Introduction
The annual mortality rate of liver cancer ranks
second among the malignant neoplasms worldwide in men. An estimated
782,500 new liver cancer cases and 745,500 mortalities occurred
worldwide during 2012, with approximately half of all cases of
liver cancer and mortality occurring in China (1). Liver cancer initially develops
without symptoms, progresses rapidly, is associated with a poor
prognosis and poses a serious threat to human health (2).
An increasing amount of evidence suggests that β
adrenergic signaling serves an essential role in progression of a
variety of tumor types (3).
Clinical epidemiological studies on the application of β receptor
blockers and the reduction of tumor metastasis demonstrated that β
receptor blockers decreased the development, metastasis, recurrence
and tumor-associated mortality rate (4) of various types of solid tumor,
including breast cancer (5,6),
thyroid cancer (7), liver
metastases (8) and pancreatic
cancer (9). This suggests that β
receptor blockers may be suitable as a novel adjuvant therapy for
tumors. However, another study observed that β receptor blockers
did not improve the survival rate of patients with breast cancer
(10).
Among the aforementioned studies, the nonselective β
receptor blocker propranolol was the most frequently investigated.
Propranolol demonstrates high safety and tolerance, has been used
in clinical practice for >40 years and it is a recommended
first-line oral medication for infantile hemangioma (11). Furthermore, propranolol has been
clinically applied in patients with liver cirrhosis and liver
cancer for a number of years, as a first-line prophylaxis for
esophageal and gastric variceal bleeding (12). The present study aimed to determine
whether propranolol, in addition to reducing the portal vein
pressure in patients with liver cirrhosis, exhibits an anti-tumor
effect in liver cancer.
In the present study, the effect of propranolol
administered at different concentrations and time intervals on the
proliferation of the human liver cancer cell lines HepG2,
HepG2.2.15 and the normal human liver cell line HL-7702 was
investigated. Experiments were also performed to determine the
effect of propranolol on the rate of apoptosis and cell cycle
distribution, detect the alterations in the expression of β
receptors on the cell membrane and determine the biological
influences exerted on the three cell lines. The results of the
present study demonstrated that propranolol inhibited liver cancer
cell proliferation and induced apoptosis and S-phase arrest, thus
exerting an anti-tumor effect on the liver cancer cells.
Materials and methods
Experimental materials and
instruments
HepG2, HepG2.2.15 and HL-7702 cell lines were
routinely maintained and subcultured in the laboratory. Propranolol
hydrochloride was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). Monoclonal antibodies against adrenergic
receptor β-1 (ADRB1; cat. no. 12271), β-2 (ADRB2; cat. no. 8513),
caspase-3 (cat. no. 9665) and poly (ADP-ribose) polymerase (PARP;
cat. no. 9532) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). β-actin rabbit polyclonal antibody (cat. no.
E12-051) was purchased from EnoGene Biotech Co., Ltd. (Nanjing,
China); Dulbecco's modified Eagle's medium (DMEM)/F12 culture
medium and premium fetal bovine serum (FBS) were purchased from
Biological Industries (Kibbutz Beit-Haemek, Israel). An Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) Apoptosis
Detection kit was purchased from EnoGene Biotech Co., Ltd.
(Nanjing, China). RNase A was purchased from Takara Bio, Inc.
(Otsu, Japan). PI dye, 4% paraformaldehyde (PFA), Triton X-100 and
DAPI dye were all purchased from Beijing Leagene Biotech Co., Ltd.
(Beijing, China). Trypsin was purchased from Sangon Biotech Co.,
Ltd. (Shanghai, China) and Alexa Fluor 546-conjugated donkey
anti-rabbit IgG secondary antibody (cat. no. A10040) was purchased
from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). The
IncuCyte ZOOM second-generation live-cell imaging and analysis
system was purchased from Essen Bioscience (Ann Arbor, MI, USA). A
BD FACSCanto II flow cytometer was purchased from BD Biosciences
(Franklin Lakes, NJ, USA). The CSP-IIventilator was purchased from
Beijing Chengwei Lab-Equipment Engineering Technology Co., Ltd.
(Beijing, China) and the MCO-20AIC carbon dioxide incubator was
purchased from SANYO Electric Co., Ltd. (Osaka, Japan). The
ViiATM7thermocycler was purchased from Applied Biosystems (Thermo
Fisher Scientific, Inc.).
Drug preparation
Propranolol hydrochloride (1.0 g) was dissolved in
20 ml of distilled water to form a working solution with a
concentration of 169 mmol/l. Subsequently, 1 ml of working solution
was added to 9.6 ml of distilled water to form a stock solution
with a concentration of 16 mmol/l. The stock solution was filtered
through a 0.22-µm microporous membrane and stored at 4°C for future
use. The stock solution was diluted to different concentrations
(2.5, 5, 10, 20, 40, 80, 160 and 320 µmol/l) to determine a
suitable concentration for the treatment of liver cancer cells.
Cell culture
HepG2, HepG2.2.15 and HL-7702 cells were cultured in
DMEM/F12 medium containing 6% FBS with 100 U/ml penicillin and 100
µg/ml streptomycin. Cells were incubated at 37°C supplemented with
5% CO2 in a humidified incubator. The culture medium was
removed at cell subculture and the cells were washed twice with
physiological saline. Trypsin was added and cells were incubated at
37°C for 3–5 min. When the cells rounded up and separated from each
other, the trypsin was removed and the cells were tapped off the
wall of the flask prior to adding fresh medium and the cells were
transferred into a centrifuge tube. The suspended cells were
centrifuged at 92 × g at 25°C for 3 min and the supernatant was
discarded. Fresh medium was added to resuspend the cells, and the
solution was thoroughly mixed prior to placing in the
aforementioned incubator. For cell freezing, l ml cell
cryopreservation solution (culture medium 9:1 DMSO) was added to
the cell pellet and mixed thoroughly prior to being transferred
into a cryotube for long-term storage in liquid nitrogen.
Observation of cell proliferation
using a live-cell imaging and analysis system
Cells in the logarithmic growth phase were obtained
and the culture medium was removed, prior to washing twice with
physiological saline. Subsequently, 0.2 ml 0.25% trypsin was added
to the washed cells and incubated at 37°C for 2–5 min, prior to
adding 2 ml DMEM/F12 medium to terminate the trypsinization. The
trypsinized cells were collected, counted, and seeded in a 96-well
plate at a density of 1×104/ml. The 96-well plate was
incubated at 37°C supplemented with 5% CO2 overnight.
The following day, the cells were observed to confirm their health,
and culture medium was removed, prior to washing twice with
physiological saline. Subsequently, culture medium containing
propranolol at a range of concentrations (0, 2.5, 5, 10, 20, 40,
80, 160 and 320 µmol/l) was added to the cells, which were then
transferred to the IncuCyte ZOOM incubator.
Two images of each well were captured every 6 h,
with a total observation period of 48 h; the 96-well plate was not
moved during this time. IncuCyte 2104A software (Essen Bioscience)
was used for the analysis of cell confluence and cell count in each
image, and growth curves were generated. The cell proliferation
ratio was calculated as the ratio between the cell confluence (%)
of cells treated with propranolol a teach concentration and cells
treated with 0 µmol/l propranolol at the same time point. The cell
proliferation ratio at 6, 12, 18, 24, 30, 36, 42 and 48 h was
calculated as the ratio between cell confluence (%) at each time
point and the time when the drug was added (0 h). Each condition
was performed in triplicate. The whole experiment was repeated
three times.
Immunofluorescence detection of ADRB1
and ADRB2 and their alterations
Cells in the logarithmic growth phase were seeded in
a 48-well plate and incubated at 37°C in 5% CO2
overnight. The cells were observed, to confirm their health, prior
to removing the culture medium and washing twice with physiological
saline. Culture medium containing 0 or 40 µmol/l propranolol was
added to the cells, which were incubated at 37°C in 5%
CO2 for 48 h. Subsequently, the culture medium was
removed and cells were washed three times with 1X PBS, for 3 min
each time.
A total of 100 µl 4% PFA was added to each well and
incubated at room temperature for 15–30 min. Subsequently, the PFA
was discarded and the cells were washed three times with 1X PBS for
3 min each time. This was followed by the addition of 100 µl 0.1%
Triton X-100 into each well and incubation at room temperature for
30 min. Following the permeabilization with Triton X-100, cells
were washed three times with 1X PBS for 3 min each time.
A total of 100 µl 1% bovine serum albumin
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was added to each
well and incubated at room temperature for 30 min. Antibodies
against ADRB1 and 2 were diluted to 1:100 in 1X PBS. A total of 100
µl ADRB1 or 2 antibody solution was added to each well and
incubated at 4°C overnight. The following day, cells were washed
three times with 1X PBS, 3 min each time. Fluorescent secondary
antibodies were diluted 1:200 in 1X PBS. Diluted secondary
antibodies (100 µl) were added to each well and incubated in the
dark at room temperature for 1 h.
DAPI (100 µl; dilution, 1:1,000) or Hoechst 33342/PI
was added to the wells and incubated in the dark at room
temperature for 2–3 min. Subsequently, DAPI was removed from the
cells and they were washed with PBS, followed by immediate imaging
using an inverted fluorescence microscope. Captured images were
analyzed using Image-Pro Plus5.0 image analysis software (Media
Cybernetics, Inc., Rockville, MD, USA). Each condition was
performed in triplicate. The whole experiment was repeated three
times.
Western blotting
Following the treatment of the three cell lines with
0, 40 or 80 µmol/l propranolol for 48 h, cells were lysed with
radioimmunoprecipitation assay lysis buffer (CW2333S; Cowin Biotech
Co., Ltd, Beijing, China), proteinase/phosphatase inhibitor
cocktail (cat. no. PP1301; Aidlab Biotechnologies Co., Ltd.,
Beijing, China) and phenylmethanesulfonyl fluoride (EnoGene,
Nanjing, China) at a 100:1:1 ratio. A bicinchoninic protein assay
kit (CW0014S; Cowin Biotech Co., Ltd., Beijing, China) was
subsequently used to determine protein concentration. A total of 20
µg protein per lane was separated by 10–12% SDS-PAGE and
electrotransferred onto polyvinylidene fluoride membranes.
Subsequently, the transferred membranes were blocked with 5%
skimmed milk for 2 h at 25°C and incubated with primary monoclonal
antibodies, including ADRB1, ADRB2, caspase-3, PARP and β-actin
(all dilutions, 1:1,000) at 4°C overnight. The membranes were then
washed with Tris-HCl and incubated with a goat anti-rabbit
IgG-horseradish peroxidase conjugated secondary antibody (dilution,
1:4,000; Abmart, Inc., Shanghai, China) at 37°C for 1.5 h.
Following incubation with an enhanced chemiluminescence reagent
(BeyoECL Star; Beyotime Institute of Biotechnology, Shanghai,
China) for 10 min at 25°C, a dark chamber was used to develop
images. The protein bands were quantified using Image J software
(version 1.46r; National Institutes of Health, Bethesda, MD,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR was used to detect the expression of ADRB2
mRNA in the three cell lines. Total RNA was extracted from the
cells using TRIzol® reagent (Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Total RNA (3
µg) was reverse transcribed to cDNA using a RevertAid First Strand
cDNA Synthesis kit (Thermo Fisher Scientific, Inc.), with 1 µl
Oligo(dT)18, 5X Reaction Buffer (4 µl), RiboLock RNase Inhibitor (1
µl), dNTP Mix 2 µl, RevertAid Reverse Transcriptase 1 µl and
ddH2O to supplement to a total volume of 20 µl. The
synthesis conditions for cDNA were 5 min at 25°C, 60 min at 42°C
and 15 min at 70°C.
SYBR Premix Ex Taq™ II (Tli RNaseH Plus; cat. no.
RR820A; Takara Bio, Inc.) was used for the PCR reaction, including
10 µl SYBR Premix Ex Taq II (Tli RNaseH Plus), 0.4 µl ROX Reference
Dye II, 0.8 µl ADRB2 forward primer (5′-GCCTGTGCTGATCTGGTCAT-3′),
0.8 µl ADRB2 reverse primer (5′-AATGGAAGTCCAAAACTCGCA-3′; primers
synthesized by Invitrogen; Thermo Fisher Scientific, Inc.), 2.0 µl
cDNA template and 6.0 µl ddH2O, in a 20 µl reaction system. The
following conditions were used for PCR: 10 min at 95°C, 40 cycles
of 15 sec at 95°C and 60 sec at 60°C. GAPDH was used as a reference
gene (forward, 5′-GACCCCTTCATTGACCTCAAC-3′ and reverse,
5′-CGCTCCTGGAAGATGGTGAT-3′; Invitrogen; Thermo Fisher Scientific,
Inc.). According to the analysis of the amplification and melting
curves, the relative expression of the ADRB2 gene was calculated
using the 2−ΔΔCq method (13).
Detection of apoptosis using flow
cytometry
Following incubation with 0, 40 or 80 µmol/l
propranolol for 48 h, culture medium was removed from the cells and
transferred into a centrifuge tube. Adherent cells were washed with
PBS once prior to trypsinization with trypsin (with 0.25% EDTA).
The trypsinized cells were added to back into the culture medium
and mixed well prior to centrifugation 92 × g for 5 min at 25°C.
The supernatant was discarded and the cell pellet was gently
resuspended in PBS and counted. A total of 4×105 cells
were then centrifuged at 92 × g for 5 min 25°C. The supernatant was
discarded and 500 µl binding buffer (apoptosis detection kit;
EnoGene Biotech Co., Ltd.) was added to gently resuspend the cells.
Subsequently, cells were filtered through a 400-mesh filter to form
a single-cell suspension, followed by the addition of 5 µl Annexin
V-FITC and 5 µl PI, with gentle mixing. The cells were incubated in
the dark at room temperature for 10 min and immediately subjected
to flow cytometry detection. The results were analyzed using FlowJo
software (version 7.6.1; Tree Star, Inc., Ashland, OR, USA). Each
condition was performed in triplicate. The whole experiment was
repeated three times.
Detection of cell cycle distribution
using flow cytometry
Following incubation with 0, 40 or 80 µmol/l
propranolol for 48 h, the cell culture medium was removed from the
cells and transferred to a centrifuge tube. Adherent cells were
washed with PBS twice, trypsinized and resuspended. The cell
suspension was centrifuged at 138 × g at 4°C for 5 min. The
supernatant was discarded and the cells were washed with precooled
1X PBS once and centrifuged at 1,000 × g at 4°C for 5 min. The
supernatant was discarded, and the cell pellet was added to 5 ml of
70% ethanol, gently mixed and incubated at 4°C overnight. The
following day, the cells were centrifuged at 138 × g for 5 min at
25°C and the supernatant was discarded. The pellet was washed three
times with precooled 1X PBS, for 3 min each time. The cell
suspension was centrifuged at 138 × g for 5 min at 25°C and the
supernatant was discarded. The pellet was added to a 0.4-ml PI
mixture (with 4 µl RNase) in the dark and stained for 30 min at
25°C prior to immediate detection using a flow cytometer. The
results were analyzed using FlowJo 7.6.1 software (Tree Star,
Inc.).
Statistical analysis
Data were statistically analyzed using SPSS 17.0
software (SPSS, Inc., Chicago, IL, USA). Quantitative results were
expressed as the mean ± standard deviation. A t-test was used to
compare differences between two groups, and a one-way analysis of
variance followed by the Least-significant difference post hoc test
was used to compare differences among multiple groups. P<0.05
was considered to indicate a statistically significant
difference.
Results
Propranolol inhibits the proliferation
of liver cancer cells
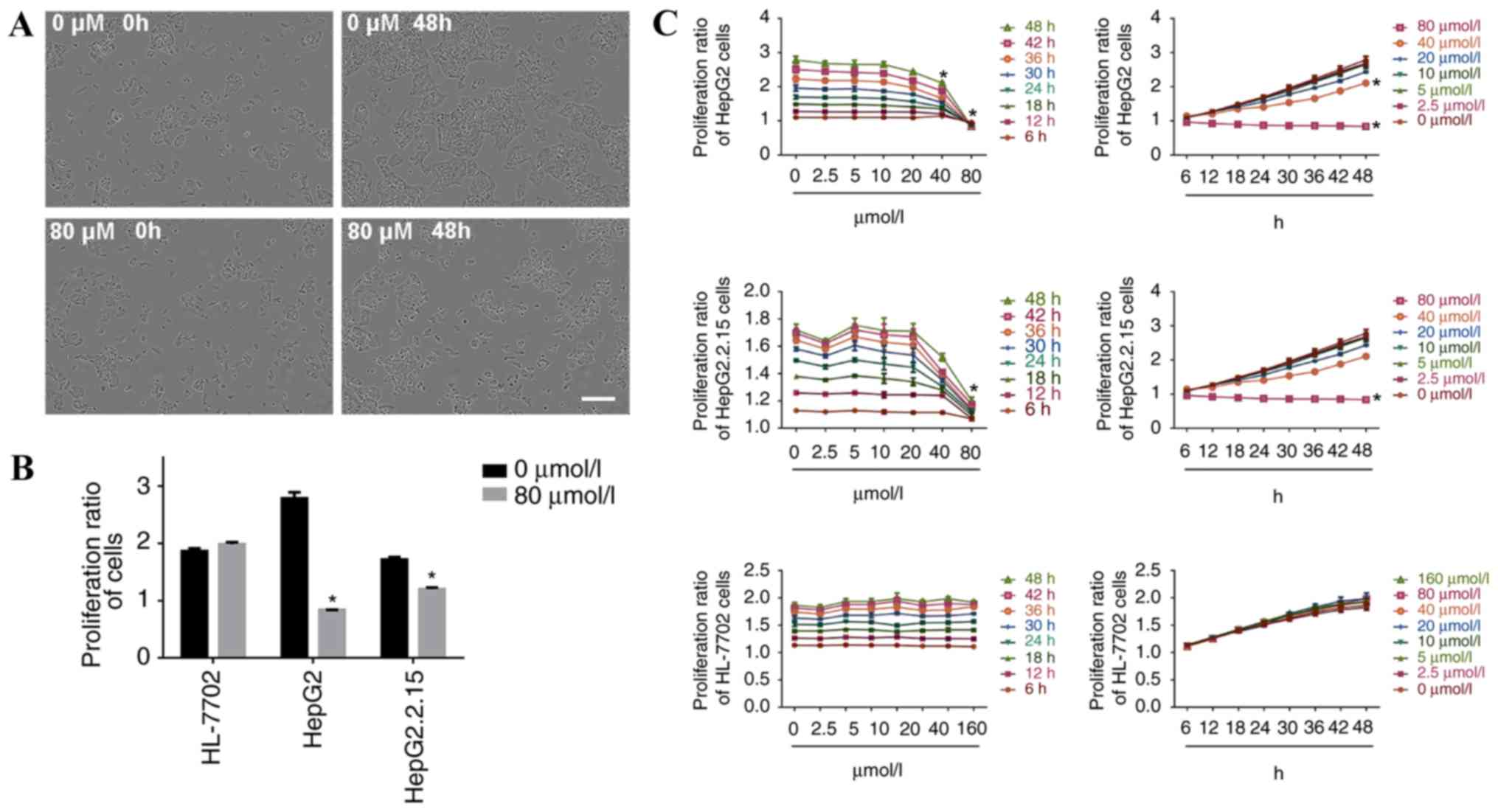
IncuCyte ZOOM phase-contrast imaging revealed the
growth rates as indicated by the cell confluence (%) of the three
investigated cell lines.
When HepG2 cells were incubated with 80 µmol/l
propranolol for 48 h, cell confluence was reduced and a small
number of cells became detached from the surrounding cells and
suspended, suggesting the inhibition of cell proliferation
(Fig. 1A). The proliferation ratio
of HepG2 and HepG2.2.15 cells was significantly decreased at 48 h
of treatment with 80 µmol/l propranolol (P<0.05), whereas the
proliferation of HL-7702 cells was unaltered (Fig. 1B). The results demonstrated that at
the same concentration, the inhibitory effect of propranolol on the
proliferation ratio of HepG2 and HepG2.2.15 cells increased with
the increase in duration of the treatment. At the same treatment
time point, the inhibitory effect was enhanced with the increase in
propranolol concentration. Therefore, propranolol hydrochloride
suppressed liver cancer cell proliferation in a time and
concentration dependent manner (Fig.
1C).
The proliferation ratio of HepG2 cells decreased
after 18 h of treatment with 20 µmol/l propranolol (P<0.05) and
after 48 h of treatment with 40 or 80 µmol/l of propranolol (both
P<0.05). The proliferation ratio of HepG2.2.15 cells decreased
when the cells were treated with 40 µmol/l propranolol for 18 h
(P<0.05), and when treated with 40 or 80 µmol/l of propranolol
for 48 h (P<0.05). No inhibitory effect was observed following
treatment with different propranolol concentrations for 48 h in
HL-7702 cells.
Following the incubation of HepG2 and HepG2.2.15
cells with 160 µmol/l propranolol, and all three cell lines with
320 µmol/l propranolol, the adherent cells visibly shrank and the
microscopic observation revealed a large number of floating cells
(data not shown); therefore, statistical analysis of the cell
proliferation ratio was not performed in the present study at these
doses of treatment.
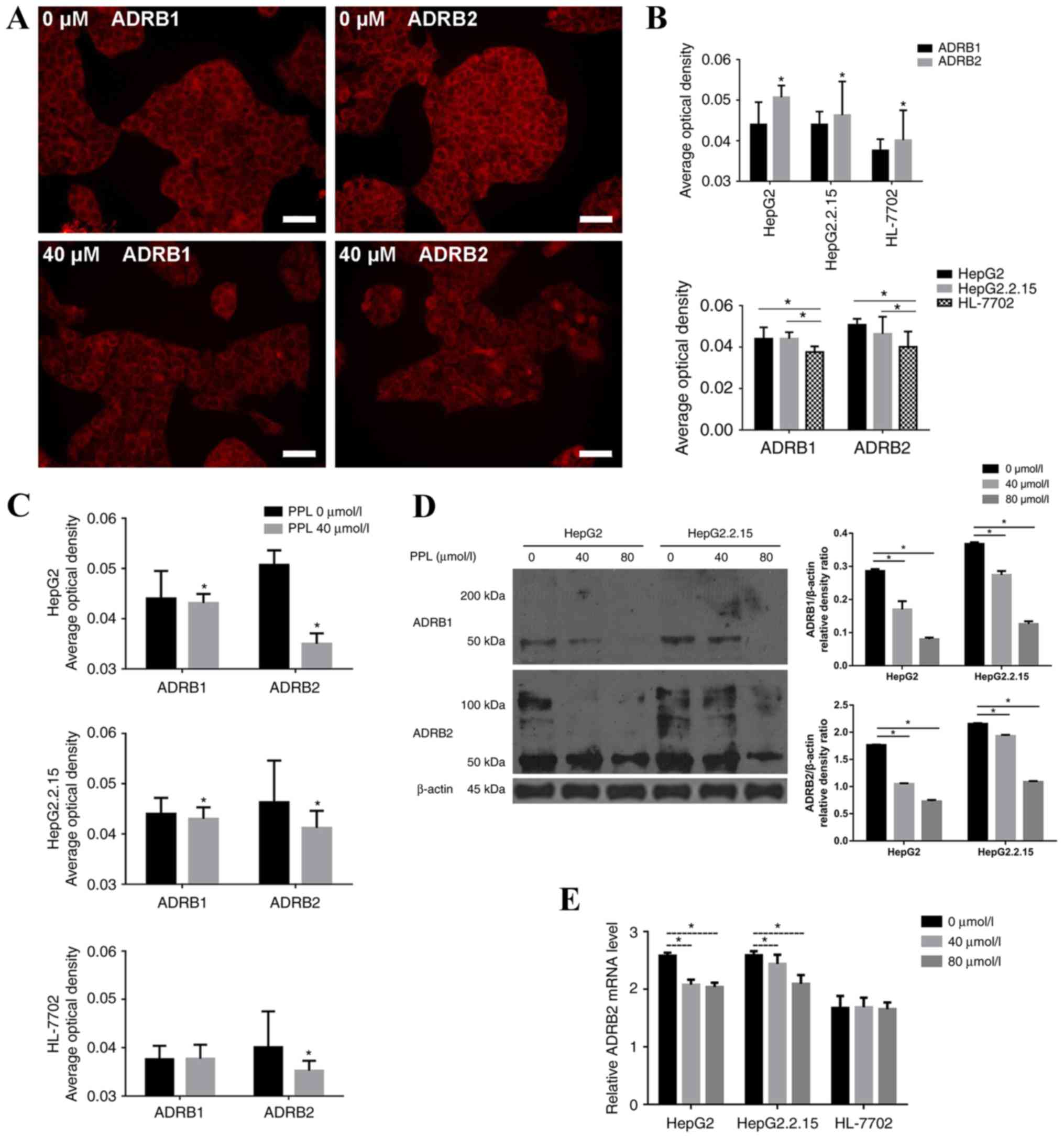
Propranolol decreases the expression
of β receptors for the three cell lines
The spatial distribution of ADRB1 and ADRB2 on
HepG2, HepG2.2.15 and HL-7702 cells was measured by
immunofluorescence. It was demonstrated that ADRB1 and ADRB2 were
expressed on the membrane of the three cell lines, and were more
highly expressed on HepG2 and HepG2.2.15 cells compared with
HL-7702 cells (P<0.05). The expression of ADRB2 was higher than
the expression of ADRB1 on all cell lines (all P<0.05; Fig. 2A and B).
Propranolol (40 µmol/l; the lowest that inhibited
the proliferation of HepG2 and HepG2.2.15 cells) was added to
HepG2, HepG2.2.15 and HL-7702 cells, and the alterations in the
expression of β receptors on the membranes of the three cell lines
were observed. The results demonstrated that treatment with
propranolol decreased the expression of ADRB1 and ADRB2 on HepG2
and HepG2.2.15 cells (all P<0.05), while only the expression of
ADRB2 was decreased on HL-7702 cells (P<0.05). The decrease in
the expression of ADRB2 on the three cell lines was more pronounced
than the expression of ADRB1 (Fig.
2C).
The protein expression of ADRB1 and ADRB2 in HepG2
and HepG2.2.15 cells decreased following treatment with 40 and 80
µmol/l propranolol for 48 h, as demonstrated using western blotting
(Fig. 2D). As propranolol
treatment most markedly decreased the level of ADRB2 protein, the
ADBR2 mRNA expression level was determined by RT-qPCR. Following
incubation with propranolol for 48 h, the relative ADRB2 mRNA level
decreased in the liver cancer cells, but not in HL-7702 cells
(Fig. 2E).
Propranolol affects the rate of
apoptosis in liver cancer cell lines
The effect of propranolol on the apoptotic
morphology of liver cancer cells was observed under a microscopeby
staining the nuclei with DAPI and Hoechst 33342/PI. The HepG2 cells
treated with propranolol for 48 h demonstrated alterations in their
nuclear chromatin that represented each stage of apoptosis,
including stages I, IIa and IIb, and the formation of apoptotic
bodies, suggesting that treatment with propranolol could induce
apoptosis in liver cancer cells (Fig.
3A). Following Hoechst 33342/PI double staining, the number of
apoptotic cells (strong blue staining) increased with the increase
in propranolol concentration, indicating that the apoptosis of
HepG2 cells occurred following the incubation with propranolol for
48 h (Fig. 3B).
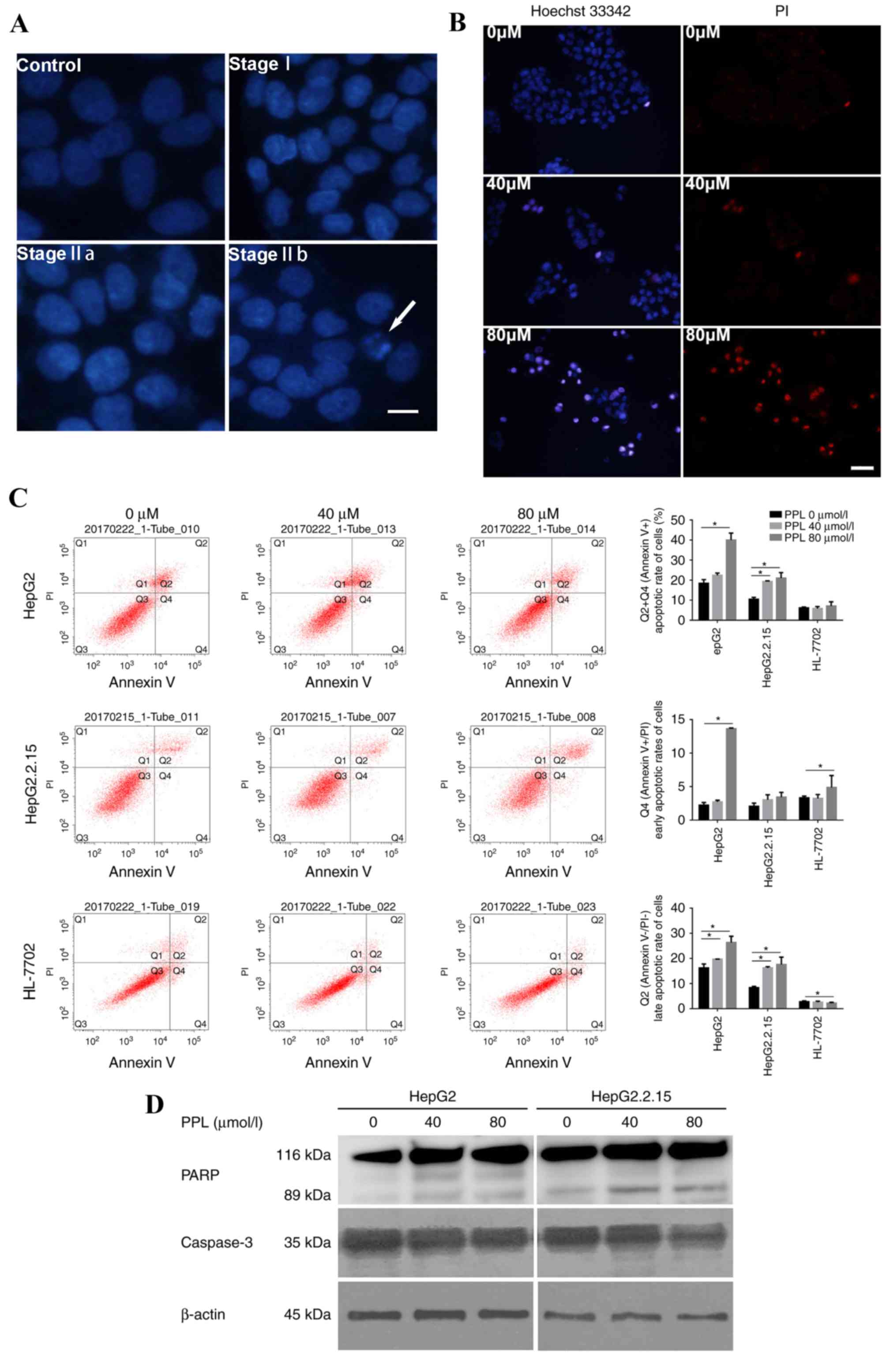 | Figure 3.PPL treatment induced the apoptosis
of HepG2 and HepG2.2.15 cells. (A) Representative DAPI staining
images of HepG2 cells cultured with or without PPL for 48 h. The
nuclei of HepG2 without PPL treatment (the control) were
homogeneously stained and regularly shaped. Following treatment
with PPL, the HepG2 cells demonstrated the characteristics of stage
I apoptosis; the nuclei of HepG2 cells were corrugated and
chromatin condensation was visible. In stage IIa, the nuclei
displayed chromatin condensation and marginalization. In stage IIb,
the nuclei of HepG2 cells were cleaved and apoptotic bodies were
formed. The white arrow indicates an apoptotic body. Scale bar, 25
µm. (B) Representative images of the Hoechst 33342/PI double
staining of HepG2 cells cultured with different concentrations of
PPL for 48 h. Incubation with 80 µmol/l PPL induced the Hoechst
33342 (blue) and PI (red) staining of HepG2 cells, which was
visibly increased compared with 0 µmol/l PPL. Scale bar, 50 µm. (C)
PPL increased the rate of apoptosis for HepG2 and HepG2.2.15 cells,
while it exhibited no significant effect on the apoptosis of
HL-7702 cells, as demonstrated by flow cytometry. Q1, Annexin
V-FITC−/PI+, necrotic cells; Q2, Annexin
V-FITC+/PI+, late apoptotic cells; Q3,
Annexin V-FITC−/PI−, viable cells; Q4,
Annexin V-FITC +/PI−, early apoptotic cells.
The apoptotic rate was calculated based on the sum of quadrants 2
and 4, which included all Annexin V-FITC+ cells.
*P<0.05. (D) In HepG2 and HepG2.2.15 cells treated with
different concentrations of PPL for 48 h, the expression of
full-length caspase3 decreased, and expression of the PARP fragment
(89 kDa) increased, as demonstrated by western blotting. PPL,
propranolol hydrochloride; PI, propidium iodide; PARP, poly
(ADP-ribose) polymerase. *P<0.05. |
Furthermore, Annexin V-FITC/PI double staining was
used to quantitatively measure the rate of apoptosis. Cells
incubated with 0, 40 and 80 µmol/l propranolol for 48 h were
subjected to flow cytometry detection. It was demonstrated that 80
µmol/l propranolol increased the rate of apoptosis of the HepG2 and
HepG2.2.15 cell lines (both P<0.05), while it exerted no
influence on the apoptotic rate of HL-7702 cells (Fig. 3C). The same gate could not be used
for all flow cytometry apoptosis assays due to alterations in the
size and morphology of cells following the incubation with
propranolol, particularly 160 µmol/l propranolol. Instead, a
positive control was used in each sample to adjust the position of
the gate. The location on the X-axis and Y-axis were then adjusted
on the basis of the positive control data.
Following incubation with 40 and 80 µmol/l
propranolol for 48 h, the extent of PARP cleavage increased,
whereas the expression of full-length caspase 3 (35 kDa) decreased
in HepG2 and HepG2.2.15 cells (Fig.
3D). These results indicated that propranolol may have induced
the apoptosis of liver cancer cells by promoting caspase-associated
signaling.
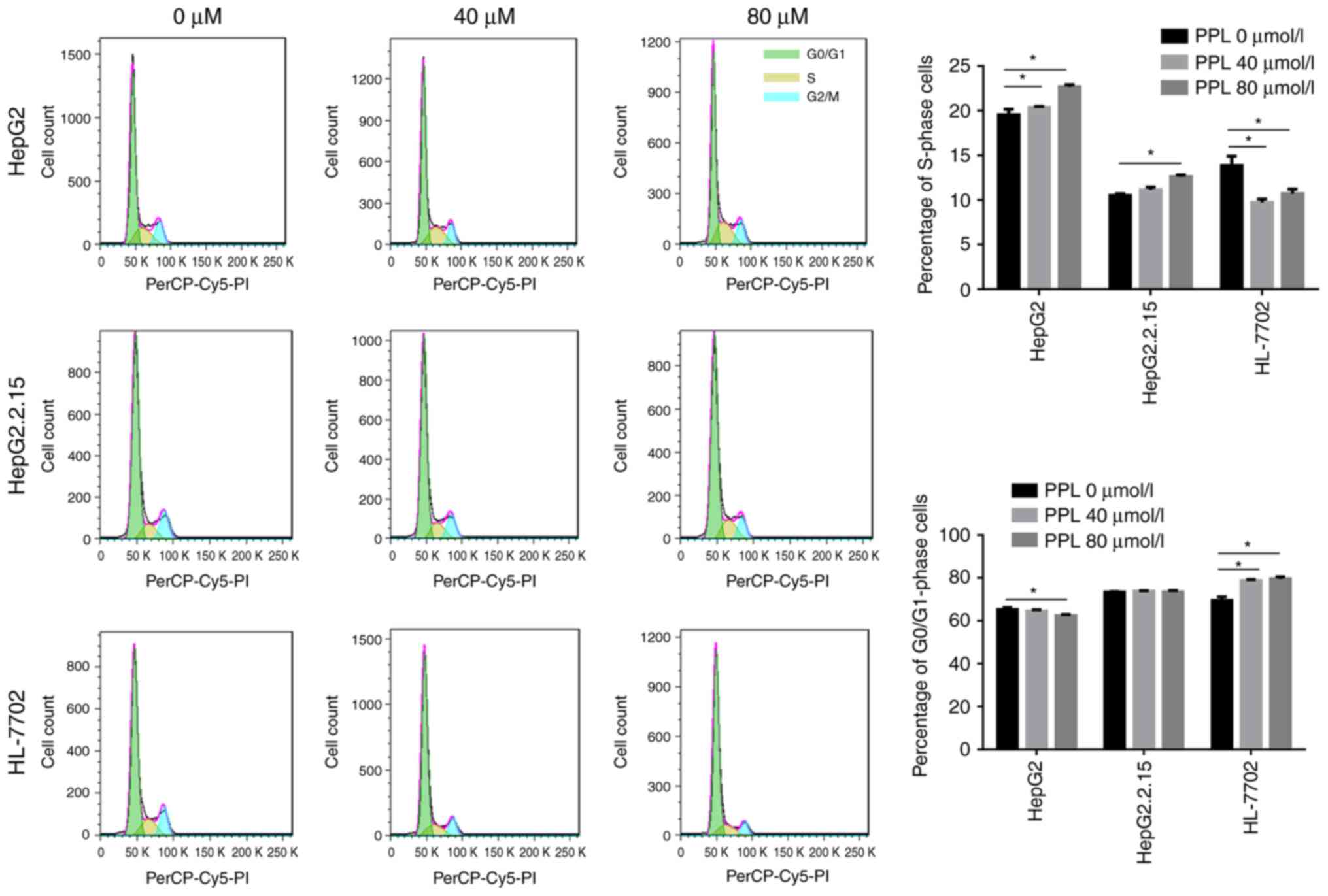
Effect of treatment with propranolol
on the cell cycle distribution of the three cell lines
Propranolol was demonstrated in the present study to
promote the apoptosis of liver cancer cells. Subsequently, flow
cytometry and PI staining were used to investigate whether cell
cycle arrest occurred during propranolol-induced liver cancer cell
apoptosis. It was demonstrated that the treatment of HepG2 and
HepG2.2.15 cells with 40 or 80 µmol/l propranololincreased the
proportion of cells in S-phase (all P<0.05), suggesting an
S-phase arrest during the cell cycle. However, the incubation of
HL-7702 cells with propranolol led to an increase in the number of
G0/G1 phase cells (both P<0.05),
suggesting that cells were arrested at the
G0/G1 phase of the cell cycle (Fig. 4).
Discussion
Recent studies have indicated that propranolol
hydrochloride treatment may reduce the progression, metastasis and
recurrence of various tumors, and therefore reduce the
tumor-associated mortality rate, through the inhibition of β
receptors (14–16), which may be suitable as a novel
clinical adjuvant therapy against tumors (17). However, there is limited data
regarding the anti-tumor effect of propranolol in liver cancer
cells. The present study demonstrated that propranolol inhibited
liver cancer cell proliferation by inhibiting the expression of
ADRB2 and inducing apoptosis by activating caspase-associated
signaling.
A previous meta-analysis reported that β receptor
blockers reduced the mortality rate of patients with liver
cirrhosis. One potential explanation for this effect is the
reduction in gastrointestinal bleeding by β receptor blockers;
another possible explanation is a reduction in the incidence of
liver cancer (18). It was also
demonstrated that propranolol reduced the 10-year cumulative
incidence of liver cancer in patients with hepatitis C-associated
cirrhosis (19). Liver cancer in
patients in China primarily develops from hepatitis B virus
(HBV)-associated liver cirrhosis and is frequently accompanied by
an increase in α-fetoprotein levels (20–22).
The HepG2 cell line, the parental cell line of HepG2.2.15, has been
reported to be misidentified; it was originally identified as a
hepatocellular carcinoma cell line, but was later revealed to be
derived from a hepatoblastoma (23). Hepatoblastoma is a tumor that
originates from cells in the liver. At present, HepG2 is widely
used to study liver cancer in vitro. Therefore, the present
study was performed with HepG2 (which secretes α-fetoprotein) and
HepG2.2.15 (which expresses HBV antigen and secretes complete HBV
particles) cells, as well as the human normal liver cell line
HL-7702, to investigate the anti-tumor effect of propranolol.
It was initially demonstrated in the present study
that β receptors were localized on the cell membrane of the liver
cancer cell lines and were more highly expressed in liver cancer
cells compared with normal liver cells, and the expression was
reduced by treatment with propranolol. Previous studies have
reported that the expression of β receptors in liver cancer
patients was elevated, particularly ADRB2 (24,25).
The results of the present study are therefore consistent with the
results of previous in vitro and in vivo studies.
Propranolol, as a nonselective receptor blocker, primarily affects
ADRB2, with a lesser effect on ADRB1 (26), suggesting an anti-tumor effect of
propranolol. The results of the present study demonstrated that
propranolol reduced the expression of the ADRB2 receptor on the
liver cancer cell membranes to a greater extent than the expression
of ADRB1, suggesting that ADRB2 may serve a more important role in
liver cancer. Previous studies indicated that the ADRB2 receptor
may be a prognostic indicator for liver cancer (27), and that the ADRB2 receptor
signaling pathway is associated with liver cancer cell
proliferation and autophagy (28),
while the underlying mechanisms remain to be elucidated.
Furthermore, the present study confirmed that
propranolol inhibited the proliferation of HepG2 and HepG2.2.15
cells. The inhibitory effect of propranolol on liver cancer cells
was enhanced with a prolonged duration of treatment or an increase
in propranolol concentration. A previous study demonstrated that
propranolol inhibited the proliferation, invasion and migration of
MCF7, HT-29 and HepG2 cells (26).
However, since propranolol inhibited tumor cell proliferation, it
was necessary to ensure that the drug did not affect normal cell
function while inhibiting tumor cell proliferation. Therefore, the
determination of the optimal dose of propranolol is necessary in
cellular and clinical studies. The effect of propranolol was
studied with eight different concentrations, ranging from 2.5 to
320 µmol/l. The results demonstrated that propranolol at low
concentrations demonstrated no significant influence on cell
proliferation and that propranolol at the highest concentrations
led to cell death. Treatment with 40 and 80 µmol/l propranolol
demonstrated significant inhibitory effect on HepG2 and HepG2.2.15
cells while demonstrating no influence on normal liver cells.
Furthermore, the present study confirmed that
propranolol induced apoptosis of HepG2 and HepG2.2.15 cells and
resulted in the S-phase arrest of these cells. A previous study
demonstrated that propranolol induced cell cycle arrest and cell
apoptosis in melanoma cells (29).
It was also identified in the present study that propranolol
induced morphological alterations in the nuclei of liver cancer
cells during the process of apoptosis, and stimulated the formation
of apoptotic bodies. The apoptotic rate of liver cancer cells
increased with the increase in the concentration of propranolol,
while the drug did not affect the apoptotic rate of HL-7702 cells.
Furthermore, it was demonstrated that propranolol induced the
apoptosis of liver cancer cells by promoting caspase-dependent
signaling, which may provide a direction for further research.
Anti-tumor drugs include cell cycle-specific and cell
cycle-non-specific drugs; the data of the present study
demonstrated that the effect of propranolol on liver cancer cells
is cell cycle-specific, and led to a significant increase in the
percentage of HepG2 and HepG2.2.15 cells in the S phase, indicating
that cells were arrested at the S phase. Clinically, commonly
available anti-tumor drugs that affect S phase progression include
fluorouracil and methotrexate. Whether the anti-tumor effect of
propranolol is also achieved by targeting the S phase requires
further investigation.
The above results indicate that 80 µmol/l is the
optimum dose of propranolol for studying anti-tumor effects in
liver cancer cells. Propranolol at 80 µmol/l inhibited cell
proliferation and induced apoptosis to the greatest extent without
affecting the biological function of HL-7702 cells.
HepG2.2.15 cells demonstrated greater resistance to
propranolol compared with the HepG2 cells. The HepG2.2.15 cell line
expresses the HBV antigen and secretes complete HBV particles, and
therefore can serve as a cellular model for HBV-associated liver
cancer. The present study demonstrated that the proliferation ratio
of HepG2 and HepG2.2.15 cells started to significantly decline
following treatment with 20 and 40 µmol/l propranolol,
respectively. Microscopic observation demonstrated that the
alterations in HepG2.2.15 were not as evident as in HepG2 cells
following incubation with the same concentration of propranolol.
Compared with HepG2 cells, the decrease in expression of ADRB2 and
the apoptotic rate were lower in HepG2.2.15 cells following the
incubation with propranolol (P<0.05). HepG2.2.15 cells required
a larger dose of propranolol to induce the same effect as in HepG2
cells. Future studies should aim to determine whether HepG2.2.15
cells are more resistant to propranolol due to the HBV infection,
whether an increased dose of propranolol will be required when
treating HBV-associated liver cancers in patients, and whether the
use of anti-HBV drugs could reduce the difficulty in treating
HBV-associated liver cancer.
There are certain limitations associated with the
present study; although it was demonstrated that propranolol could
inhibit the proliferation of liver cancer cells and induce their
apoptosis, and that the apoptosis induced by propranolol maybe
activated by caspase-dependent signaling, the mechanism underlying
the anti-tumor effect of propranolol was not elucidated. Therefore,
the mechanisms of the anti-tumor effect of propranolol should be
elucidated in future studies.
In conclusion, the expression of β receptors,
primarily ADRB2, in HepG2 and HepG2.2.15 cells was markedly higher
than in HL-7702 cells; this was reduced by treatment with
propranolol. Propranolol inhibited proliferation, and induced
apoptosis and S-phase arrest in liver cancer cells. Further
investigation of the role of the inhibition of β adrenergic
receptor signaling in liver cancer cells is required. The possible
mechanisms underlying the anti-tumor effect of propranolol and the
pathogenesis of liver cancer need to be elucidated to provide novel
insights into the development of adjuvant therapies for liver
cancer.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81673651
and 81273552), the Major Projects of the Ministry of Science and
Technology of China (grant no. 2017ZX10302202) and Tianjin
Municipal Health and Family Planning Commission (grant nos. 16KG151
and 2014KY03).
Glossary
Abbreviations
Abbreviations:
|
HBV
|
hepatitis B virus
|
|
PFA
|
paraformaldehyde
|
|
PI
|
propidium iodide
|
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cole SW and Sood AK: Molecular pathways:
Beta-adrenergic signaling in cancer. Clin Cancer Res. 18:1201–1206.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maccari S, Buoncervello M, Rampin A, Spada
M, Macchia D, Giordani L, Stati T, Bearzi C, Catalano L, Rizzi R,
et al: Biphasic effects of propranolol on tumour growth in B16F10
melanoma-bearing mice. Br J Pharmacol. 174:139–149. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Montoya A, Amaya CN, Belmont A, Diab N,
Trevino R, Villanueva G, Rains S, Sanchez LA, Badri N, Otoukesh S,
et al: Use of non-selective beta-blockers is associated with
decreased tumor proliferative indices in early stage breast cancer.
Oncotarget. 8:6446–6460. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilson JM, Lorimer E, Tyburski MD and
Williams CL: β-Adrenergic receptors suppress Rap1B prenylation and
promote the metastatic phenotype in breast cancer cells. Cancer
Biol Ther. 16:1364–1374. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wei WJ, Shen CT, Song HJ, Qiu ZL and Luo
QY: Propranolol sensitizes thyroid cancer cells to cytotoxic effect
of vemurafenib. Oncol Rep. 36:1576–1584. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sorski L, Melamed R, Matzner P, Lavon H,
Shaashua L, Rosenne E and Ben-Eliyahu S: Reducing liver metastases
of colon cancer in the context of extensive and minor surgeries
through β-adrenoceptors blockade and COX2 inhibition. Brain Behav
Immun. 58:91–98. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Partecke LI, Speerforck S, Kading A,
Seubert F, Kuhn S, Lorenz E, Schwandke S, Sendler M, Kessler W,
Trung DN, et al: Chronic stress increases experimental pancreatic
cancer growth, reduces survival and can be antagonised by
beta-adrenergic receptor blockade. Pancreatology. 16:423–433. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cardwell CR, Pottegard A, Vaes E, Garmo H,
Murray LJ, Brown C, Vissers PA, O'Rorke M, Visvanathan K,
Cronin-Fenton D, et al: Propranolol and survival from breast
cancer: A pooled analysis of European breast cancer cohorts. Breast
Cancer Res. 18:1192016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hoeger PH, Harper JI, Baselga E, Bonnet D,
Boon LM, Ciofi Degli Atti M, El Hachem M, Oranje AP, Rubin AT,
Weibel L and Leaute-Labreze C: Treatment of infantile haemangiomas:
Recommendations of a European expert group. Eur J Pediatr.
174:855–865. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garcia-Tsao G, Sanyal AJ, Grace ND and
Carey W: Practice Guidelines Committee of the American Association
for the Study of Liver D, Practice Parameters Committee of the
American College of G. Prevention and management of
gastroesophageal varices and variceal hemorrhage in cirrhosis.
Hepatology. 46:922–938. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rico M, Baglioni M, Bondarenko M, Laluce
NC, Rozados V, Andre N, Carre M, Scharovsky OG and Menacho Marquez
M: Metformin and propranolol combination prevents cancer
progression and metastasis in different breast cancer models.
Oncotarget. 8:2874–2889. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hwa YL, Shi Q, Kumar SK, Lacy MQ, Gertz
MA, Kapoor P, Buadi FK, Leung N, Dingli D, Go RS, et al:
Beta-blockers improve survival outcomes in patients with multiple
myeloma: A retrospective evaluation. Am J Hematol. 92:50–55. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang PY, Huang WY, Lin CL, Huang TC, Wu
YY, Chen JH and Kao CH: Propranolol reduces cancer risk: A
population-based cohort study. Medicine (Baltimore). 94:e10972015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pantziarka P, Bouche G, Sukhatme V, Meheus
L, Rooman I and Sukhatme VP: Repurposing drugs in oncology
(ReDO)-propranolol as an anti-cancer agent. Ecancermedicalscience.
10:6802016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thiele M, Wiest R, Gluud LL, Albillos A
and Krag A: Can non-selective beta-blockers prevent hepatocellular
carcinoma in patients with cirrhosis? Med Hypotheses. 81:871–874.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Herrera I, Pascual S, Zapater P, Carnicer
F, Bellot P and María Palazón J: The use of β-blockers is
associated with a lower risk of developing hepatocellular carcinoma
in patients with cirrhosis. Eur J Gastroenterol Hepatol.
28:1194–1197. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu HH, Liu J, Lin YL, Luo WS, Chu YJ,
Chang CL, Jen CL, Lee MH, Lu SN, Wang LY, et al: The rs2296651
(S267F) variant on NTCP (SLC10A1) is inversely associated with
chronic hepatitis B and progression to cirrhosis and hepatocellular
carcinoma in patients with chronic hepatitis B. Gut. 65:1514–1521.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cavuoto J: Neurotech report.
Neuromodulation. 14:1032011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee HS, Chung YH and Kim CY: Specificities
of serum alpha-fetoprotein in HBsAg+ and HBsAg-patients in the
diagnosis of hepatocellular carcinoma. Hepatology. 14:68–72. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lopez-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: Hep G2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kassahun WT, Guenl B, Ungemach FR, Jonas S
and Abraham G: Expression and functional coupling of liver
β2-adrenoceptors in the human hepatocellular carcinoma.
Pharmacology. 89:313–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bevilacqua M, Norbiato G, Chebat E, Baldi
G, Bertora P, Regalia E, Colella G, Gennari L and Vago T: Changes
in alpha-1 and beta-2 adrenoceptor density in human hepatocellular
carcinoma. Cancer. 67:2543–2551. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Iseri OD, Sahin FI, Terzi YK, Yurtcu E,
Erdem SR and Sarialioglu F: Beta-Adrenoreceptor antagonists reduce
cancer cell proliferation, invasion and migration. Pharm Biol.
52:1374–1381. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang ZF, Feng XS, Chen H, Duan ZJ, Wang
LX, Yang D, Liu PX, Zhang QP, Jin YL, Sun ZG and Liu H: Prognostic
significance of synergistic hexokinase-2 and beta2-adrenergic
receptor expression in human hepatocelluar carcinoma after curative
resection. BMC Gastroenterol. 16:572016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu FQ, Fang T, Yu LX, Lv GS, Lv HW, Liang
D, Li T, Wang CZ, Tan YX, Ding J, et al: ADRB2 signaling promotes
HCC progression and sorafenib resistance by inhibiting autophagic
degradation of HIF1α. J Hepatol. 65:314–324. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou C, Chen X, Zeng W, Peng C, Huang G,
Li X, Ouyang Z, Luo Y, Xu X, Xu B, et al: Propranolol induced
G0/G1/S phase arrest and apoptosis in melanoma cells via AKT/MAPK
pathway. Oncotarget. 7:68314–68327. 2016.PubMed/NCBI
|