors. Seizure. 44:93–97. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Falco-Walter J, Owen C, Sharma M, Reggi C,
Yu M, Stoub TR and Stein MA: Magnetoencephalography and new imaging
modalities in epilepsy. Neurotherapeutics. 14:4–10. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Loscher W, Hirsch LJ and Schmidt D: The
enigma of the latent period in the development of symptomatic
acquired epilepsy-Traditional view versus new concepts. Epilepsy
Behav. 52:78–92. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang I, Alizadeh D, Liang J, Zhang L, Gao
H, Song Y, Ren H, Ouyang M, Wu X, D'Apuzzo M and Badie B:
Characterization of arginase expression in glioma-associated
microglia and macrophages. PLoS One. 11:e01651182016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Eyo UB, Murugan M and Wu LJ:
Microglia-neuron communication in epilepsy. Glia. 65:5–18. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luan G, Gao Q, Zhai F, Chen Y and Li T:
Upregulation of HMGB1, toll-like receptor and RAGE in human
Rasmussen's encephalitis. Epilepsy Res. 123:36–49. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Iori V, Maroso M, Rizzi M, Iyer AM,
Vertemara R, Carli M, Agresti A, Antonelli A, Bianchi ME, Aronica
E, et al: Receptor for advanced glycation endproducts is
upregulated in temporal lobe epilepsy and contributes to
experimental seizures. Neurobiol Dis. 58:102–114. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
van Beijnum JR, Buurman WA and Griffioen
AW: Convergence and amplification of toll-like receptor (TLR) and
receptor for advanced glycation end products (RAGE) signaling
pathways via high mobility group B1 (HMGB1). Angiogenesis.
11:91–99. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang Z, Zhong Z, Zhang L, Wang X, Xu R,
Zhu L, Wang Z, Hu S and Zhao X: Down-regulation of HMGB1 expression
by shRNA constructs inhibits the bioactivity of urothelial
carcinoma cell lines via the NF-κB pathway. Sci Rep. 5:128072015.
View Article : Google Scholar : PubMed/NCBI
|