Introduction
Severe sepsis and septic shock account for 20% of
all admissions to intensive care units and remains the most common
cause of mortality resulting from nosocomial infections (1,2).
Severe sepsis is characterized by acute organ dysfunction,
including heart, lung and liver. Cardiac dysfunction is conferred
to impaired myocardial function and collapsed circulation, and has
been demonstrated to be the highest risk factor for severe
sepsis-linked mortality (3). The
mechanisms underlying severe sepsis-induced acute cardiac
dysfunction are considered to involve an excessive inflammatory
response leading to the overexpression and release of
proinflammatory cytokines, in addition to neutrophil hyperactivity
(4). It has been reported that
injured cardiomyocytes release excessive proinflammatory cytokines,
including tumor necrosis factor (TNF)-α, interleukin (IL)-1 and
IL-6, thus leading to marked neutrophil aggregation and filtration
in the heart in severe sepsis (4,5).
Toll-like receptor (TLR) 4 is a transmembrane
pattern-recognition receptor, which is a key component of the
innate immune system and is involved in the modulation of the
sepsis-induced inflammatory response. TLR4 detects
pathogen-associated molecular patterns and then binds to bacterial
lipopolysaccharide (LPS). Activation of TLR4 has been reported to
induce inflammatory responses involved in the impairment of cardiac
contractility. Therefore, TLR4 has been proposed as a potential
therapeutic target to control the inflammatory response and improve
cardiac function (6). Numerous
studies revealed that TLR4 promotes cardiac dysfunction, induced by
severe sepsis, particularly in the presence of high-dose endotoxin
(7,8). Severe sepsis is characterized by
numerous bacterial infections and can be mimicked in animal models.
However, accumulating evidence has demonstrated that the inhibition
of TLR4 during inflammation may alleviate heart failure by
suppressing inflammatory responses mediated by the TLR4-myeloid
differentiation primary response 88 (MyD88) signaling pathway and
toll or interleukin-1 receptor-domain-containing adapter-inducing
interferon-β (TRIF), another adaptor signal, which is also
associated with this inflammatory response. Therefore, the
mechanisms of TLR4 in heart dysfunction during severe sepsis
require further investigation.
Additional studies investigated the apoptotic
pathway which is activated in cardiomyocytes by inflammatory
mediators in septic cardiomyopathy (9,10).
Activation of apoptosis regulatory factors, including caspase 3,
have been reported to account for cardiomyopathy following septic
challenge (10). Evidence of these
studies revealed that the apoptotic pathway is associated with a
partially reversible decrease in cardiac myocyte fractional
shortening and cytokine decrease (11). However, few reports have indicated
that TLR4 is associated with septic heart apoptosis. Therefore, the
present study aimed to investigate the effects of TLR4 deletion on
myocardial apoptosis following cecum ligation and puncture
(CLP).
In the present study, a modified procedure of CLP
was employed to establish severe sepsis models on wild type (WT)
and TLR4 deficient (TLR4-KO) mice to investigate the role of TLR4
signaling pathways in cardiac dysfunction during severe sepsis.
Materials and methods
Animal models
WT and TLR4-KO male mice (n=80), weighing 20–25g and
aged 6–8 weeks, were purchased from the Model Animal Research
Center of Nanjing University (Stock: J003752; Nanjing, China).
TLR4-KO mice (C57BL/10ScNJNju) were progenies of C57BL/10ScN from
the Jackson Laboratory (Ben Harbor, ME, USA), harboring a II12rb2
allele deletion. Animals were separately housed at 26°C by sex and
maintained in a specific pathogen free and humid (50%) environment
exposed to a 12 h light/dark cycle; animals had ad libitum
access to food and water. All experimental procedures were approved
by the medical ethical committee of the Second Xiangya Hospital of
Central South University. Bowel perforation (CLP) was used to
establish severe sepsis. Briefly, all mice were anesthetized with
1.5% pentobarbital sodium [40 mg/kg, intraperitoneal (i.p).;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany]. A 1.0 cm long
incision was performed on the abdomen and the cecum was exposed,
ligated by silk 4-0 below the ileocecal valve and punctured twice
with a 20-gauge needle. The sham group underwent laparotomy however
without CLP. A total of 16 mice were divided randomly into two
groups (n=8 each) for observation of survival rate, 64 mice were
divided randomly into four groups (n=16 each, 8 for Langendorff
system analysis and 8 for serum and heart sample analysis):
WT-sham, TLR4-KO-sham, WT-CLP, and TLR4-KO-CLP group. All surgeries
were performed by operators blinded to the genotype
information.
All mice were anesthetized with 1.5% pentobarbital
sodium (40 mg/kg, i.p.; Sigma-Aldrich; Merck) and cardiac function
was evaluated using a S3000 ultrasound scanner (Acuson S3000,
Siemens Healthcare, Erlangen, Germany) coupled with an 18.0 MHz
linear transducer (Siemens Healthcare). All images were collected
by a single experienced operator who was blinded to experimental
design. Fractional shortening (FS) was calculated using M-mode
method at the mid-papillary level in the parasternal short-axis
view. Strain was obtained in the middle of the posterior wall on
short-axis views during ≥3 consecutive heartbeats. Strain was
analyzed online using Software Velocity Vector Imaging (VVI, 3.5,
Siemens Healthcare).
Langendorff system
Left ventricular (LV) function of the hearts
isolated from septic or sham mice were measured 12 h following the
surgical procedure using a Langendorff perfusion apparatus as
previously described (7,12). Briefly, mice were heparinized
(1,000 IU/kg) and anesthetized pentobarbital sodium, 40 mg/kg,
i.p.). The hearts were excised and immersed immediately in cold
(4°C) perfusion fluid (Sigma-Aldrich; Merck KGaA). The aortas were
cannulated and retrograde-perfusion was performed at a constant
flow rate (3 ml/min) with modified Krebs-Henseleit buffer
(Sigma-Aldrich; Merck KGaA), while the heart was paced at 7 Hz (420
beats/min). Following 20 min of coronary perfusion, LV end-systolic
pressure (LVESP), LV end-diastolic pressure (LVEDP) and the heart
rate were recorded for ≤30 min. LV developed pressure (LVDP) was
calculated as follows: LVDP=LVESP-LVEDP; +dP/dtmax was
defined as peak rate of left ventricular pressure rise.
Measurement of serum cardiac troponin
I (cTnI)
Blood samples were collected via the inferior vena
cava of the mice 12 h following CLP under anesthesia with
pentobarbital sodium (40 mg/kg, i.p.). Mice were then sacrificed
via cervical dislocation. Subsequently, the blood samples were
centrifuged at 589 × g for 10 min at 4°C to obtain the supernatant,
which was immediately stored at −20°C until further analysis.
Troponin I (cTnI) levels in serum were measured by ELISA
(Quantikine Mouse kit, KT29998, MSK Biotechnology Co., Ltd., Wuhan,
China) according to the manufacturer's protocols.
Quantification of expression levels of
inflammatory cytokines (IL-1, IL-6, TNF-α) and MyD88, TRIF, nuclear
factor-κB (NF-κB) in heart tissues
Following euthanasia, heart tissues of mice were
harvested. Total RNA was purified from heart tissue using
TRIzol® reagent (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the manufacture's protocols. Reverse
transcription (RT) and PCR were performed to amplify mouse IL-1,
IL-6, TNF-α, MyD88, TRIF, NF-κB and β-actin mRNA. Using 2 µl
reverse transcriptase (Promega Corporation, Madison, WI, USA),
reactions were performed with a final volume of 20 µl using
gene-specific primers. Additionally, the expression of the selected
genes was normalized to that of β-actin as an internal control. PCR
amplification was conducted at 94°C for 4 min and products were
evaluated by 1.7% agarose gel electrophoresis and stained with 0.5
ug/l ethidium bromide at 50–60°C. The integral optical density
(IOD) of the electrophoretic bands was quantified. Therefore, the
data in the figures was the ratio of IOD of target gene to the IOD
of reference gene. Results were interpreted using Image-Pro Plus
6.0 (Media Cybernetics, Inc., Rockville, MD, USA).
Myeloperoxidase (MPO) assay
The heart sample were excised and washed with
ice-cold saline. The ventricles were weighed, minced and
homogenized to 5% heart tissue homogenate (weigh proportion, 1:19)
in a solution containing 0.5% hexa-decyltrimethyl-ammonium bromide
dissolved in 60 ml PBS. Then, 0.9 ml tissue homogenate was mixed
well with 0.1 ml MPO reagent III (Jiancheng Bioengineering
Institute, Nanjing, China). The mixture was incubated for 15 min at
37°C and then incubated in a 60°C water-bath for 10 min, during
which the colorimetric ware (Jiancheng Bioengineering Institute)
and H2O2 were added to the resulting mixture.
Subsequently, the rate of alteration in absorbance at a wavelength
of 460 nm was measured using a spectrophotometer (CE 9000; Cecil
Instruments, Ltd., Cambridge, UK). MPO activity was expressed as
the content of MPO in the tissue homogenate per liter (U/l)
Histopathological examinations
Samples of heart were dissected and fixed in 10%
buffered formalin (Rongbo Bioengineering Institute, Shanghai,
China) at 26°C for 24 h, and subsequently embedded in paraffin.
Then, the tissue sections were dewaxed, hydrated, incubated with
EDTA antigen retrieval buffer solution (pH 9.0) for 8 min at 100°C
and treated with 3% bovine serum albumin (BSA; Sigma-Aldrich; Merck
KGaA) for 30 min at room temperature. Sections were rehydrated in
PBS and 0.1% BSA for 15 min. Samples were cut to 5 µm thickness and
stained with hematoxylin (5 min at 26°C) and eosin (40 sec at 26°C)
by two separate pathologists. To assess the neutrophil accumulation
and macrophages in heart tissues, the sections were incubated with
rabbit polyclonal anti-Gr-1 antibody (1:200, ab25377, Abcam,
Cambridge, UK) and rabbit polyclonal anti-cluster of
differentiation 45 (CD45) antibody (1:200, ab3638, Abcam),
respectively, overnight at 4°C. Following rinsing, the sections
were incubated with biotinylated goat anti-rabbit immunoglobulin G
(1:200; G23303; Jackson ImmunoResearch Laboratories, Inc., West
Grove, PA, USA) for 50 min at room temperature. The tissue sections
were treated with a 3,3′-diaminobenzidine staining system (Dako;
Agilent Technologies, Inc., Santa Clara, CA, USA) according to the
manufacturer's protocols. The slides were observed under a light
microscope (Zeiss AG, Oberkochen, Germany) at magnifications of
×200 and ×400. The Image-Pro P1us 6.0 image analysis system (Media
Cybernetics, Inc.) was used to analyze the images.
Quantification of caspase-3, Fas cell
surface death receptor (FAS)/Fas ligand (FASL) mRNA in heart
tissue
Caspase-3, FAS/FASL mRNA were measured using the
aforementioned RT-PCR procedure.
Statistical analysis
Data are presented as the mean ± standard deviation
organized by GraphPad Prism 5.0 software (GraphPad Software, Inc.,
La Jolla, CA, USA). Data was analyzed by two-way analysis of
variance followed by a Bonferroni post hoc test for statistical
significance between groups. Survival rate analysis was estimated
by log-rank test. For all tests, P<0.05 was considered to
indicate a statistically significant difference.
Results
WT mice exhibit decreased survival
rates compared with TLR4-KO mice during severe sepsis
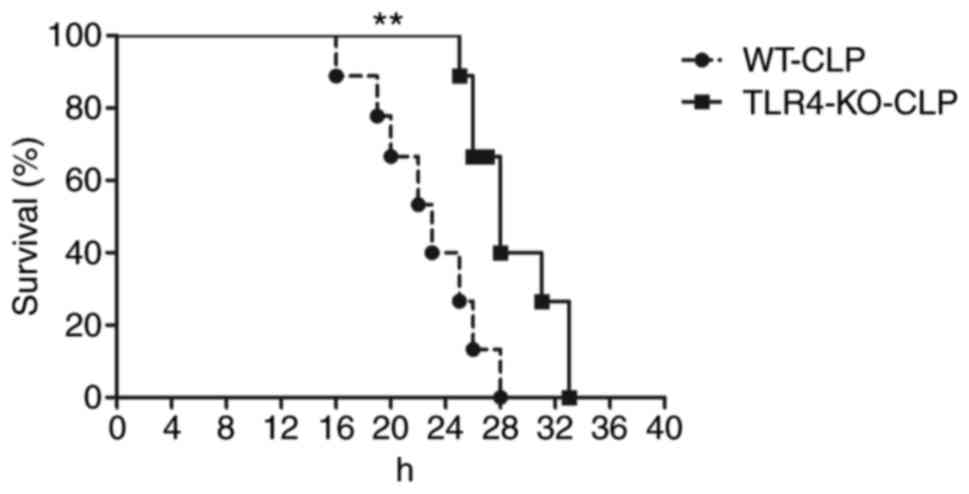
A total of 12 h following CLP, WT mice revealed
septic symptoms, including ruffled hair, slow physical actions,
shivering and low temperature. The survival rate at 24 h was 40%,
whereas TLR4-KO mice presented moderate unhealthy activities
throughout the observation period and exhibited no mortality at 24
h following CLP (Fig. 1).
Additionally, hemodynamic analysis was preformed to
further investigate the effect of TLR4 signaling to cardiovascular
function during severe sepsis. As presented in Table I, WT and TLR4-KO mice demonstrated
hypotension despite fluid resuscitation following CLP surgery.
Compared with sham mice, a 22% decrease in blood pressure in
TLR4-KO-CLP mice was observed compared with a 60% decrease in
WT-CLP mice. There was no difference between WT-sham and
TLR4-KO-sham mice with respect to subtle hemodynamic alterations
during the sham operation.
 | Table I.Serial echocardiographic measurements
prior to and following CLP. |
Table I.
Serial echocardiographic measurements
prior to and following CLP.
|
| Sham | CLP |
|---|
|
|
|
|
|---|
| Measurement | Baseline | 6 h | 12 h | 24 h | Baseline | 6 h | 12 h | 24 h |
|---|
| Heart rate,
bpm |
|
|
|
|
|
|
|
|
| WT | 598±11 | 612±12 | 622±13 | 618±9 | 601±14 | 512±19 | 493±25b | 480±30b |
|
TLR4-KO | 601±10 | 609±14 | 614±11 | 615±8 | 604±12 | 598±11 | 615±12 | 590±19 |
| Mean blood pressure
(mm Hg) |
|
|
|
|
|
|
|
|
| WT | 84±2.0 | 84±2.0 | 84±3.0 | 83±3.2 | 94±3.2 | 72±4.3a | 55±6.8b | 37±6.0b |
|
TLR4-KO | 86±3.0 | 86±2.6 | 84±2.0 | 85±2.3 | 88±2.2 | 80±3.0a | 71±4.5b | 68±6.1b |
| Strain |
|
|
|
|
|
|
|
|
| WT | 19.6±1.7 | 20.3±2.0 | 19.3±2.4 | 19.1±2.1 | 20.3±2.2 |
16.6±1.7a |
14.5±2.0b |
13.4±2.0b |
|
TLR4-KO | 20.2±1.5 | 20.4±2.3 | 20.4±1.4 | 19.4±2.2 | 19.8±2.4 |
18.7±2.2c |
17.6±2.5a,c |
16.3±1.8a,c |
TLR4-KO mice maintain better cardiac
function compared with WT mice in severe sepsis
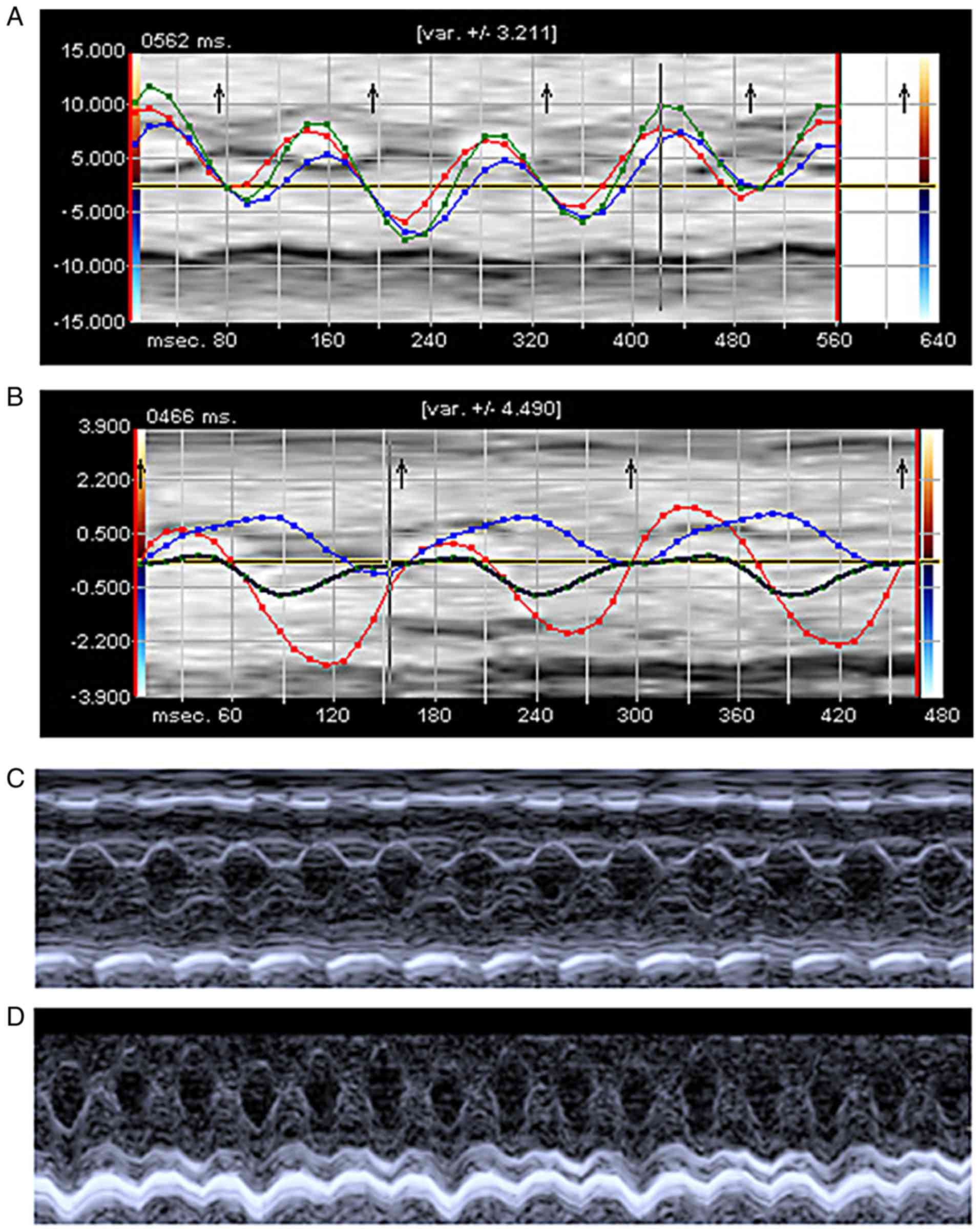
The VVI technique was used to measure cardiac
function of mice at 6, 12, and 24 h following sham or CLP
operation. There was a significant deterioration of LV function in
WT-CLP mice compared with WT-sham mice. At 6 h post-CLP, there was
a marked attenuation of strain (16.6 vs. 18.7%) in WT-CLP mice
compared with TLR4-KO-CLP group. In TLR4-KO-CLP mice, the global
longitudinal strain was significantly increased compared with
WT-CLP mice at 12 and 24 h following operation (14.5 vs. 17.6%;
13.4 vs. 16.3%; Fig. 2A and B;
Table I). TLR4-KO-CLP mice
revealed a similar level of FS to TLR4-KO -sham mice at 6 h
following CLP (P>0.05), and increased FS at 12 and 24 h than
WT-CLP mice (Fig. 2C and D;
Table I). In addition, LV function
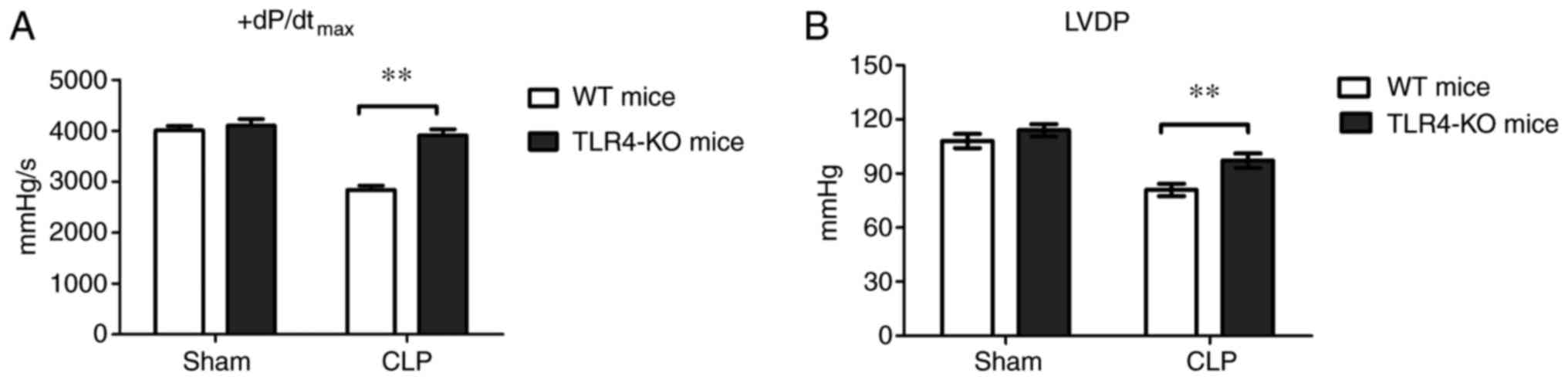
of the hearts isolated from sham or septic mice was assessed ex
vivo. The isolated hearts were perfused in a Langendorff system
with a constant preload. The results demonstrated that there was no
difference in LVDP and +dP/dtmax between WT-sham and
TLR4-KO-sham mice (Fig. 3);
however, following CLP surgery, TLR4-KO mice presented increased
LVDP and +dP/dtmax compared with WT mice (Fig. 3).
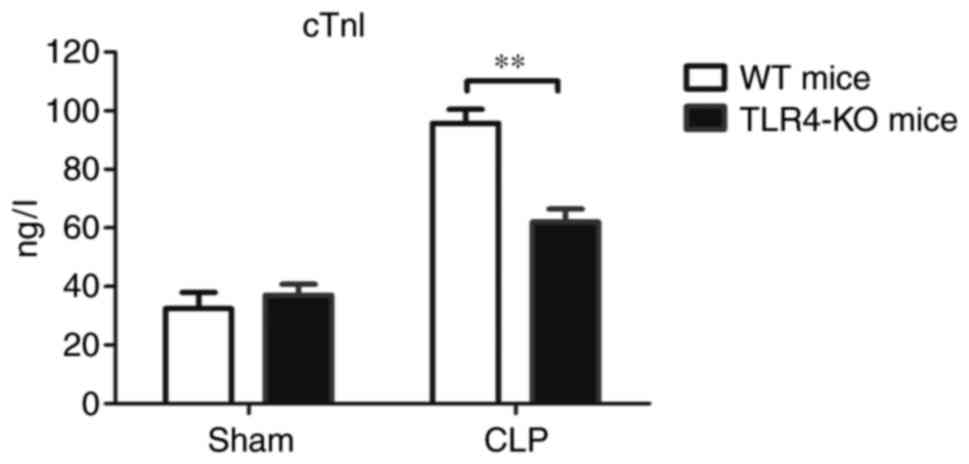
Serum levels of cTnI, a cardiac injury biomarker,
were analyzed in mice of the four experimental groups 12 h
post-CLP. The results revealed that the circulating levels of cTnI
in WT-CLP mice were significantly increased compared with
TLR4-KO-CLP mice (Fig. 4).
TLR4-KO mice have reduced levels of
proinflammatory cytokines compared with WT mice during severe
sepsis
To determine the impact of TLR4 on the induction of
inflammatory cytokines, including TNF, IL-1, and IL-6 in severe
sepsis, RT-PCR was conducted to measure cytokine mRNA expression
levels in heart tissue. As presented in Table II, high tissue concentrations of
TNF mRNA were detected in WT mice following CLP compared with
WT-sham mice. However, in the TLR4-KO-CLP group, there were
significantly decreased levels of TNF mRNA expression compared with
in the WT-CLP group. Similarly, tissue expression levels of IL-1
and IL-6 mRNA were significantly upregulated in WT-CLP mice
compared with in TLR4-KO-CLP mice, respectively (Fig. 5; Table II).
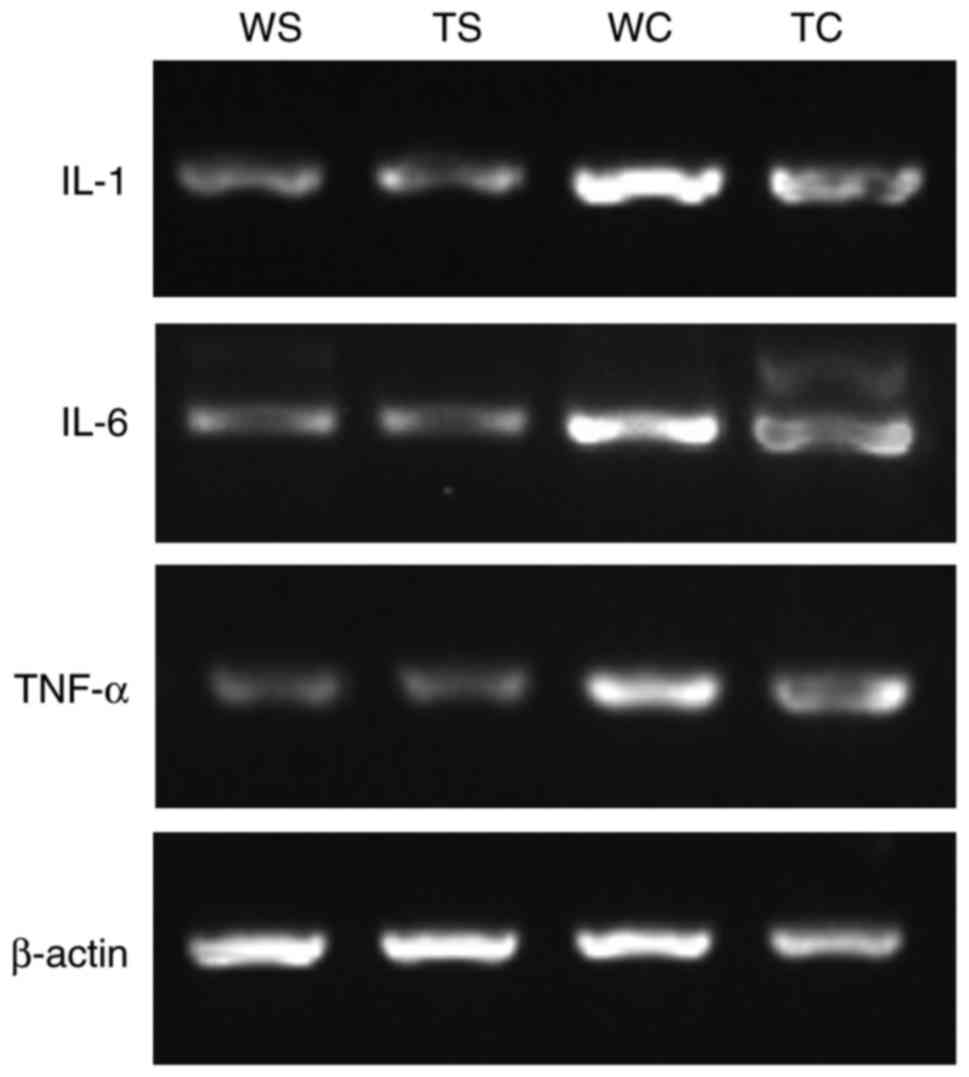 | Figure 5.IL-1, IL-6 and TNF-α mRNA expression
levels in heart following CLP. Representative image of mRNA
expression levels detected by reverse transcription-semi
quantitative polymerase chain reaction. CLP, cecum ligation and
puncture; IL, interleukin; TNF-α, tumor necrosis factor-α. TLR4-KO,
Toll-like receptor 4 knockout; WT, wild-type; WS, WT-Sham; TC,
TLR4-KO-CLP; TS, TLR4-KO-Sham; WC, WT-CLP. |
| Table II.IL-1, IL-6 and TNF-α mRNA expression
levels in the heart following CLP. |
Table II.
IL-1, IL-6 and TNF-α mRNA expression
levels in the heart following CLP.
| Group | IL-1/actin | IL-6/actin | TNF-α/actin |
|---|
| Sham-WT | 0.455±0.082 | 0.337±0.045 | 0.327±0.038 |
| Sham-TLR4-KO | 0.460±0.051 | 0.331±0.600 | 0.322±0.042 |
| CLP-WT |
0.878±0.040a |
0.670±0.450a |
0.652±0.051a |
| CLP-TLR4-KO |
0.654±0.047b |
0.584±0.480b | 0.470±0.033 |
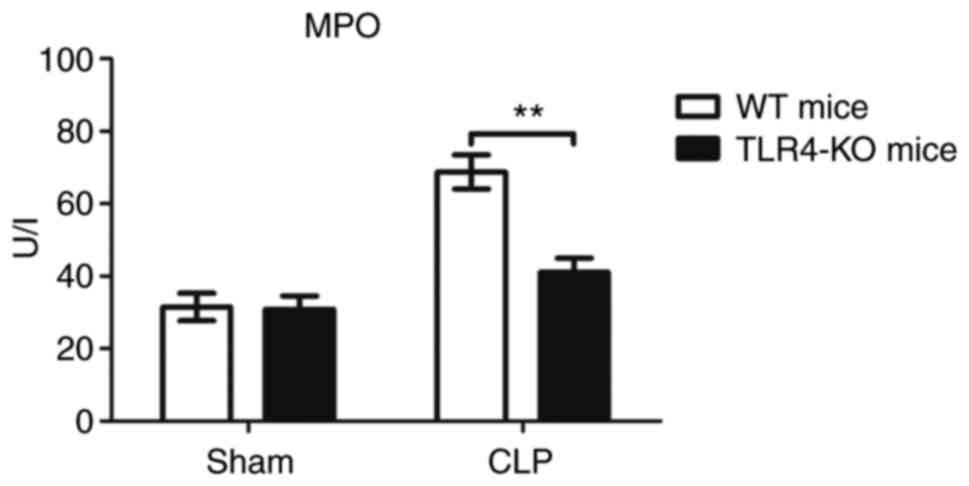
Knockout of TLR4 inhibits neutrophil
activation by severe polymicrobial sepsis
To evaluate the degree of neutrophil infiltration in
myocardium of these four groups, MPO activity was determined in the
heart. As presented in Fig. 6,
there was no significant difference in MPO activity between the
WT-sham and TLR4-KO-sham groups; however, there was a significant
decrease in MPO activity in the myocardial tissue of TLR4-KO-CLP
mice compared with in WT-CLP mice (Fig. 6).
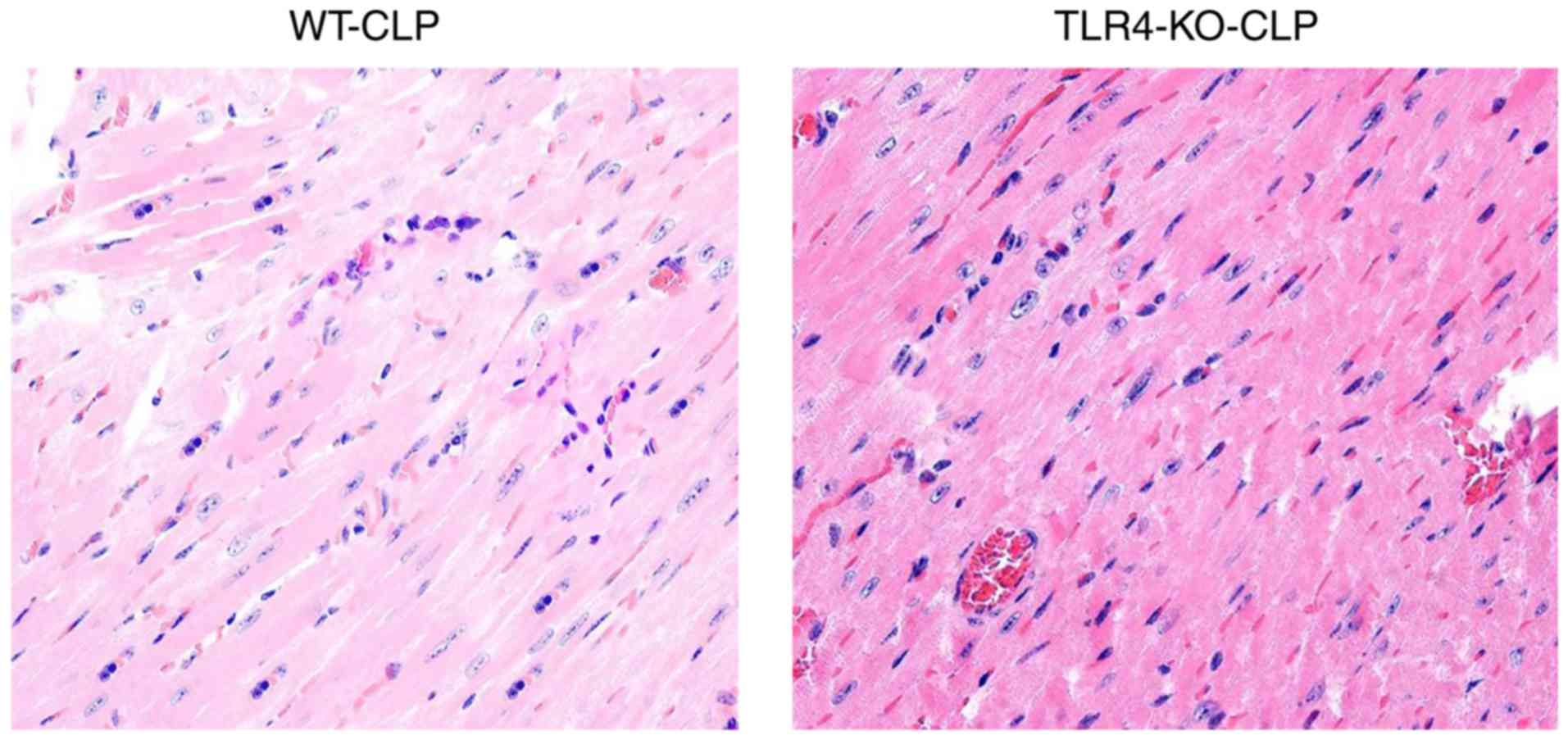
TLR4-KO mice exhibit a better
myocardium structure and less neutrophil and macrophage
infiltration compared with WT mice during severe sepsis
In the TLR4-KO-CLP groups, myocardial fibers were
arranged regularly with distinct striations and no apparent
degeneration or necrosis was observed; however, the myocardium of
WT-CLP mice revealed edema and karyopyknosis, along with abundant
fibroblastic hyperplasia in part of myocardium (Fig. 7).
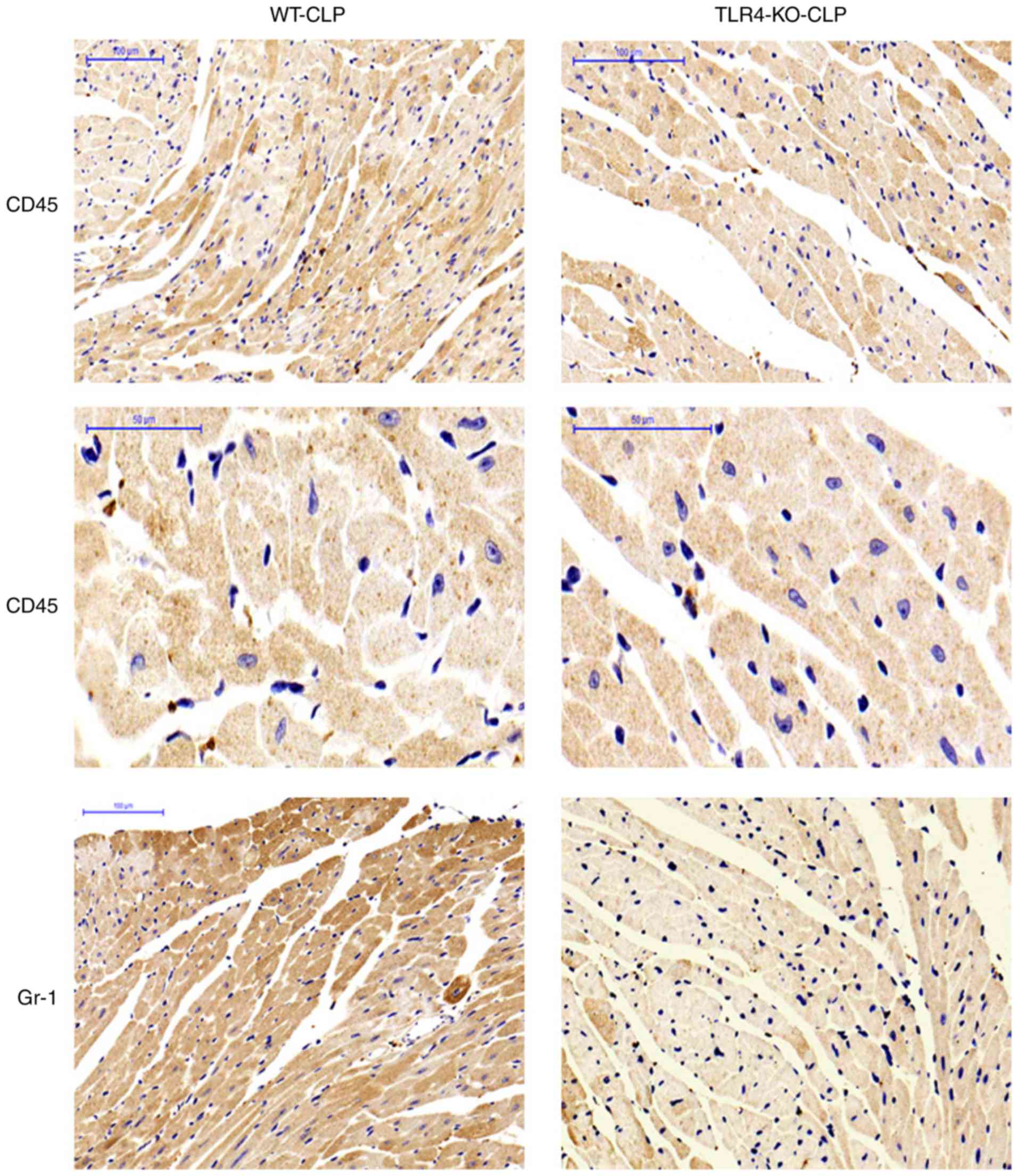
Neutrophils and macrophages were detected in cardiac
myocytes by Gr-1 and CD45 immunohistochemical staining and
represented by a pervasive brown color. As presented in Fig. 8, the numbers of neutrophils and
macrophages in the TLR4-KO mice heart tissue were significantly
decreased following CLP compared with the WT mice. This finding is
consistent with the data of myocardial MPO results.
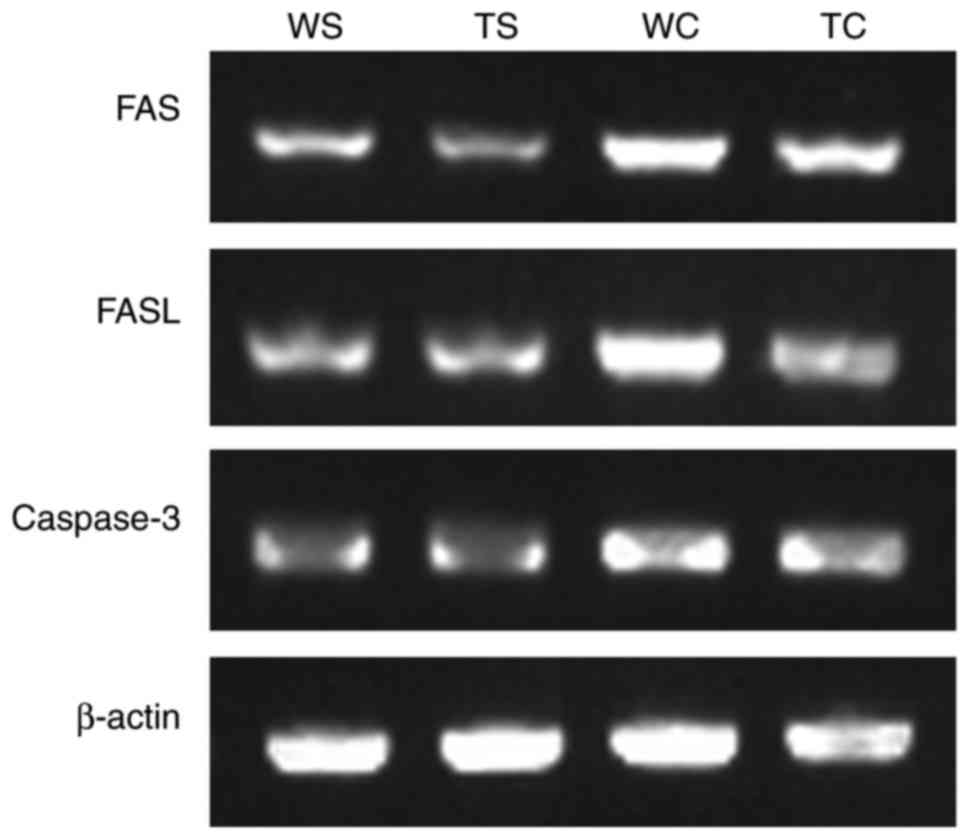
TLR4-KO mice leads to attenuated
myocardial apoptosis during severe polymicrobial sepsis
In contrast to WT-sham mice, the WT-CLP mice
revealed a marked increase in FAS/FASL and caspase-3 expression;
however, TLR4-KO mice exhibited lower levels of FAS/FASL and
caspase-3 expression levels compared with in WT-CLP mice (Fig. 9; Table III).
| Table III.FAS/FASL and caspase-3 mRNA
expression levels in the heart following CLP. |
Table III.
FAS/FASL and caspase-3 mRNA
expression levels in the heart following CLP.
| Group | FAS/actin | FASL/actin |
Caspase-3/actin |
|---|
| Sham-WT | 0.43±0.08 | 0.45±0.07 | 0.46±0.05 |
| Sham-TLR4-KO | 0.38±0.04 | 0.40±0.06 | 0.44±0.07 |
| CLP-WT |
0.91±0.13a |
0.95±0.09a |
0.75±0.04a |
| CLP-TLR4-KO |
0.71±0.05a,b |
0.66±0.10a,b |
0.52±0.06b |
Expression of myocardial MyD88, TRIF
and NF-κB following CLP procedure
Expression of MyD88, TRIF and NF-κB in mice heart
increased following the CLP procedure in WT and TLR4-KO group;
however, compared with WT-CLP mice, TLR4-KO-CLP mice expressed
significantly decreased level of myocardial MyD88, TRIF and NF-κB
mRNA (P<0.05; Fig. 10;
Table IV).
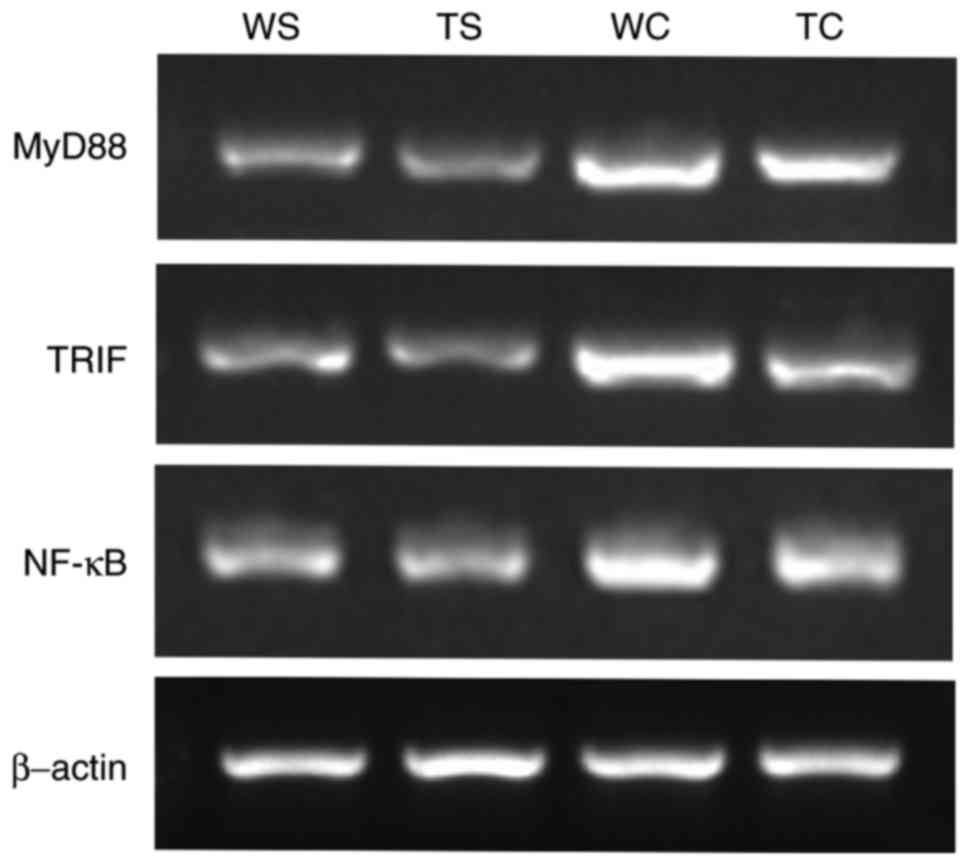 | Figure 10.MyD88, TRIF and NF-κB mRNA expression
levels in the heart following CLP. Representative image of mRNA
expression levels detected by reverse transcription-semi
quantitative polymerase chain reaction. CLP, cecum ligation and
puncture; WT, wild-type; TLR4-KO, Toll-like receptor 4 knockout;
MyD88, myeloid differentiation factor 88; NF-κB, nuclear factor-κB;
TC, TLR4-KO-CLP; TRIF, toll or interleukin-1
receptor-domain-containing adaptor-inducing interferon-β; TS,
TLR4-KO-Sham; WS, WT-Sham; WC, WT-CLP. |
| Table IV.MyD88, TRIF and NF-κB mRNA expression
levels in heart following CLP. |
Table IV.
MyD88, TRIF and NF-κB mRNA expression
levels in heart following CLP.
| Group | MyD88/actin | TRIF/actin | NF-κB/actin |
|---|
| Sham-WT | 0.36±0.03 | 0.40±0.03 | 0.50±0.05 |
| Sham-TLR4-KO | 0.31±0.04 | 0.34±0.10 | 0.48±0.06 |
| CLP-WT |
0.83±0.06a |
0.89±0.12a |
0.82±0.02a |
| CLP-TLR4-KO |
0.60±0.06b |
0.56±0.08b |
0.64±0.08b |
Discussion
Severe sepsis is defined as a systemic
hyperinflammatory response with multiple organ failure (13) of which cardiovascular disorder is a
primary associated complication (14). Cardiac dysfunction in severe sepsis
is the manifestation of unregulated inflammatory reactions
(15) and cardiomyocyte apoptosis
(16). The findings of the present
study revealed that TLR4 is involved in the development of severe
sepsis-induced myocardial dysfunction, in part via activation of
proinflammatory cytokines and promoting myocardial neutrophil
infiltration. Knockout of TLR4 resulted in protection of
sepsis-induced myocardial apoptosis.
The mechanisms primarily involved in inappropriate
proinflammatory response constitute excessive cytokine secretion,
including IL-1, IL-6, and TNF-α, and inappropriate neutrophil
infiltration. Increased levels of proinflammatory cytokines and
neutrophil infiltration may injure the endothelium of the blood
vessel, promote platelet aggregation and adhesion to endothelium,
and block the blood flow, subsequently resulting in myocardial
ischemic injury. In addition, they contribute to myocardial
depression by producing numerous myocardial depressant substances.
They also have significant cardiotoxic effects (17), resulting in calcium ion leakage and
left ventricular impairment (18,19).
It has previously been demonstrated that TLR4 is a
key mediator in the signal transduction of systemic inflammatory
response syndrome (20,21). TLR4 also recognizes LPS; the
combination of TLR4 and LPS results in TLR4 dimerization and
induces intracellular signaling pathways that lead to the
activation of cytosolic nuclear factor NF-κB, which increases the
transcription of the aforementioned proinflammatory cytokines
(7,8,15,22).
In lethal endotoxic sepsis, TLR4 has been
demonstrated to serve a critical role in cardiac depression
(23). In the present study, a CLP
model was employed to induce severe sepsis; the results revealed
that in severe sepsis, TLR4-KO-CLP mice exhibited increased
survival rates and more efficient cardiac function, including
improved echocardiographic parameters in vivo, better
isolated heart pump function in vitro, and lower levels of
cTnI compared with WT-CLP mice. However, in non-lethal models of
sepsis, which present low mortality and ineffective host defense,
TLR4 appears to protect cardiac function from septic damage
(24). The role of TLR4 signaling
in the pathogenesis of sepsis is complex and may well depend on the
severity of sepsis. In sublethal sepsis, where inflammatory
suppression dominates the underlying pathology, TLR4 may induce an
effective innate immune defense to protect against myocardial
injury; however, the key underlying the severe sepsis is systemic
hyperinflammation featured by an inflammatory cascade response and
the deletion of TLR4, which mediates the harmful hyperinflammatory
response. In accordance with this theory, the present study
provided compelling evidence that elevated myocardial levels of
proinflammatory cytokines are closely associated with the levels of
cardiac stress biomarkers and heart depression in WT-CLP mice,
whereas the significant decrease in concentration of these factors
in TLR4-KO-CLP mice may be associated with improved cardiac
function (5). The findings of the
present study support the notion that high levels of
proinflammatory cytokines are potentially associated with impaired
cardiac function in severe sepsis and may be effectively protected
by knocking out the TLR4 gene.
Increasing evidence suggests that during severe
sepsis, apoptotic pathways are stimulated within the myocardium and
are closely associated with myocardial depression (25). Previous studies reported that
FAS/FASL and caspase-3 may serve a role in regulating cardiac
contraction and sarcomere disarray (26,27).
The present study reported that the CLP procedure evokes FAS/FASL
and caspase 3 production, subsequently resulting in myocardial
injury, and may be prevented by the deletion of TLR4. However, the
present study did not investigate the associated pathway of
FAS/FASL and caspase-3.
CLP induces severe sepsis in the mice via TLR4
signaling mediated by the TLR4-MyD88 and TLR4-TRIF signaling
pathways (28). NF-κB is closely
associated with the inflammatory reaction. Recruitment of MyD88
leads to the activation of cytosolic NF-κB to regulate
proinflammatory cytokine gene expression (29). The TLR4-TRIF-dependent pathway
induces type I interferon (IFN) regulatory factor 3, IFN-b and
slower NF-κB activation, and regulates the production of various
cytokines, including inflammatory cytokines and
apoptosis-associated inducing factors, which represent the primary
host antiviral mechanism (30). It
was reported in the present study that MyD88, TRIF and NF-κB mRNA
expression levels were decreased following the deletion of TLR4.
MyD88 and TRIF may serve important roles in preventing cardiac
impairment during severe sepsis; however, further investigation is
required.
In conclusion, it was demonstrated that knockout of
TLR4 gene improved survival and cardiac function in severe sepsis
induced by CLP by decreasing the myocardial levels of inflammatory
cytokines, weakening neutrophil infiltration in myocardium, and
attenuating the heart apoptosis. Targeting the TLR4 signaling
pathway may be a potential therapeutic treatment for severe
sepsis-associated myocardial dysfunction in clinical practice.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81201096 and
81401431), Hunan Provincial Natural Science Foundation of China
(grant no. 2017JJ3443) and the Hunan Province Science &
Technology program (grant no. 2013SK3041). The funders had no role
in study design, data collection and analysis, decision to publish,
or preparation of the manuscript.
References
|
1
|
Angus DC, Linde-Zwirble WT, Lidicker J,
Clermont G, Carcillo J and Pinsky MR: Epidemiology of severe sepsis
in the United States: Analysis of incidence, outcome, and
associated costs of care. Crit Care Med. 29:1303–1310. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Natanson C, Hoffman WD, Suffredini AF,
Eichacker PQ and Danner RL: Selected treatment strategies for
septic shock based on proposed mechanisms of pathogenesis. Ann
Intern Med. 120:771–783. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Parrillo JE, Parker MM, Natanson C,
Suffredini AF, Danner RL, Cunnion RE and Ognibene FP: Septic shock
in humans. Advances in the understanding of pathogenesis,
cardiovascular dysfunction, and therapy. Ann Intern Med.
113:227–242. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Antonucci E, Fiaccadori E, Donadello K,
Taccone FS, Franchi F and Scolletta S: Myocardial depression in
sepsis: From pathogenesis to clinical manifestations and treatment.
J Crit Care. 29:500–511. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baumgarten G, Knuefermann P, Nozaki N,
Sivasubramanian N, Mann DL and Vallejo JG: In vivo expression of
proinflammatory mediators in the adult heart after endotoxin
administration: The role of toll-like receptor-4. J Infect Dis.
183:1617–1624. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Topkara VK, Evans S, Zhang W, Epelman S,
Staloch L, Barger PM and Mann DL: Therapeutic targeting of innate
immunity in the failing heart. J Mol Cell Cardiol. 51:594–599.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang E, Feng Y, Zhang M, Zou L, Li Y, Buys
ES, Huang P, Brouckaert P and Chao W: Toll-like receptor 4
signaling confers cardiac protection against ischemic injury via
inducible nitric oxide synthase- and soluble guanylate
cyclase-dependent mechanisms. Anesthesiology. 114:603–613. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Binck BW, Tsen MF, Islas M, White DJ,
Schultz RA, Willis MS, Garcia JV, Horton JW and Thomas JA: Bone
marrow-derived cells contribute to contractile dysfunction in
endotoxic shock. Am J Physiol Heart Circ Physiol. 288:H577–H583.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bergmann MW, Loser P, Dietz R and von
Harsdorf R: Effect of NF-kappa B Inhibition on TNF-alpha-induced
apoptosis and downstream pathways in cardiomyocytes. J Mol Cell
Cardiol. 33:1223–1232. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Carlson D, Maass DL, White DJ, Tan J and
Horton JW: Antioxidant vitamin therapy alters sepsis-related
apoptotic myocardial activity and inflammatory responses. Am J
Physiol Heart Circ Physiol. 291:H2779–H2789. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fauvel H, Marchetti P, Obert G, Joulain O,
Chopin C, Formstecher P and Nevière R: Protective effects of
cyclosporin A from endotoxin-induced myocardial dysfunction and
apoptosis in rats. Am J Respir Crit Care Med. 165:449–455. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zou L, Feng Y, Chen YJ, Si R, Shen S, Zhou
Q, Ichinose F, Scherrer-Crosbie M and Chao W: Toll-like receptor 2
plays a critical role in cardiac dysfunction during polymicrobial
sepsis. Crit Care Med. 38:1335–1342. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wichterman KA, Baue AE and Chaudry IH:
Sepsis and septic shock-a review of laboratory models and a
proposal. J Surg Res. 29:189–201. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Levy MM, Fink MP, Marshall JC, Abraham E,
Angus D, Cook D, Cohen J, Opal SM, Vincent JL and Ramsay G;
SCCM/ESICM/ACCP/ATS/SIS, : 2001 SCCM/ESICM/ACCP/ATS/SIS
International Sepsis Definitions Conference. Crit Care Med.
31:1250–1256. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fallach R, Shainberg A, Avlas O, Fainblut
M, Chepurko Y, Porat E and Hochhauser E: Cardiomyocyte Toll-like
receptor 4 is involved in heart dysfunction following septic shock
or myocardial ischemia. J Mol Cell Cardiol. 48:1236–1244. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hsu SP, Chen CC and Chien CT: Pretreatment
of sialic acid efficiently prevents lipopolysaccharide-induced
acute renal failure and suppresses TLR4/gp91-mediated apoptotic
signaling. Kidney Blood Press Res. 41:267–277. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hunter JD and Doddi M: Sepsis and the
heart. Br J Anaesth. 104:3–11. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Casey LC, Balk RA and Bone RC: Plasma
cytokine and endotoxin levels correlate with survival in patients
with the sepsis syndrome. Ann Intern Med. 119:771–778. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Duncan DJ, Yang Z, Hopkins PM, Steele DS
and Harrison SM: TNF-alpha and IL-1beta increase Ca2+ leak from the
sarcoplasmic reticulum and susceptibility to arrhythmia in rat
ventricular myocytes. Cell Calcium. 47:378–386. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Remick DG, Bolgos G, Copeland S and
Siddiqui J: Role of interleukin-6 in mortality from and physiologic
response to sepsis. Infect Immun. 73:2751–2757. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alves-Filho JC, de Freitas A, Russo M and
Cunha FQ: Toll-like receptor 4 signaling leads to neutrophil
migration impairment in polymicrobial sepsis. Crit Care Med.
34:461–470. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nemoto S, Vallejo JG, Knuefermann P, Misra
A, Defreitas G, Carabello BA and Mann DL: Escherichia coli
LPS-induced LV dysfunction: Role of toll-like receptor-4 in the
adult heart. Am J Physiol Heart Circ Physiol. 282:H2316–H2323.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tavener SA, Long EM, Robbins SM, McRae KM,
Van Remmen H and Kubes P: Immune cell Toll-like receptor 4 is
required for cardiac myocyte impairment during endotoxemia. Circ
Res. 95:700–707. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang M, Zou L, Feng Y, Chen YJ, Zhou Q,
Ichinose F and Chao W: Toll-like receptor 4 is essential to
preserving cardiac function and survival in low-grade polymicrobial
sepsis. Anesthesiology. 121:1270–1280. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li X, Luo R, Jiang R, Meng X, Wu X, Zhang
S and Hua W: The role of the Hsp90/Akt pathway in myocardial
calpain-induced caspase-3 activation and apoptosis during sepsis.
BMC Cardiovasc Disord. 13:82013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ruetten H, Badorff C, Ihling C, Zeiher AM
and Dimmeler S: Inhibition of caspase-3 improves contractile
recovery of stunned myocardium, independent of apoptosis-inhibitory
effects. J Am Coll Cardiol. 38:2063–2070. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ren J, Ren BH and Sharma AC:
Sepsis-induced depressed contractile function of isolated
ventricular myocytes is due to altered calcium transient
properties. Shock. 18:285–288. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iwasaki A and Medzhitov R: Toll-like
receptor control of the adaptive immune responses. Nat Immunol.
5:987–995. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen C, Feng Y, Zou L, Wang L, Chen HH,
Cai JY, Xu JM, Sosnovik DE and Chao W: Role of extracellular RNA
and TLR3-Trif signaling in myocardial ischemia-reperfusion injury.
J Am Heart Assoc. 3:e0006832014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey
DR, Visintin A, Latz E, Monks B, Pitha PM and Golenbock DT:
LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll
adapters TRAM and TRIF. J Exp Med. 198:1043–1055. 2003. View Article : Google Scholar : PubMed/NCBI
|