Introduction
Lipopolysaccharide (LPS), present in the external
part of the cell wall of Gram (−) bacteria, can induce sepsis
characterized by an uncontrolled hyper-inflammatory response that
frequently results in multiple organ failure (1). The liver serves a role in the
reduction of inflammation and is responsible for the initiation of
multiple organ failure (2,3). Upon exposure of the liver to LPS,
several inflammatory responses are initiated, including the release
of inflammatory cytokines, and the activation of the
renin-angiotensin (Ang) system (RAS) and the associated Ang
converting enzyme (ACE)-Ang II-Ang I type 1 receptor (AT1R)
(4–6).
Ang-(1–7) is currently recognized as a
biologically active component of non-classic RAS, which,
counteracts Ang II-AT1R by upregulating the production of nitric
oxide and prostaglandins, and mediating anti-fibrosis, vasodilation
and anti-diuretic responses (7–11).
It has also been demonstrated that Ang-(1–7)
decreases LPS-induced inflammatory responses in macrophages
(12). However, the
anti-inflammatory effect of Ang-(1–7) on
LPS-induced liver injury and the underlying molecular mechanism
remain to be elucidated.
Tumor necrosis factor-α (TNF-α) is a cytokine
involved in the progression of numerous inflammatory diseases,
including LPS-induced liver injury (4,13).
Activator protein (AP)-1 is a transcription factor that regulates
the expression of TNF-α in cells (14). In addition, p38 mitogen activated
protein kinase (p38MAPK) serves a role in mediating inflammatory
responses from the extracellular space to the cytoplasm and nucleus
(15). p38MAPK is activated in the
liver by LPS via the phosphorylation and activation of components
of the MAPK signaling pathway, that in turn promote the activation
of AP-1 (16–18). Therefore, targeted inhibition of
the MAPK/AP-1 signaling pathway has been hypothesized to serve a
role in potential anti-inflammatory therapeutic approaches.
In the present study, the anti-inflammatory effect
of Ang-(1–7) was investigated in LPS-induced
hepatocytes, in order to elucidate whether the anti-inflammatory
effect of Ang-(1–7) is mediated via the modulation of the
p38MAPK/AP-1 signaling pathway.
Materials and methods
Cell culture
Immortalized rat liver BRL cells were cultured in
Dulbecco's modified Eagle's medium (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% (v/v) fetal
bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.), 100
IU/ml penicillin and 100 µg/ml streptomycin (Beijing Suolaibao
Biotechnology Co. Ltd., Beijing, China). Cells were incubated at
37°C in a humidified atmosphere containing 5% CO2. All
experiments were carried out following 24 h once cells were seeded.
The cell number in each 25 ml culture flask was
4–5×106.
Treatment groups
All cell treatments were performed at 37°C.LPS
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was used to induce
inflammation in BRL cells as a model of sepsis. Initially, 10 µg/ml
LPS was applied for 0, 6, 12 and 24 h to determine the optimal time
point to induce inflammation in BRL cells. Subsequently, as the
maximum effect was recorded following 12 h of stimulation, 12 h of
LPS stimulation was applied in the following experiments. The cells
were divided into the following 5 groups: i) Control-untreated
cells; ii) LPS-treated cells (10 µg/ml LPS for 12 h); iii) the
LPS+Ang-(1–7) group, in which, cells were treated
with 10−7, 10−6 or 10−5 mol/l
Ang-(1–7) for 30 min followed by incubation with
10 µg/ml LPS for 12 h; iv) the LPS+A779 group, in which, cells were
treated with 10−7, 10−6 and 10−5
mol/l of the Ang-(1–7) antagonist, A779, for 30 min followed
by incubation with 10 µg/ml LPS for 12 h; and v) the
LPS+Ang-(1–7)+SB 203580 group, in which, cells were
pretreated with 10−5 mol/l Ang-(1–7) and
10−5 mol/l of the p38MAPK inhibitor, SB 203580, for 30
min followed by incubation with 10 µg/ml LPS for 12 h. Cells in
each group were harvested 12 h following LPS stimulation.
Ang-(1–7) and A779 were supplied by Sigma-Aldrich
(Merck KGaA) and SB 203580 was obtained from Beyotime Institute of
Biotechnology (Haimen, China).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cultured BRL cells
using the TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Reverse
transcription was performed using random primers, M-MLV reverse
transcriptase and RNase inhibitor (Fermentas; Thermo Fisher
Scientific, Inc.). Expression levels of all transcripts were
normalized to the expression level of GAPDH. qPCR was performed
using the Power SYBR Green PCR Master Mix and an ABI 7500
instrument (both Applied Biosystems; Thermo Fisher Scientific,
Inc.) with the following primers: AP-1 (forward:
5′-CTACAAACTCCTGAAACCCACC-3′, reverse: 5′-TCTGATCCCTGACCCGAAA-3′);
phosphorylated (p-)p38MAPK (forward: 5′-GGACCTAAAGCCCAGCAA−3′,
reverse: 5′-CAGCCCACGGACCAAATA−3′); TNF-α (forward:
5′-GGTGCCTATGTCTCAGCCTCTT-3′; reverse: 5′-GCACCTCCACTTGGTGGTTT-3′),
and GAPDH (forward: 5′-GGCACAGTCAAGGCTGAGAATG-3′; reverse:
5′-ATGGTGGTGAAGACGCCAGTA-3′). The following thermocycling
conditions were used for PCR: 95°C for 5 min, then 40 cycles of
95°C for 15 sec and 60°C for 1 min. Data were analyzed according to
the 2−ΔΔCq method, as previously described (19).
Western blot analysis
Cultured BRL cells were homogenized in
radioimmunoprecipitation assay buffer (150 mM NaCl, 50 mM Tris, 1
mM PMSF, 1 mM Na3VO4, 1% NP-40, 0.1% SDS, 0.5% deoxycholic acid, 1%
protease inhibitor cocktail, pH 7.5) and centrifuged (room
temperature, 400 × g and for 5–10 min). The supernatant was
collected from whole-cell lysates. Total protein concentration was
determined using a Bicinchoninic Acid Protein Assay kit (Pierce;
Thermo Fisher Scientific, Inc.). Proteins were separated by 8%
SDS-PAGE and transferred by electroblotting to polyvinylidene
fluoride membranes (the quantity of protein loaded per lane was 15
µg). The membranes were separately incubated for 3 h with 5%
skimmed dry milk at 37°C, followed by an overnight incubation at
4°C with primary antibodies against rabbit-p38 (1:800; CST
Biological Reagents Co., Ltd., Shanghai, China; cat no. 8690),
rabbit-p-p38 (1:800; Abcam, Cambridge, UK; cat no. ab4822),
rabbit-AP-1 (1:800; CST Biological Reagents Co., Ltd.; cat no.
9165) and mouse β-actin monoclonal antibody (1:800; Abcam; cat no.
ab8226). Following the overnight incubation, membranes were further
incubated for 1 h at room temperature in Tris-buffered saline with
1% Tween (TBS-T) with peroxidase-conjugated goat anti-mouse
secondary antibodies (cat no. KC-MM-035) or peroxidase-conjugated
goat anti-rabbit secondary antibodies (cat no. KC-RB-035) (both
1:6,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). Enhanced
chemiluminescence (EMD Millipore, Billerica, MA, USA) was used to
detect immune reactive bands. Finally, densitometric analysis of
the bands was performed using Image Labä software (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Data analysis was performed using SPSS software
(version 17.0; SPSS Inc., Chicago, IL, USA) and differences between
multiple groups were evaluated using one-way analysis of variance
with Bonferroni correction for post hoc comparison. The difference
in gene expression levels was evaluated by Student's t-test. The
results are expressed as the mean ± standard deviation (n=3).
P<0.05 was considered to indicate a statistically significant
difference.
Results
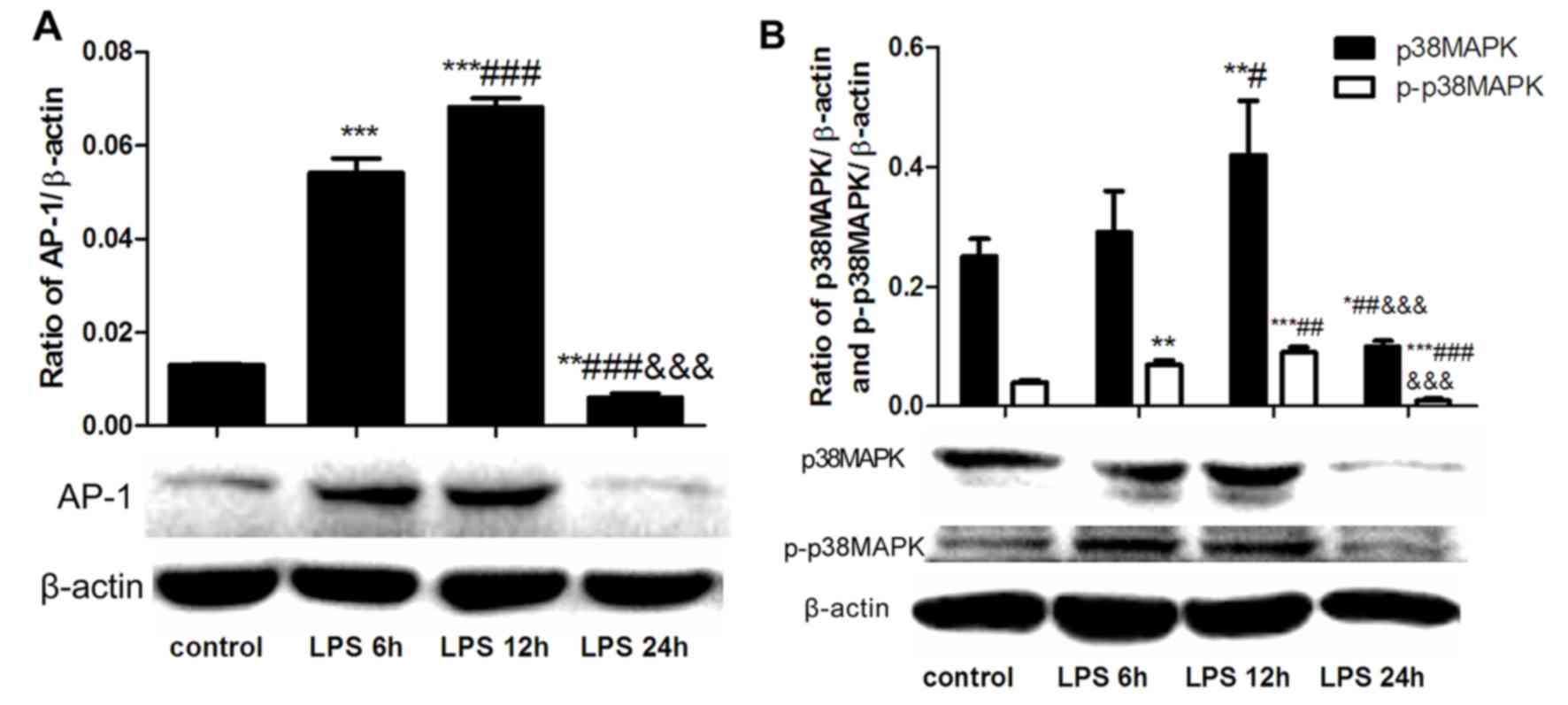
LPS treatment induces time-dependent
alterations in AP-1 and p38MAPK expression in hepatocytes
To characterize the effect of LPS on signaling
pathways mediating inflammation, BRL cells were stimulated with LPS
for 6, 12 and 24 h. When compared with the control-untreated cells,
LPS significantly increased the expression levels of AP-1 (Fig. 1A), and p38MAPK and p-p38MAPK
(Fig. 1B), following 6 and 12 h of
stimulation. By contrast, LPS demonstrated no effect on the
expression levels of these proteins following 24 h of stimulation
(Fig. 1). As the maximum LPS
effect was recorded following 12 h of stimulation, this time point
was used for subsequent experiments.
Inhibitory effect of Ang-(1–7) on
LPS-induced AP-1 and p-p38MAPK expression
Activation of p-p38MAPK and AP-1 serves an important
role in the production of TNF-α (14,17).
Therefore, the aim of the present study was to determine the
inhibitory effect of Ang-(1–7)
treatment on the activation of p-p38MAPK and AP-1. BRL cells were
pretreated with different concentrations (10−7,
10−6 and 10−5 mol/l) of Ang-(1–7) for
30 min and were subsequently stimulated with 10 µg/ml LPS for 12 h.
Pretreatment with Ang-(1–7) neutralized the effect of LPS (Fig. 2). Protein and mRNA levels of AP-1
(Fig. 2A and B), and p38MAPK and
p-p38MAPK (Fig. 2C and D) were
significantly reduced following pretreatment with Ang-(1–7) in a
concentration-dependent manner, compared with the LPS-treated
cells.
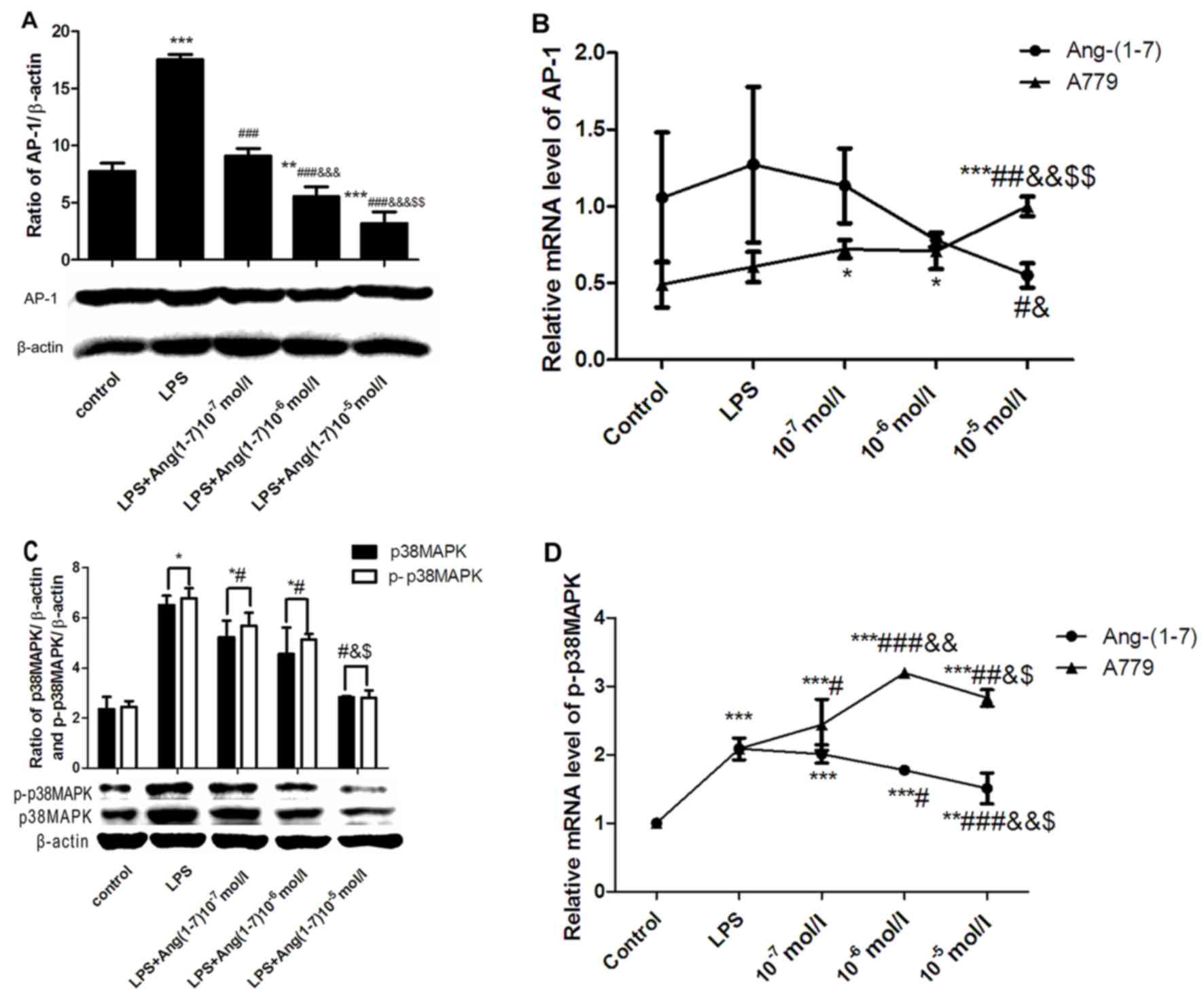 | Figure 2.Dose-dependent effects of
Ang-(1–7) on LPS-induced expression of AP-1,
p38MAPK and p-p38MAPK. (A) AP-1 protein expression in BRL cells
treated with or without Ang-(1–7) at
10−7, 10−6 and 10−5 mol/l. (B)
AP-1 mRNA expression in BRL cells treated with or without
Ang-(1–7) or A779 at 10−7,
10−6 and 10−5 mol/l. (C) p38MAPK and
p-p38MAPK protein expression in BRL cells treated with or without
Ang-(1–7) at 10−7, 10−6 and
10−5 mol/l. (D) p-p38MAPK mRNA expression in BRL cells
treated with or without Ang-(1–7) or
A779 at 10−7, 10−6 and 10−5 mol/l.
The control cells were untreated (culture medium only) and those in
the LPS group were treated with 10 µg/ml LPS only. Data are
presented as the mean ± standard deviation. *P<0.05, **P<0.01
and ***P<0.001 vs. the control group; #P<0.05,
##P<0.01 and ###P<0.001 vs. the LPS
group; &P<0.05, &&P<0.01
and &&&P<0.001 vs. the LPS+
[Ang-(1–7) or A779] 10−7 mol/l group;
$P<0.05 and $$P<0.01 vs. the LPS+
[Ang-(1–7) or A779] 10−6 mol/l group.
LPS, lipopolysaccharide; AP-1, activator protein 1; p-p38MAPK,
phosphorylated-p38 mitogen activated protein kinase; A779, Mas
receptor selective antagonist; Ang-(1–7),
Angiotensin-(1–7). |
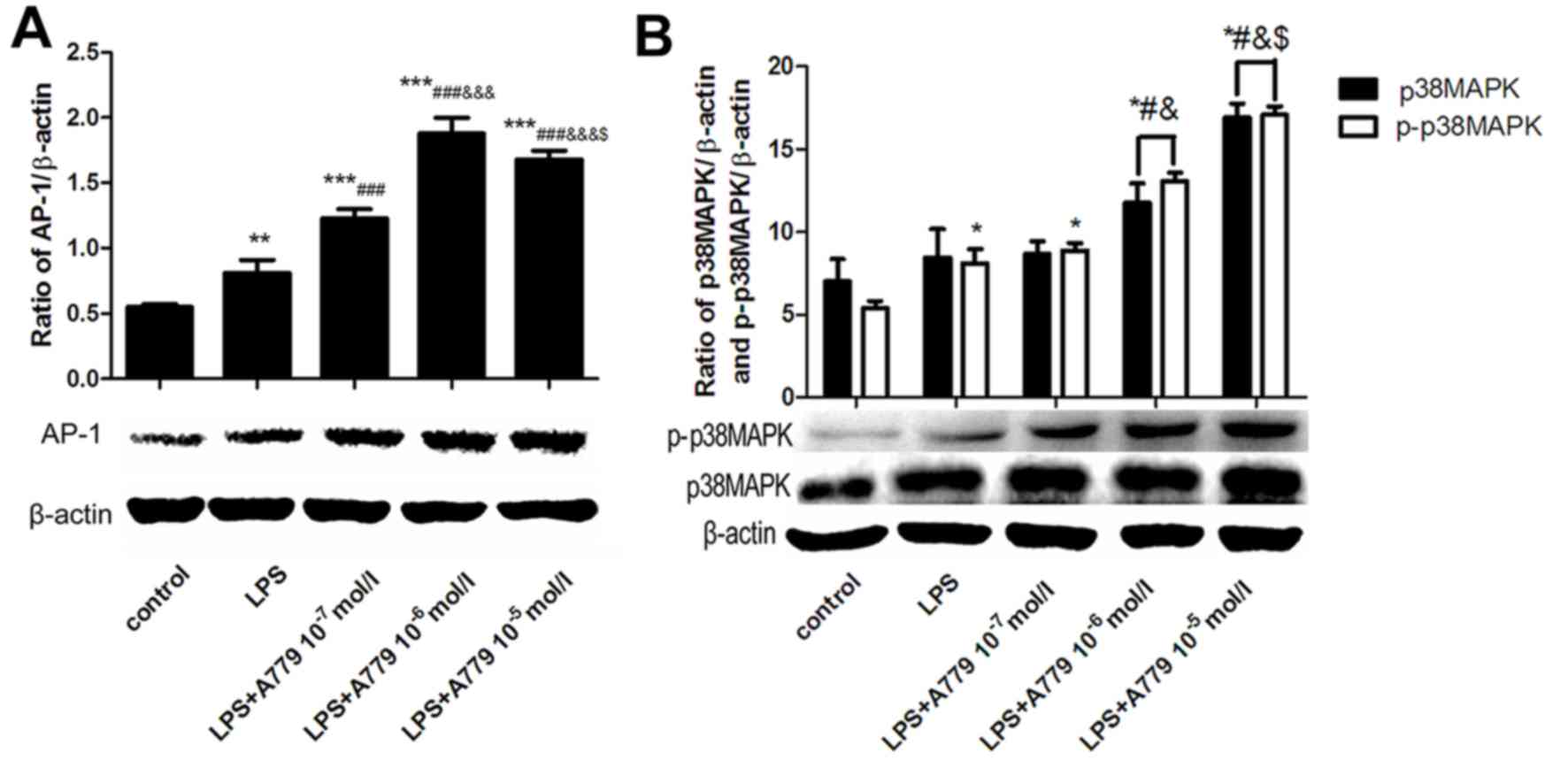
A779 further upregulates LPS-induced
AP-1 and p-p38MAPK expression
In order to further elucidate the protective
anti-inflammatory role of Ang-(1–7), the
effect of the Ang-(1–7) proto-oncogene Mas (Mas) receptor
(MasR) selective antagonist, A779, on AP-1 and p-p38MAPK expression
was evaluated. BRL cells were pretreated with 10−7,
10−6 and 10−5 mol/l A779 for 30 min and then
stimulated with 10 µg/ml LPS for 12 h. AP-1 mRNA expression was
significantly increased by treatment with A779 in a
concentration-dependent manner, with the peak mRNA level observed
at 10−5 mol/l, when compared with the control-untreated
cells and cells stimulated with LPS only (Fig. 2B). A779 at 10−6 mol/l
induced the most elevated protein level of AP-1 (Fig. 3A). A779 at concentrations between
10−7-10−5 mol/l enhanced the LPS-induced
increase in the mRNA levels of p-p38MAPK, with peak levels observed
with A779 at 10−6 mol/l (Fig. 2D). Pretreatment with A799 resulted
in a significant increase in the protein levels of p38MAPK and
p-p38MAPK, when administered at concentrations of 10−6
and 10−5 mol/l, when compared with the control or LPS
groups (Fig. 3B). However, A779 at
10−7 mol/l did not affect p38MAPK and p-p38MAPK protein
levels.
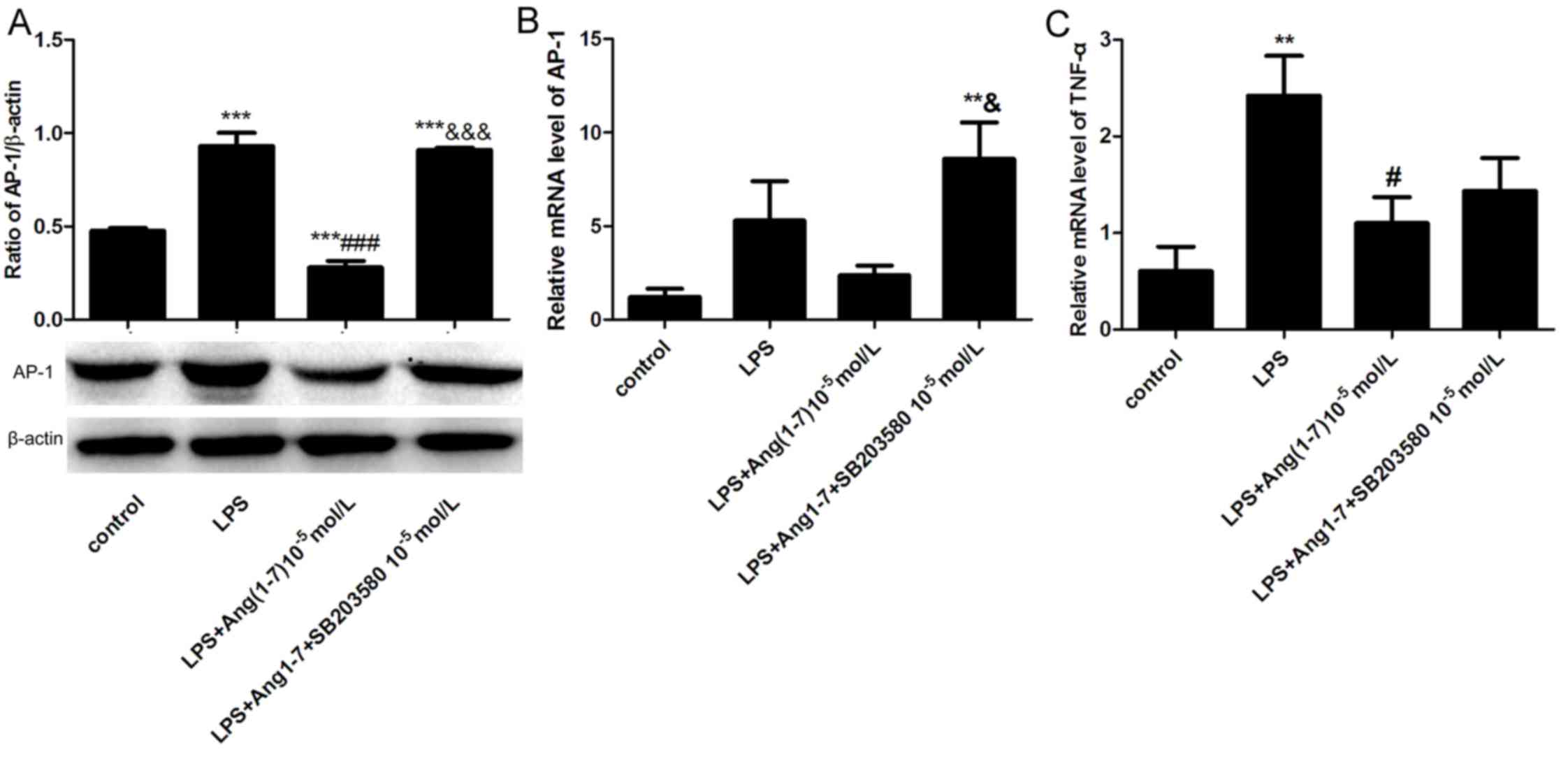
SB-203580 eliminates the inhibitory
effects of Ang-(1–7) on the LPS-induced expression of AP-1
and TNF-α
SB-203580, a specific p38MAPK signaling pathway
inhibitor, was used to further verify the anti-inflammatory effect
of Ang-(1–7) on MAPK signaling. Ang-(1–7) was
added to BRL cells alone or with SB-203580 prior to stimulation
with LPS. When compared with the LPS group, incubation with
LPS+Ang-(1–7) significantly decreased the protein
expression of AP-1by 69.89% at 12 h; whereas, incubation with LPS
in combination with Ang-(1–7) and
SB-203580 significantly increased the protein expression of AP-1 by
3.25-fold compared with the LPS+Ang-(1–7)
group (both P<0.05; Fig. 4A).
In addition, LPS+Ang-(1–7)+SB-203580 treatment increased the mRNA
expression of AP-1, when compared with the control and
Ang-(1–7)+LPS groups (Fig. 4B). Ang-(1–7)
inhibited the LPS-induced expression of TNF-α compared with the LPS
group, which was marginally ameliorated by treatment with SB 203580
(Fig. 4C). The above results
suggested that Ang-(1–7) may be protective against LPS-induced
liver injury through the inhibition of the p38MAPK signaling
pathway.
Discussion
In the present study, LPS induced a significant
upregulation of the inflammatory response in hepatic cells. A
significant increase was observed in the expression levels of
transcriptional regulatory factors, AP-1 and p38MAPK. The BRL cell
model was used to investigate the anti-inflammatory role of
Ang-(1–7) in hepatocytes. Ang-(1–7)
significantly reduced the LPS-induced inflammatory response.
However, the Ang-(1–7) MasR antagonist, A779, inhibited the
protective effect of Ang-(1–7) on
LPS-induced inflammatory responses. Furthermore, the protective
effects of Ang-(1–7) were abolished by treatment with the
p38MAPK inhibitor. Therefore, the results of the present study
suggest that the p38MAPK/AP-1 signaling pathway may be an important
molecular mechanism underlying the liver-protective effects of
Ang-(1–7) against LPS-mediated injury.
A previous study on the pathological mechanism of
LPS-induced liver injury demonstrated that a number of inflammatory
mediators and cytokines, including TNF-α, nitric oxide, interleukin
(IL)-1, IL-6, transforming growth factor-β and reactive oxygen
species, are released as a result of upregulated MAPK signaling in
activated hepatic Kupffer cells (16). In addition, LPS activated RAS and
associated ACE-Ang II-AT1 to affect hepatic blood flow
redistribution, microcirculation disturbances and the production of
pro-inflammatory cytokines (5,6).
Ang-(1–7) functions as a ligand for the G
protein-coupled receptor Mas and has been demonstrated to
antagonize the activity of Ang II (20,21).
Specifically, Ang-(1–7) has demonstrated anti-inflammatory
properties through its antagonistic effects on the pro-inflammatory
factor Ang II (22,23). A previous study also revealed the
anti-inflammatory effect of Ang-(1–7) on
LPS-induced macrophages (12).
Furthermore, it has been indicated previously that Ang-(1–7)
prevents acute respiratory distress syndrome in rats following
intratracheal administration of LPS, and that Ang-(1–7)
receptor Mas deficiency exacerbates LPS-induced cerebral and
systemic inflammation in mice (24,25).
A recent study indicated that Ang-(1–7)
inhibits LPS-induced acute lung inflammation in alveolar epithelial
cells (26).
In the present study, the anti-inflammatory activity
of Ang-(1–7) was demonstrated in hepatic cells
stimulated with LPS. Since TNF-α is a pro-inflammatory mediator of
liver damage in response to LPS, the inhibitory effects of
Ang-(1–7) on TNF-α production were determined by
investigating the activation of AP-1, an upstream transcriptional
regulator of TNF-α (27). The
results revealed that Ang-(1–7)
reduced the expression of TNF-α when administered at a
concentration of 10−5 mmol/l and reduced AP-1 expression
in a concentration-dependent manner. A779 increased AP-1 mRNA
expression when administered at concentrations of
10−7-10−5 mmol/l to LPS-stimulated
hepatocytes. The above results suggested that the inhibitory effect
of Ang-(1–7) on TNF-α production may be associated
with AP-1.
p38MAPK serves an important role in the regulation
of LPS-induced inflammation by controlling AP-1 activation and has
been associated with LPS-induced liver injury (4,28).
In the present study, pretreatment of hepatocytes with
Ang-(1–7) significantly inhibited the effect of
LPS by reducing the expression of p38MAPK and p-p38MAPK. However,
this inhibitory effect wascounteractedby the Ang-(1–7)
antagonist, A779, and the p38 MAPK inhibitor, SB 203580. The
results of the present study are supported by those previously
obtained by Zhou et al (29) and Akhtar et al (30). The above results suggest that
Ang-(1–7) may inhibit AP-1 and TNF-α activation
by inhibiting the p38MAPK signaling pathway in LPS-induced
hepatocytes.
In conclusion, the present study demonstrated that
Ang-(1–7) significantly reduced the levels of
LPS-induced pro-inflammatory cytokines, and the expression of MAPK
and AP-1 in hepatocytes. Inhibition of the p38MAPK/AP-1 signaling
pathway serves a role in the anti-inflammatory effect of
Ang-(1–7) treatment. Therefore, Ang-(1–7)
could potentially be used for the hepatoprotective treatment of
LPS-induced liver injury.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant. no. 81101400).
References
|
1
|
Riedemann NC, Guo RF and Ward PA: Novel
strategies for the treatment of sepsis. Nat Med. 9:517–524. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen YR, Wang X, Templeton D, Davis RJ and
Tan TH: The role of c-Jun N-terminal kinase (JNK) in apoptosis
induced by ultraviolet C and gamma radiation. Duration of JNK
activation may determine cell death and proliferation. J Biol Chem.
271:31929–31936. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bone RC: The pathogenesis of sepsis. Ann
Intern Med. 115:457–469. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu LM, Liang DY, Ye CG, Tu WJ and Zhu T:
The UII/UT system mediates upregulation of proinflammatory
cytokines through p38 MAPK and NF-κB pathways in LPS-stimulated
Kupffer cells. PLoS One. 10:e01213832015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miyoshi M, Nagata K, Imoto T, Goto O,
Ishida A and Watanabe T: ANG II is involved in the LPS-induced
production of proinflammatory cytokines in dehydrated rats. Am J
Physiol Regul Integr Comp Physiol. 284:R1092–R1097. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iwashita M, Nakatsu Y, Sakoda H, Fujishiro
M, Kushiyama A, Fukushima T, Kumamoto S, Shinjo T, Kamata H,
Nishimura F and Asano T: Valsartan restores inflammatory response
by macrophages in adipose and hepatic tissues of LPS-infused mice.
Adipocyte. 2:28–32. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Santos RA, Ferreira AJ and Simões E Silva
AC: Recent advances in the angiotensin-converting enzyme
2-angiotensin(1–7)-Mas axis. Exp Physiol. 93:519–527. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ferreira AJ, Oliveira TL, Castro MC,
Almeida AP, Castro CH, Caliari MV, Gava E, Kitten GT and Santos RA:
Isoproterenol-induced impairment of heart function and remodeling
are attenuated by the nonpeptide angiotensin-(1–7) analogue AVE
0991. Life Sci. 81:916–923. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Santos RA, Ferreira AJ, Pinheiro SV,
Sampaio WO, Touyz R and Campagnole-Santos MJ: Angiotensin-(1–7) and
its receptor as a potential targets for new cardiovascular drugs.
Expert Opin Investig Drugs. 14:1019–1031. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sampaio WO, Souza dos Santos RA,
Faria-Silva R, da Mata Machado LT, Schiffrin EL and Touyz RM:
Angiotensin-(1–7) through receptor Mas mediates endothelial nitric
oxide synthase activation via Akt-dependent pathways. Hypertension.
49:185–192. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pinheiro SV, Simões e Silva AC, Sampaio
WO, de Paula RD, Mendes EP, Bontempo ED, Pesquero JB, Walther T,
Alenina N, Bader M, et al: Nonpeptide AVE 0991 is an
angiotensin-(1–7) receptor Mas agonist in the mouse kidney.
Hypertension. 44:490–496. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Souza LL and Costa-Neto CM:
Angiotensin-(1–7) decreases LPS-induced inflammatory response in
macrophages. J Cell Physiol. 227:2117–2122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Oh YC, Jeong YH, Ha JH, Cho WK and Ma JY:
Oryeongsan inhibits LPS-induced production of inflammatory
mediators via blockade of the NF-kappaB, MAPK pathways and leads to
HO-1 induction in macrophage cells. BMC Complement Altern Med.
14:2422014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cho HY, Morgan DL, Bauer AK and Kleeberger
SR: Signal transduction pathways of tumor necrosis factor-mediated
lung injury induced by ozone in mice. Am J Respir Crit Med.
175:829–839. 2007. View Article : Google Scholar
|
|
15
|
Robinson MJ and Cobb MH: Mitogen-activated
protein kinase pathways. Curr Opin Cell Biol. 9:180–186. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Scott MJ and Billiar TR:
Beta2-integrin-induced p38 MAPK activation is a key mediator in the
CD14/TLR4/MD2-dependent uptake of lipopolysaccharide by
hepatocytes. J Biol Chem. 283:29433–29446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Klintman D, Li X, Santen S, Schramm R,
Jeppsson B and Thorlacius H: p38 mitogen-activated protein
kinase-dependent chemokine production, leukocyte recruitment, and
hepatocellular apoptosis in endotoxemic liver injury. Ann Surg.
242:830–839. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu T, Li YJ, Bian AH, Zuo HB, Zhu TW, Ji
SX, Kong F, Yin DQ, Wang CB, Wang ZF, et al: The regulatory role of
activating transcription factor 2 in inflammation. Mediators
Inflamm. 2014:9504722014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta DeltaC(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lambert DW, Hooper NM and Turner AJ:
Angiotensin-converting enzyme 2 and new insights into the
renin-angiotensin system. Biochem Pharmacol. 75:781–786. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Santos RA, Ferreira AJ, Verano-Braga T and
Bader M: Angiotensin-converting enzyme 2, angiotensin-(1–7) and
Mas: New players of the renin-angiotensin system. J Endocrinol.
216:R1–R17. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chappell MC: Angiotensin-converting enzyme
2 autoantibodies: Further evidence for a role of the
renin-angiotensin system in inflammation. Arthritis Res Ther.
12:1282010. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hanafy S, Tavasoli M and Jamali F:
Inflammation alters angiotensin converting enzymes (ACE and ACE-2)
balance in rat heart. Inflammation. 34:609–613. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wösten-van Asperen RM, Lutter R, Specht
PA, Moll GN, van Woensel JB, van der Loos CM, van Goor H, Kamilic
J, Florquin S and Bos AP: Acute respiratory distress syndrome leads
to reduced ratio of ACE/ACE2 activities and is prevented by
angiotensin-(1–7) or an angiotensin II receptor antagonist. J
Pathol. 225:618–627. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oliveira-Lima OC, Pinto MC, Duchene J,
Qadri F, Souza LL, Alenina N, Bader M, Santos RA and
Carvalho-Tavares J: Mas receptor deficiency exacerbates
lipopolysaccharide-induced cerebral and systemic inflammation in
mice. Immunobiology. 220:1311–1321. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma X, Xu D, Ai Y, Zhao S, Zhang L, Ming G
and Liu Z: Angiotensin-(1–7)/mas signaling inhibits
lipopolysaccharide-induced ADAM17 shedding activity and apoptosis
in alveolar epithelial cells. Pharmacology. 97:63–71. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chan ED and Riches DW: IFN-gamma + LPS
induction of iNOS is modulated by ERK, JNK/SAPK, and p38(mapk) in a
mouse macrophage cell line. Am J Physiol Cell Physiol.
280:C441–C450. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Veres B, Radnai B, Gallyas F Jr, Varbiro
G, Berente Z, Osz E and Sumegi B: Regulation of kinase cascades and
transcription factors by a poly(ADP-ribose) polymerase-1 inhibitor,
4-hydroxyquinazoline, in lipopolysaccharide-induced inflammation in
mice. J Pharmacol Exp Ther. 310:247–255. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou L, Xue H, Wang Z, Ni J, Yao T, Huang
Y, Yu C and Lu L: Angiotensin-(1–7) attenuates high glucose-induced
proximal tubular epithelial-to-mesenchymal transition via
inhibiting ERK1/2 and p38 phosphorylation. Life Sci. 90:454–462.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Akhtar S, Yousif MH, Dhaunsi GS,
Chandrasekhar B, Al-Farsi O and Benter IF: Angiotensin-(1–7)
inhibits epidermal growth factor receptor transactivation via a Mas
receptor-dependent pathway. Br J Pharmacol. 165:1390–1400. 2012.
View Article : Google Scholar : PubMed/NCBI
|