Introduction
White matter injury (WMI) is the most common type of
brain injury and the major cause of sequelae of the nervous system
in premature infants. In the USA, ~60,000 extremely low birth
weight infants are born every year (1) and the incidence rate of WMI in these
infants is 50% (2,3). In addition, ~90% of WMI patients
survive with sequelae to varying extents, of which 5–10%
suffercerebral palsy and 50% experience behavior and/or attention
deficit (4,5). The majority of patients with serious
sequelae demonstrate periventricular leukomalacia (PVL) as observed
by magnetic resonance imaging (MRI), pathologically manifesting as
local periventricular white matter necrosis, myelinization
disorders, and gliocyte proliferation and microglial cell
activation in adjacent white matter (6,7).
The pathogenesis of WMI remains to be fully
elucidated, although infection and hypoxia are recognized to be the
primary inducing factors. Infection and hypoxia may lead to the
release of glutamates, oxygen-derived free radicals and
inflammatory cytokines, and thus activate microglial cells.
Microglial cells may further release glutamates, oxygen-derived
free radicals and inflammatory cytokines which may aggravate WMI;
activated microglial cells may induce the death of oligodendrocyte
precursor cells (OPCs) and astrocytes, which has been previously
demonstrated (8,9). Additionally, OPCs account for 90% of
the total number of oligodendrocytes in the white matter of
premature infants. Animal experiments have demonstrated that, based
on the maturation-dependent vulnerability of oligodendrocytes,
immature oligodendrocytes are more vulnerable during a specific
window and are easily injured by glutamates, oxygen-derived free
radicals and inflammatory cytokines, thus leading to apoptosis,
necrosis and myelinization delay (10–12).
However, in the recent years, follow-ups of patients
with PVL have demonstrated that these patients frequently exhibit
such sequelae as all-round cognitive deficit, attention deficit,
human communication disorders, language retardation and autism,
which cannot be reasonably explained by WMI alone; these sequelae
are closely associated with neuronal/axonal injury. With the
development of neuropathology and imaging techniques, the focus of
previous research has been directed to gray matter injury which has
been long-neglected; a number of investigators have proposed that
the pathological alteration sand neurodevelopmental outcomes of PVL
may be not only limited to oligodendrocyte absence and
myelinization disorders (13–15).
MRI volumetric analysis indicates that the volume of gray matter in
the cerebral cortex and subcortex is decreased in premature infants
with PVL. Andiman et al (13) demonstrated by autopsy that the
density of granular neurons of patients with PVL was markedly
decreased compared with that of controls. In 2005, Volpe et
al (14) from Harvard
University (Cambridge, MA, USA) first proposed a concept of
‘encephalopathy of prematurity (EP)’, and they hypothesized that
brain injury may not be accurately defined by WMI in premature
infants and that the role of neuronal injury in brain injury was
relevant. However, there are few preclinical studies into neuron
injury in PVL. In the present study, neuronal alterations were
investigated using a previously established hypoxia-induced PVL
model (15) to clarify the impact
of neuronal injury and its mechanism.
Materials and methods
Experimental animals and grouping
Ethical approval for the present study was provided
by Shengjing Hospital of China Medical University Ethics Committee
(Shenyang, China). A total of 80 Sprague-Dawley (SD) rats
[postnatal day 3 (P3); weight, 10.2–12.1 g] were randomly assigned
to the control group (normoxic exposure; n=40) or the model group
(hypoxic exposure; n=40). The rats were raised under the following
conditions: The temperature was 18–26°C, the humidity was 47–70%,
day light for 10 h and dark for 14 h, the food and water was
supplied ad libitum. The body weight of rats was 11.5±1.3 g
in the control group (male, 22) and 11.4±1.4 g in the model group
(male, 21) and no statistical difference was identified between the
weight and sex of the two groups. All rats were provided by the
Experimental Animal Department, Shengjing Hospital of China Medical
University. A hypoxia-ischemia-induced PVL rat model established
previously by our research group was used and the modeling method
was same as that for a hypoxia model established by Mizuno et
al (16). The neonatal SD rats
(P3) were weighed and numbered prior to surgery. Animals were
anesthetized by isoflurane inhalation and fixed on the bench in a
supine position. Following conventional disinfection, a 0.7–0.8 cm
incision was made close to the tracheal midline under a dissecting
microscope, and the right common carotid artery was exposed and
ligated. Rats were postoperatively sent to the female rat cages
with a room temperature and normoxic exposure for 2 h recovery, and
were subsequently subjected to 2 h hypoxic exposure (6–8% oxygen).
Rats in the control group were only subjected to the dissection of
the right common carotid artery, without ligation and hypoxic
exposure. At 1, 3, 7 and 14 days post-surgery, the brain tissues of
experimental rats were collected.
Hematoxylin and eosin (H&E)
staining
The brain tissues were fixed with 4%
paraformaldehyde for 48 h at room temperature, dehydrated with
gradient ethanol (70% ethanol for 2 h, 80% ethanol over night, 90%
ethanol for 2 h, 100% I ethanol for 1 h, 100% II ethanol for 1 h)
vitrified with xylene, waxed, embedded with paraffin and
conventionally sliced into 5-µm tissue sections. Subsequently, the
sections were stained with hematoxylin (ZLI-9609; Beijing Zhongshan
Goldenbridge Biotechnology, Beijing, China) for 10 min, washed with
water for 15 min, then eosin for 1 min (ZLI-9612; Beijing Zhongshan
Goldenbridge Biotechnology), and washed with water for 10 sec. A
total of six tissue sections from each group were selected randomly
at various time-points, and were observed in 5 random fields under
a light microscope at magnification, ×400.
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay
The paraffin-embedded sections of brain tissues were
conventionally dewaxed, treated with 50 µl 0.1% Triton X-100
(prepared with 0.1% sodium citrate) and preserved at room
temperature for 8 min for vitrification, washed with PBS for 5 min
three times, treated with 3% H2O2 at room
temperature for 10 min to block peroxidase and washed three times
with PBS for 5 min. TUNEL reaction solution (cat. no. 11684817910;
Roche Diagnostics GmbH, Mannheim, Germany) was prepared with enzyme
solution and label solution at a dilution of 1:9 (freshly prepared
on ice). The sections were wiped dry, 50 µl TUNEL reaction solution
was added, and sections were incubated in a humidified atmosphere
away from light at 37°C for 60 min, followed by washing with PBS
for 5 min three times. Thereafter, the sections were wiped dry, 50
µl converter-POD was added, and sections were incubated in a
humidified atmosphere at 37°C for 30 min, and washed with PBS for 5
min three times. The sections were removed and wiped dry, 50 µl
diaminobenzidine substrate (DAB; 1:20; zli-9017; OriGene
Technologies, Inc.) was added at room temperature for 10 sec, and
sections were rapidly placed into water to terminate the reaction
when the color had just become deep. The sections were subsequently
counterstained with hematoxylin at room temperature for 10 min,
dehydrated with ethanol, vitrified with xylene and mounted. Instead
of TUNEL reaction solution, PBS was used in the negative controls,
and all procedures were same as the above. The cells with
brown-yellow particles deposited in the cytoplasm were assessed to
be positive cells. A total of six tissue sections from each group
were selected randomly at various time-points, and they were
observed in 5 random fields under a light microscope at
magnification, ×500.
Double-labeling immunofluorescence
staining
The frozen sections (10-µM thick; frozen at −80°C)
of brain tissues were defrosted, fixed with acetone at 4°C for 30
min, and washed with PBS for 5 min three times. Antigen retrieval
was performed by boiling in antigen retrieval solution I at 100°C
for 5 min, and washed with PBS for 5 min three times. Sections were
blocked with bovine serum albumin (BSA; cat. no. bs043a; Biosharp,
Shandong, China) at room temperature for 1 h. When the blocking
solution was removed, the sections were incubated with primary
antibodies against Beclin 1 (rabbit anti-rat antibody; 1:1,000;
cat. no. ab55878) and neuronal nuclei (NeuN; mouse anti-rat
antibody; 1:1,000; cat. no. ab104224) (both from Abcam, Cambridge,
UK) at 4°C overnight. The following day, the sections were rewarmed
at room temperature for 1 h and washed three times with PBS for 5
min. Secondary antibodies [cat. no. ta130022; fluorescein
isothiocyanate (FITC) conjugated goat anti-rabbit IgG, 1:64; and
ta130012, Cy3-conjugated goat anti-mouse IgG, 1:50; OriGene
Technologies, Inc.] were added and incubated away from light, at
room temperature for 4 h, followed by washing in the dark with PBS
for 5 min three times. Nuclear staining was performed with DAPI
(OriGene Technologies, Inc.) for 5 min, and sections were washed in
the dark with PBS for 5 min three times. Tissues were observed and
photographed under a fluorescence microscope at magnification, ×400
(MTC-600; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Western blotting
Brain tissues were collected from 3 rats in each
group at each time-point. Tissues were added into radio
immunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Haimen, China) at a dilution of 1:10, sheared into
pieces using scissors, broken up with ultrasonication by 20 kHz
frequency on ice for 5 min, and centrifuged at 4°C at 12,000 × g
for 30 min. The supernatant was removed to separate the proteins,
which were transferred into a 0.5 ml centrifuge tube. The protein
concentration was measured using a bicinchoninic acid assay. The
samples were adjusted to the same concentration, sample buffer was
added, and samples were boiled for 5 min and preserved at −80°C.
Proteins were mixed with 7.5% SDS-PAGE sample loading buffer and
boiled for 5 min to achieve protein denaturation. The protein were
loaded for 40 µg per lane in a preset sequence, and the
electrophoresis apparatus was connected to the power source (80 V;
the current was directed towards the anode) and powered off when
bromphenol blue was migrated to a position 0.5 cm away from the
bottom of the separation gel. The gel glass plate was unloaded from
the electrophoresis apparatus and washed clean with deionized
water. Two filter papers and a polyvinylidenedifluoride (PVDF)
membrane were prepared. The cut PVDF membrane was soaked in
methanol for 10 sec and transferred into the buffer solution with
filter papers for soaking. A piece of sponge was placed on a
splint, and two soaked filter papers were placed on the sponge and
aligned accurately. The rinsed gel was placed on the top filter
paper and air bubbles were removed with a glass tube. Thereafter,
the PVDF membrane was placed on the gel aligning accurately, and 3
filter papers were placed on PVDF membrane. A piece of sponge was
placed on the top filter paper, and the splint was clamped tightly
when the air bubbles had been removed. One side of the gel was
attached to the cathode and one side of filter membrane to the
anode, and 100 V electrotransfer was conducted for 80 min.
Subsequently, the filter membrane was stained with ponceau at room
temperature for 7 min to check whether protein transfer was fully
completed. The PVDF membrane was placed into the BSA to block
nonspecific binding, slowly shaken on the shaking table at room
temperature for 1 h, and removed. The PVDF membrane was placed into
a hybridization bag containing 10 ml Beclin 1 primary antibody
(1:1,000), NeuN primary antibody (1:5,000), active caspase-3
primary antibody (rabbit anti-rat antibody; 1:200; cat. no. ab2302;
Abcam), and β-actin primary antibody (rabbit anti-rat antibody;
1:2,000; cat. no. ab8227; Abcam), diluted with the blocking
solution, sealed, and slowly shaken on the shaking table at 4°C
overnight. Following discarding of the blocking solution containing
primary antibody, the PVDF membrane was placed into a hybridization
bag containing 10 ml horseradish peroxidase-labeled secondary
antibody (goat anti-rabbit IgG 1:5,000; cat. no. zb-2301; OriGene
Technologies, Inc., for the PVDF membrane incubated with Beclin 1
antibody, caspase-3 antibody, and β-actin antibody; goat anti-mouse
IgG 1:5,000; zb-2305; OriGene Technologies, Inc., for NeuN
antibody) diluted with the blocking solution, sealed, and slowly
shaken on the shaking table at room temperature for 1 h. Following
discarding of the blocking solution containing secondary antibody,
the PVDF membrane was rinsed with Tris-buffered saline and Tween-20
(TBST) for 15 min four times. Solution A and solution B of the
enhanced chemiluminescence (ECL) reagents (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) were mixed 1:1 in a dark room
(as calculated by 0.125 ml ECL mixture/1 cm2 membrane).
The PVDF membrane was gently dried with filter paper and placed on
a fresh-keeping film with the side containing protein facing
upward. ECL mixture was evenly added onto the PVDF membrane to
react for 1 min, and the PVDF membrane was lifted, dried gently
with a filter paper and covered with the fresh-keeping film.
Protein bands were scanned using Chemi Imager 5500 v2.03 software
(ProteinSimple, San Jose, CA, USA), and the results were analyzed
using a computerized image analysis system (FluorChem 2.0,
ProteinSimple; Bio-Techne, Minneapolis, MN, USA). β-actin was used
as the internal reference for comparison among groups. A higher
relative gray scale indicated that the protein content was
greater.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
A total of 1 ml TRIzol (Takara Biotechnology Co.,
Ltd., Dalian, China) was added into 1–2 g frozen tissue and
homogenized. Subsequently, the tissue homogenate was treated with
0.2 ml chloroform, heavily shaken, preserved at room temperature
for 10 min, and centrifuged at 4°C (12,000 × g; 15 min). The upper
water phase was pipetted into an RNase Free Eppendorf tube,
precooled 0.5 ml isopropanol was added and mixed evenly, and the
sample was preserved at room temperature for 10 min, followed by
centrifugation at 4°C (12,000 × g; 10 min). The supernatant was
discarded, and the sediment was added with 1 ml precooled 75%
alcohol, mixed evenly and centrifuged at 4°C (7,500 × g; 5 min).
The supernatant was discarded, and the sediment was dried at room
temperature for 10–15 min, added with 0.02 ml RNase Free water for
dissolution, and cryopreserved at −80°C for use. The random primers
and RNA template were denatured at 70°C for 5 min, the reaction
system was reverse transcribed using the Prime Script™
RT reagent kit with gDNA Eraser (Perfect Real Time) (cat. no.
RR047A; Takara Bio, Inc.) at 37°C for 1.5 h, inactivated at 70°C
for 10 min and soaked in an ice bath for 5 min, and cDNA was
preserved at −20°C for use. In order to control the sample loading
errors during experimental procedures and the difference of
amplification efficacy among various reaction tubes, GAPDH was set
as internal reference, and the cycle was controlled in the linear
phase of amplification reaction. The premiers were as follows:
GAPDH forward primer, 5′-GCAAGTTCAACGGCACA-3′ and reverse primer,
5′-CATTTGATGTTAGCGGGAT-3′; caspase-3 forward primer,
5′-GAAGAGTTGGAGCACTGTAGCAC-3′ and reverse primer,
5′-TGGATCGTAGCACCCTGTCG-3′; and Beclin 1 forward primer,
5′-ACTGATGGCTGTAACGGTCTA-3′ and reverse primer,
5′-CCAAGCAGATGGCACAGAGG-3′. SYBR® Premix Ex
Taq™ (TliRNaseH Plus) (RR420A; Takara Bio, Inc.) was
used for DNA amplification. The total volume of the mixture was 20
µl, and the reaction conditions were as follows: 95°C for 15 sec;
60°C for 1 min; 45 cycles in total. The expression of target genes
was calculated using the 2−ΔΔCq method (17).
Statistical analysis
SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA)
was used for the processing and statistical analysis of data. The
data are presented as the mean ± standard deviation. Statistical
analysis was performed by two-way analysis of variance, followed by
multiple comparison least significant difference (LSD) tests.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Morphological alterations
Cells were observed under a light microscope and it
was demonstrated that in the control group, cells were arranged
tightly and regularly, with a normal structure, had a morphology
same as that of brain tissues from healthy rats, and appeared
normal. In the model group, cell karyopyknosis and necrosis were
observed at day 1; at day 3, cells were unclear in hierarchy and
structure, and periventricular white matter had a porous, malacotic
structure; at day 7 periventricular white matter was loose and
demonstrated net necrosis, and there was gliosis; at day 14,
cerebral white matter and the corpus callosum became thinner, and
fibers were disordered. However, no obvious gray matter nuclei and
apparent cerebral cortex injury was observed by H&E staining
(Fig. 1).
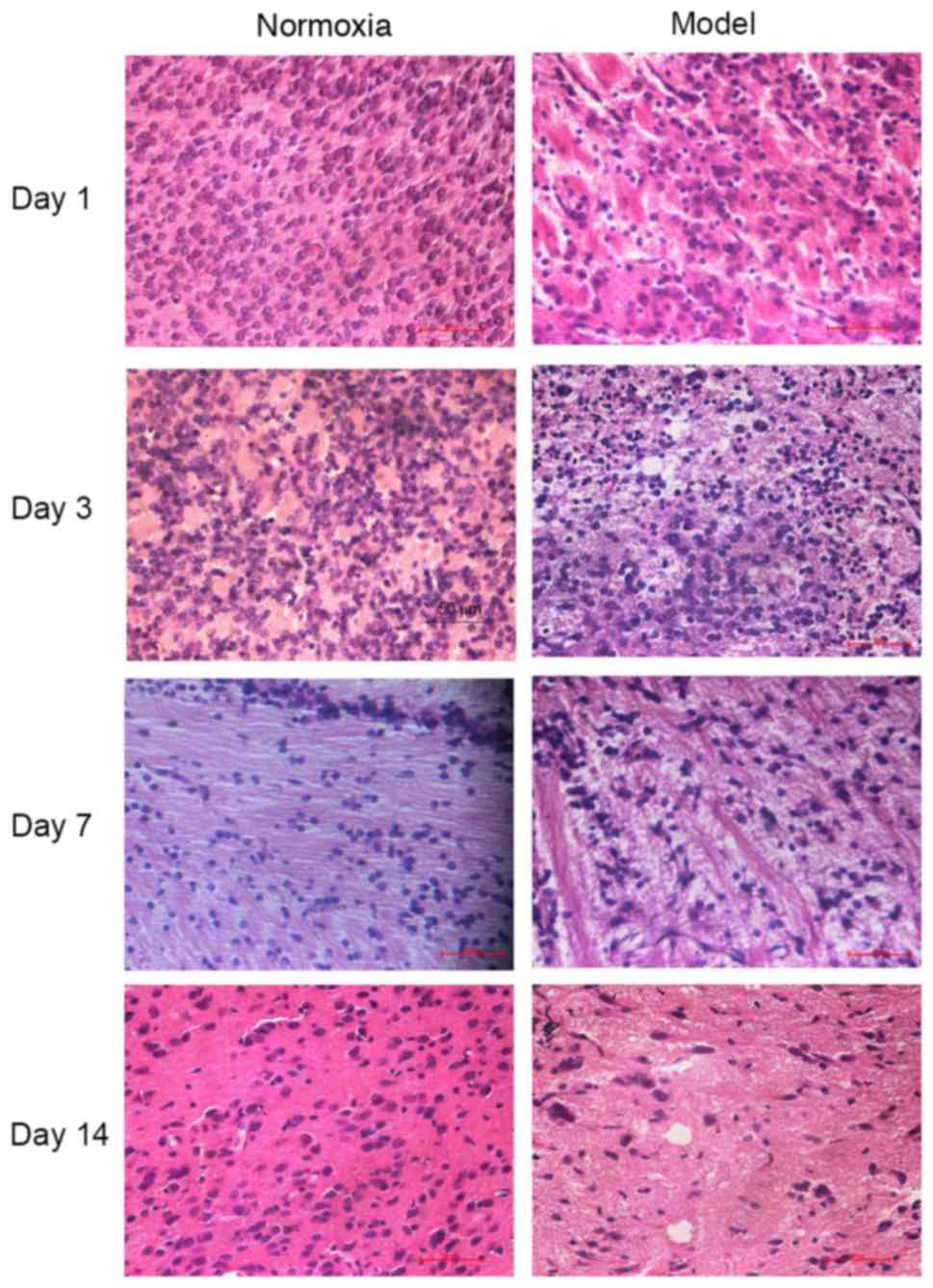 | Figure 1.Alterations in brain tissues at
different time-points following hypoxia-ischemia-induced injury
observed by hematoxylin and eosin staining. Magnification, ×400.
The histomorphology was normal and cells were arranged tightly and
regularly in the control group. In the model group, cell
karyopyknosis and necrosis were observed at day 1. At day 3, cells
were unclear in hierarchy and structure, and periventricular white
matter had a porous, malacotic structure, periventricular white
matter became malacic, cellular necrosis was observed, and there
was proliferation of gliocytes; at day 7, periventricular white
matter was loose and demonstrated net necrosis, edema was
decreased, and there was gliosis; at day 14, cerebral white matter
and corpus callosum became thinner, fibers were disordered in a net
or strip shape, and necrotic cells were decreased in number. |
Decreased expression of NeuN
In the control group, NeuN mRNA expression was
increased over the experimental time period. Compared with the
control group, NeuN mRNA expression in the model group exhibited no
significant change at day 1, although it was significantly
decreased and reached a trough at day 3, followed by an increase at
days 7 and 14; however, it remained lower compared with the
respective control group (P<0.05; Fig. 2A). In the model group, NeuN protein
expression was decreased compared with the control group. The trend
observed was consistent with the mRNA expression (Fig. 2B).
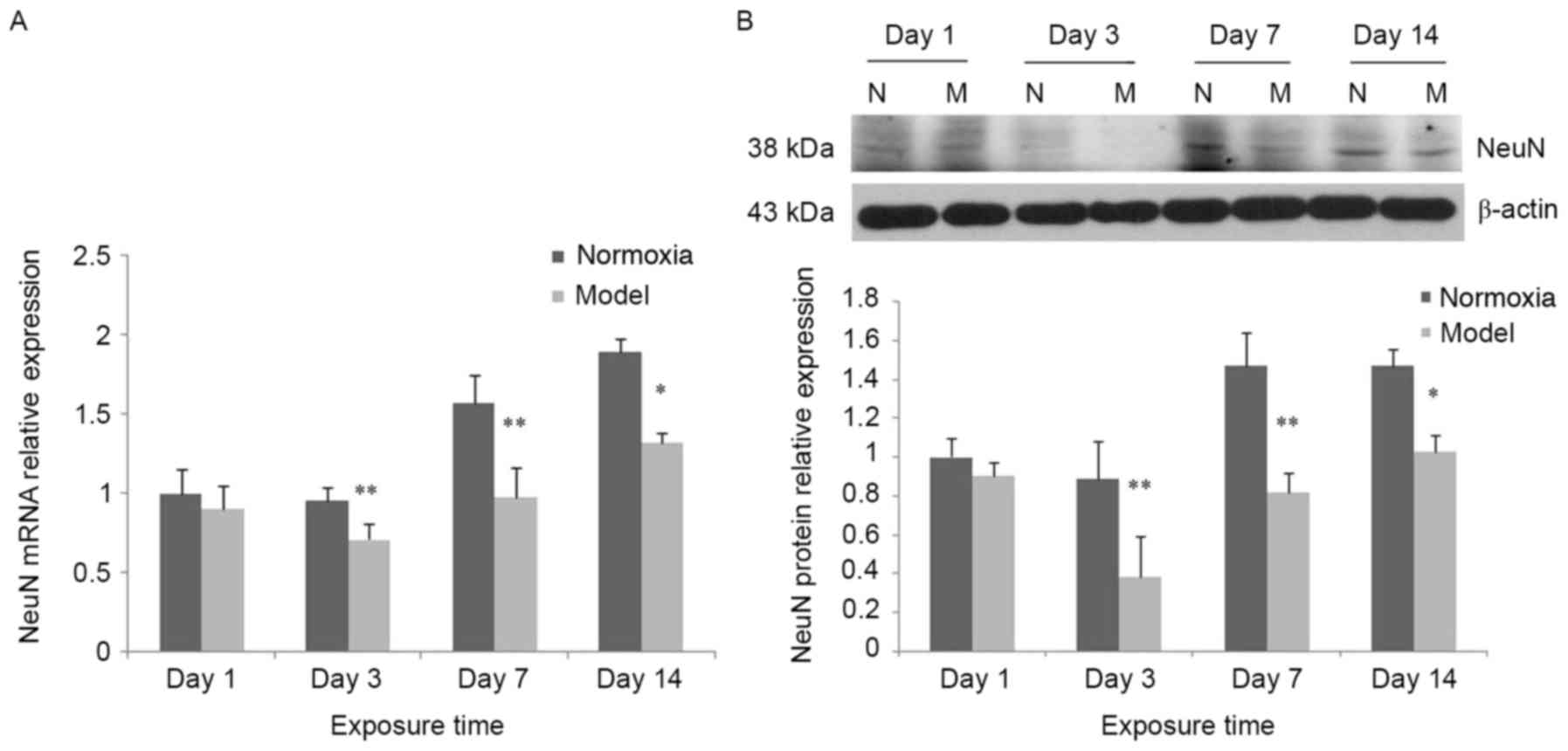 | Figure 2.Alteration of NeuN expression at
different time-points following hypoxia-ischemia-induced injury.
(A) In the hypoxia-ischemia-induced injury model, NeuN mRNA
expression in brain tissues was decreased. In the control group,
NeuN protein expression was significantly increased at days 7 and
14 compared with days 1 and 3. Compared with the control group,
NeuN mRNA expression in the model group was not significantly
decreased on day 1, although it was markedly decreased and reached
a trough at day 3, and remained decreased at days 7 and 14; in the
model group, NeuN mRNA expression at days 7 and 14 was increased
compared with day 3. (B) In the hypoxia-ischemia-induced injury
model, NeuN protein expression in brain tissues was decreased. In
the control group, NeuN protein expression was significantly
increased at days 7 and 14 compared with days 1 and 3. Compared
with the control group, NeuN protein expression in the model group
was the model group was not significantly decreased on day 1,
although it was markedly decreased and reached a trough at day 3,
and remained decreased at days 7 and 14. In the model group, NeuN
protein expression at days 7 and 14 was markedly increased compared
with day 3. Data were obtained by densitometry and were normalized
using β-actin as a loading control. Values are expressed as
relative optical density and are presented as the mean ± standard
deviation. For each column, n=3. *P<0.05, **P<0.01 vs.
thenormoxia group. N, normoxia; M, model; NeuN, neuronal
nuclei. |
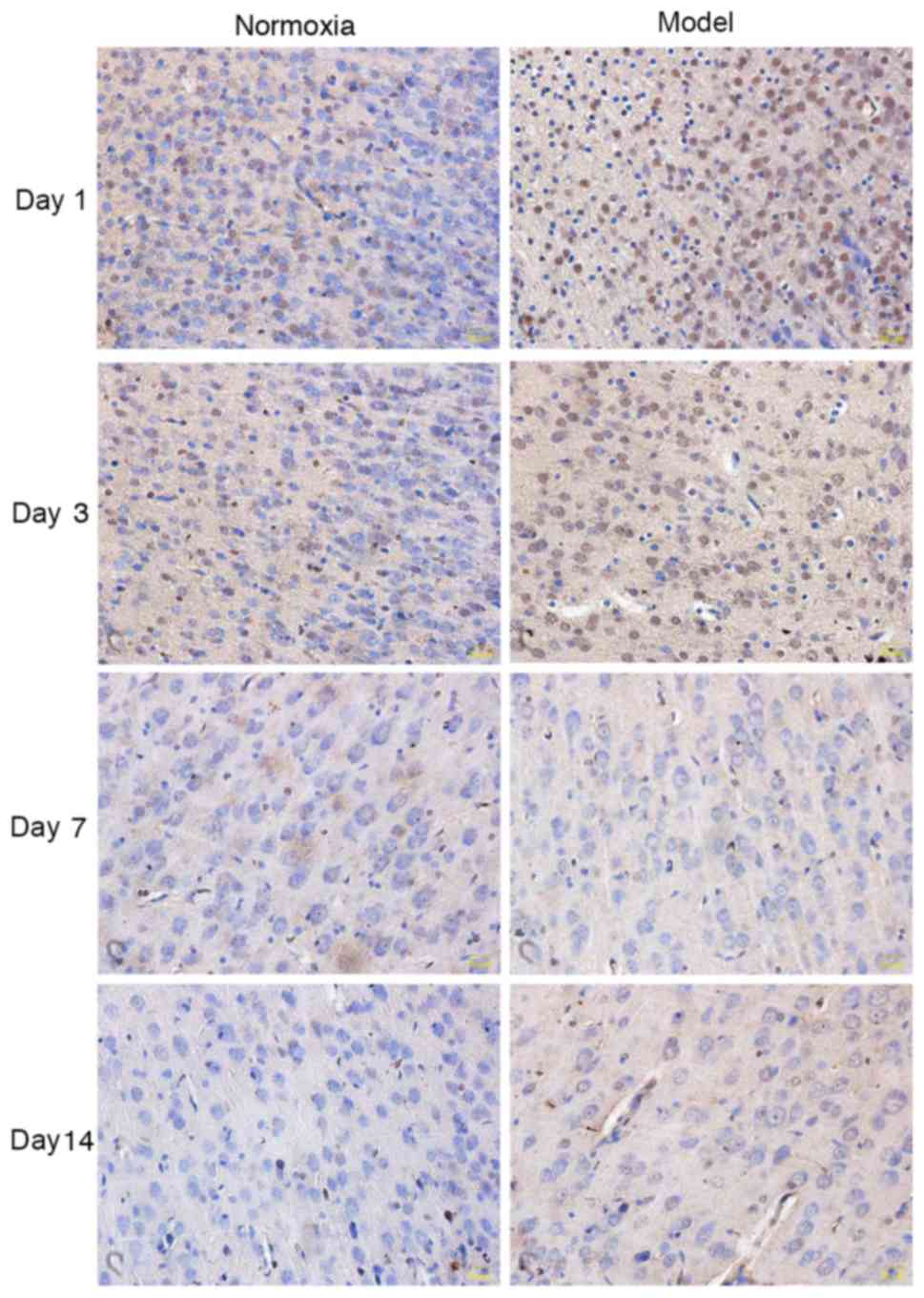
Apoptosis of neurons
Alterations in neuronal apoptosis
detected by TUNEL
In the model group, the nuclei of TUNEL-positive
cells were stained in a scattered spot pattern and shaped
irregularly. Neuronal TUNEL-positive cells were notably increased
and widely present in periventricular white matter, corpus
callosum, hippocampus and cerebral cortex. A small number of
positive cells were observed in the control group. At days 1 and 3,
the number of stained positive cells in the model group was
markedly increased compared with the control group; at days 7 and
14, no difference was identified between the two groups (Fig. 3).
Increased expression of caspase-3
In the control group and model group, caspase-3 mRNA
expression was significantly increased at day 3 compared with day 1
(P<0.01), although it was decreased at days 7 and 14 compared
with day 1. At days 1, 3 and 7, caspase-3 mRNA expression in the
model group was increased compared with the control group
(P<0.05); at day 14, caspase-3 mRNA expression in the model
group was increased compared with the control group, although the
difference between the two groups was not statistically significant
(P>0.05; Fig. 4A). In the
control group and the model group, cleaved caspase-3 protein
expression was significantly increased at day 3 compared with day 1
(P<0.01), and significantly decreased at days 7 and 14 compared
with day 1. At all time-points, cleaved caspase-3 protein
expression in the model group was greater when compared with the
control group (P<0.05; Fig.
4B).
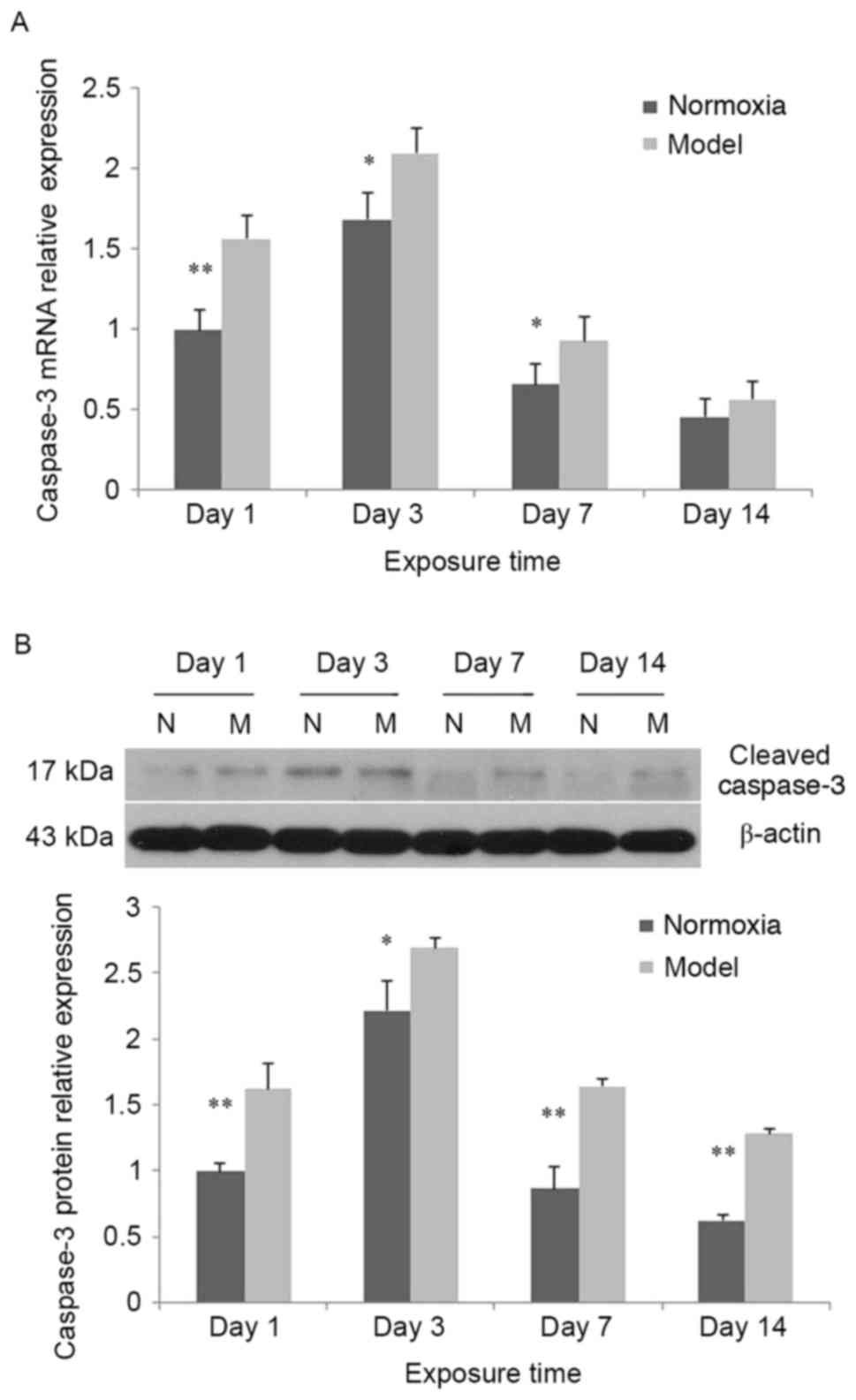 | Figure 4.Alterations in caspase-3 expression
at different time-points following hypoxia-ischemia-induced injury.
(A) In the hypoxia-ischemia-induced injury model, caspase-3 mRNA
expression in brain tissues was increased. In the control and model
groups, caspase-3 mRNA expression was significantly increased at
day 3 compared with day 1, and decreased at days 7 and 14 compared
with day 1. At days 1, 3 and 7, caspase-3 mRNA expression in the
model group was increased compared with the control group; at day
14, caspase-3 mRNA expression in the model group was not
significantly increased compared with the control group. (B) In the
hypoxia-ischemia-induced injury model, cleaved caspase-3 protein
expression in brain tissues was increased. In the control and model
groups, cleaved caspase-3 protein expression was significantly
increased at day 3 compared with day 1, and decreased at days 7 and
14 compared with day 1. At all time-points, cleaved caspase-3
protein expression in the model group was increased compared with
the control group. Values are expressed as relative optical density
and are presented as the mean ± standard deviation. For each
column, n=3. *P<0.05, **P<0.01 vs. the normoxia group. N,
normoxia; M, model. |
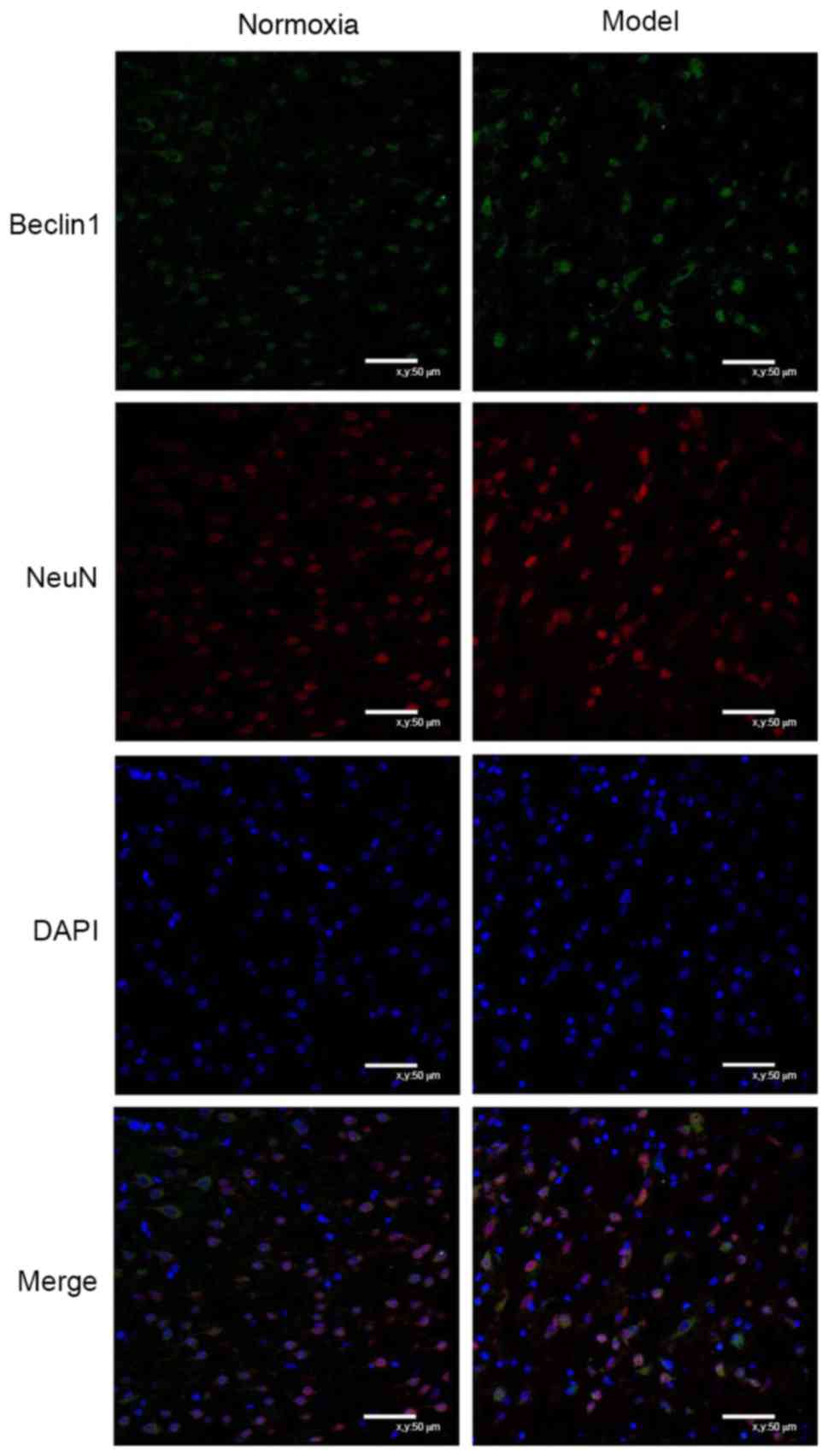
Neuronal autophagy
Double-labeling immunofluorescence
staining of Beclin 1 and NeuN
Beclin1 was expressed in the cytoplasm, while NeuN
was primarily expressed in the nuclei, exhibiting limited
expression in the cytoplasm. At day 7, cells were arranged
regularly in the control group and irregularly in the model group;
the co-expression of Beclin 1 and NeuN in the same cells was
observed in the two groups, and the number of cells with such
co-expression was increased in the model group compared with the
control group (Fig. 5).
Increased expression of Beclin 1
In the control group, Beclin 1 mRNA expression was
significantly increased at days 7 and 14 compared with days 1 and 3
(P<0.05). Compared with the control group, Beclin 1 mRNA
expression in the model group was significantly increased at days
1, 3 and 7 (P<0.01; Fig. 6A).
In the model group, Beclin 1 protein expression was increased
compared with the control group. The trend was consistent with the
mRNA expression (Fig. 6B).
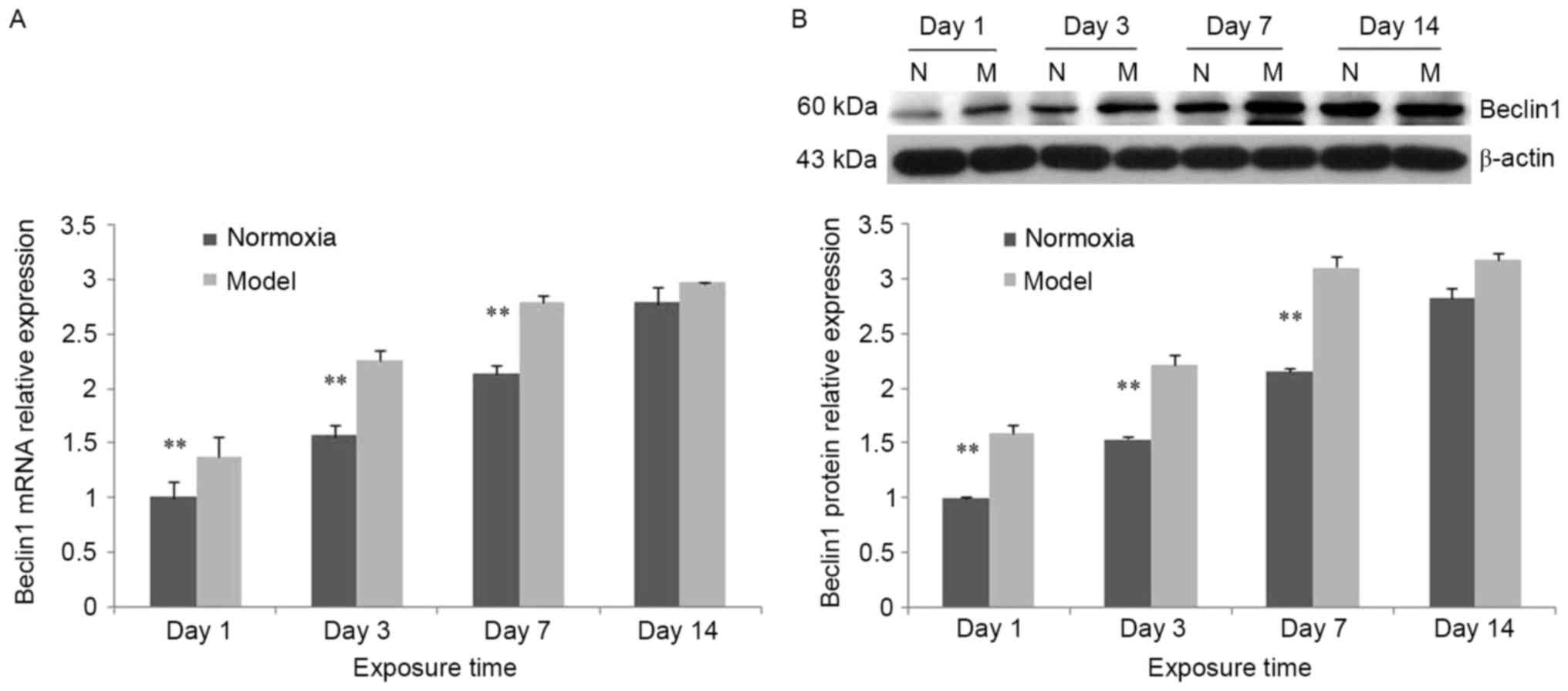 | Figure 6.Alterations in Beclin 1 expression at
different time points following hypoxia-ischemia-induced injury.
(A) In the hypoxia-ischemia-induced injury model, Beclin 1 mRNA
expression in brain tissues was increased. In the control and model
groups, Beclin 1 mRNA expression was increased between days 1 and
14. Compared with the control group, Beclin 1 mRNA expression in
the model group was significantly increased at days 1, 3 and 7. (B)
In the hypoxia-ischemia-induced injury model, Beclin 1 mRNA
expression in brain tissues was increased. In the control group and
model group, Beclin 1 mRNA expression was increased from day 1 to
day 14. Compared with the control group, Beclin 1 protein
expression in the model group was significantly increased at days
1, 3 and 7. Values are expressed as relative optical density and
are presented as the mean ± standard deviation. For each column,
n=3. *P<0.05, **P<0.01 vs. the normoxia group. N, normoxia;
M, model. |
Discussion
Perinatal brain injury, one of the most common
diseases in neonates and, particularly, premature neonates, may
lead to altered development of the neonatal nervous system and
result in varying degrees of sequelae. The most frequently reported
manifestation is PVL (local or diffuse), which is the primary cause
of cerebral palsy and may induce cognitive dysfunction, motor
dysfunction or visual impairment (18,19).
PVL is most frequently observed in premature infants and maybe
caused by a number of factors, including perinatal infection and
asphyxia. Previous studies of PVL have focused on oligodendrocyte
involvement, myelinization disorders and microglial cell
activation. OPCs are the primary targets organ for PVL (20,21).
In general, PVL occurs in premature infants at a gestational age of
23–32 weeks; OPCs account for ~90% of the total number of
oligodendrocytes in the cerebral white matter of these infants,
although they are markedly decreased in >32-week infants, which
is consistent with a reduction in the incidence rate of PVL. A
number of previous studies have demonstrated the increased
vulnerability of immature oligodendrocytes during a specific window
(22,23). Animal experiments have confirmed
that, based on the maturation-dependent vulnerability of
oligodendrocytes, OPCs are easily injured by glutamates,
oxygen-derived free radicals and inflammatory cytokines, thus
leading to apoptosis, necrosis and myelinization delay (24,25).
Hypoxia increases the phagotrophic and oxidative stress
injury-promoting functions of microglial cells through mediation of
inflammatory cytokines, including γ-interferon, tumor necrosis
factor-α, interleukin-1 and lipopolysaccharide causing microglia
cells to release excitatory glutamates (26,27).
Accordingly, the expression of
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid and
N-methyl-D-aspartic acid receptors (NR1 and NR2A-D) has been
observed to be increased in microglial cells (28). Activated microglial cells may
induce the death of OPCs and astrocytes, which has been widely
confirmed (29,30).
However, neuronal injury has also been frequently
observed in PVL as the understanding of PVL has been increasing.
Volpe et al (14), first
proposed the concept of EP, and they observed non-negligible
neuronal injury among brain injuries in premature infants. Andiman
et al (13), reported a 38%
decrease in the density of pyramidal neurons in the fifth layer of
the cerebral cortex of premature infants with PVL. In addition,
neuropathologists have previously demonstrated that the time at
which neurons in the germinative zone migrated towards the cerebral
cortex via the periventricular white matter coincided with the time
when periventricular WMI occurred, and the toxicities of
inflammatory cytokines and excitatory amino acids may injure these
migrating neurons while damaging periventricular white matter
(31). Such neurons (for example,
subplate neurons) may exhibit the same pathogenesis as OPCs in
white matter (32), express a
number of glutamate receptors, and demonstrate selective
vulnerability similar to OPCs (33,34).
In the present study, neuronal injury in premature infants
following hypoxia-ischemia was investigated using a
hypoxia-ischemia-induced PVL model. The model was successfully
established in a previous PVL study completed by the present
research group (15). NeuN is a
soluble nucleoprotein and a marker of mature neurons, and its
molecular weight is 46–48 kDa. In the present study, NeuN
expression was reduced and mature neurons were reduced in the model
group, which indicated that the number of mature neurons was
decreased in the rat model of hypoxia-ischemia-induced PVL; the
presence of neuronal injury in PVL was confirmed in the animal
models. NeuN expression reached a trough in the model group at day
3, suggesting that neuronal injury was most severe at day 3
following hypoxic exposure. NeuN protein expression was increased
at day 7 to exceed the level at day 1; however, it remained low
compared with the level observed in the control group, which
demonstrated that a certain repair mechanism to achieve neuronal
regeneration or transformation following neuronal injury was
induced by hypoxia in premature infants, although NeuN protein
expression was not able to be recovered to the same level as prior
to the injury. The trough of NeuN expression in the model group was
observed at day 3, perhaps due to the repair mechanism not having
been initiated while tissue injury was ongoing. The present study
further investigated the apoptosis and autophagy of injured
neurons, in an attempt to elucidate the injury and repair
mechanisms.
The following types of cell death have been
recognized: Necrosis, apoptosis (type I apoptosis) and autophagic
apoptosis (type II apoptosis) (35). Necrosis is a type of non-apoptotic
cell death caused by external injury, and its morphological
characteristics are different from those of apoptosis; generally,
necrosis is accompanied by inflammation (36). Caspase-3 is a member of the caspase
family and an important protein to detect in apoptosis, as there
are caspase-3-dependent and caspase-3-independent forms of
apoptosis. Cleaved caspase-3 is an active form of sliced caspase-3,
and its protein expression elevation may indicate apoptosis
increase (37). In the present
study, caspase-3 expression was increased and reached a peak at day
3 (when NeuN expression reached at rough) in the model group, and
thereafter it was decreased (mean while NeuN expression was
increased). Therefore, it may be inferred that caspase-3-dependent
apoptosis may be an important cause of neuronal injury. A number of
studies have demonstrated that hypoxia may lead to an increase in
neuronal apoptosis and result in brain injury (38–40).
The peak expression of caspase-3 in the control group was
additionally observed at day 3 (6 days following birth). There are
previous studies investigating hypoxia-ischemia-induced brain
injury in P5-7 rats (equal to full-term or nearly full-term human
infants) (41,42). Therefore, it was considered that
P5-7 maybe a window for neuronal injury and P5-7 rats may
demonstrate the most marked apoptosis and the least tolerance to
hypoxia. The TUNEL assay results in the present study further
confirmed that neuronal apoptosis in the cerebral cortex was
increased in the hypoxia-ischemia-induced PVL model. The results of
the present study demonstrated that in the hypoxia-ischemia-induced
PVL model, in addition to injury to oligodendrocytes, there was a
decrease in the number of neurons and an increase in neuronal
apoptosis, which validates the hypothesis that neuronal apoptosis
is an important cause of neuronal injury in this model.
Autophagic programmed cell death is embodied by the
appearance of numerous vacuolar structures, encapsulating the
cytoplasm and organelles within the cytoplasm, and the degradation
of their contents by lysosomes. In autophagic programmed cell
death, intermediate filaments and microfilaments are redistributed,
although not degraded, and actin remains in a polymeric form, so
the cytoskeletal system is well-maintained. Conversely, in
apoptotic programmed cell death, the depolymerization of actin and
the degradation of intermediate filaments occur early on, and the
cytoskeletal system is consequently damaged. Autophagy exerts an
important role in the growth and development of cells, and the
occurrence of diseases. A number of studies have reported that
autophagy and apoptosis may antagonize or promote one another in
certain cases, and they may occur successively or co-exist in the
same cells (35,43–45).
Beclin1 was the first autophagy-associated gene identified in
mammals, and the Beclin 1 protein is able to bind to type III
phosphatidylinositol 3-kinase (PI3K) to regulate autophagy
(46). The results of the present
study demonstrated that Beclin 1 expression was gradually increased
in the model group, with a trend coincident to the increase of NeuN
expression and opposite to the decrease of caspase-3 expression.
Therefore, it was hypothesized that autophagy may be involved in
the inhibition of apoptosis and even the repair process of neuronal
injury, which requires validation in subsequent experiments.
Balduini et al (38)
suggested that autophagy may be part of a pro-survival signal
containing the PI3K-Rac-α serine/threonine protein kinase-protein
kinase mTOR axis, and that its activation mechanisms included drug
effects, infection and hypoxia-ischemia. Carloni et al
(47) demonstrated that rapamycin
administration prior to hypoxia-ischemia markedly increased the
expression of Beclin 1 and microtubule-associated proteins 1A/1B
light chain 3B (autophagy-associated proteins), and decreased
neuronal injury. These previous results indicated that facilitating
autophagy prior to hypoxia-ischemia may have a certain protective
effect on neurons, while interrupting autophagy may inhibit such
protection. It was previously reported that blocking autophagy at 3
h post-ischemia/reperfusion had a neuroprotective role (48). Therefore, autophagy may be used as
a target for the protective treatment of the nervous system. This
opposite result demonstrates the double role of autophagy:
Autophagy is protective in the early stage sand harmful in the late
stages of nerve degeneration, which depends on the time of drug
administration. Therefore, autophagy has a double effect on cells
and neurons: In hypoxia-ischemia-induced injury, it may protect
cells and prevent cell death, or may become one of the factors
causing cell death, specifically depending on different phases of
disease progression, alterations to the pericellular environment,
and different intervention measures. The present study demonstrated
that Beclin 1 was expressed in neurons, and that Beclin 1
expression was increased in the model group, close to the level of
the control group at day 14 following hypoxia-induced injury; the
time of the Beclin 1 expression increase coincided with the time
window of repair following neuronal injury. Therefore, it may be
inferred that cellular autophagy may exert a protective effect on
neurons in the hypoxia-ischemia-induced PVL rat model, which
requires confirmation in further experiments.
In conclusion, there are alterations to
oligodendrocytes and neurons in a hypoxia-ischemia-induced PVL
model. In the model group, the trough of neuronal cell expression
was observed at the time-point when cellular apoptosis was in the
most active state, which proves that neuronal apoptosis is an
important cause of neuronal injury. Neuronal autophagy is
additionally involved in this pathological process, although it is
increased at the later stages of injury. Cellular autophagy may
exhibit protective effect in neurons, although its specific role in
this process has not been confirmed. The present study may promote
a novel target for the treatment of PVL.
Acknowledgements
We gratefully acknowledge Professors Dongyan Liu and
Guifeng Zhao for invaluable help during the study. We would like to
thank the Center Laboratory and Department of Animal of Shengjing
Hospital of China Medical University for expert assistance.
Funding
Not applicable.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
LQ: Conceptualization of the study, performance of
assays, data curation, formal analysis, investigation, validation,
writing at the original draft and review and editing stage. JF:
Conceptualization of the study, data curation, performance of
assays, validation and writing at the review and editing stage. XX:
Conceptualization of the study, data curation, performance of
assays, validation and supervision. YS: Conceptualization of the
study and performance of assays. LY: Conceptualization of the study
and performance of assays. WH: Data curation and formal analysis.
JL: Data curation and formal analysis. DZ: Investigation. NL:
Investigation. XT: Formal analysis and software operation. YD:
Performance of assays and writing of the original draft. YP:
Performance of assays and writing of the original draft.
Ethics approval and consent to
participate
Ethical approval for the present study was provided
by Shengjing Hospital of China Medical University Ethics Committee
(Shenyang, China).
Consent for publication
Not applicable.
Competing interests
All authors declared that they have no conflict of
interest with regard to this study.
References
|
1
|
Deng W, Pleasure J and Pleasure D:
Progress in periventricular leukomalacia. Arch Neuro. l65:1–1295.
2008.
|
|
2
|
Hamilton BE, Miniño AM, Martin JA,
Kochanek KD, Strobino DM and Guyer B: Annual summary of vital
statistics: 2005. Pediatrics. 119:345–360. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Volpe JJ: Cerebral white matter injury of
the premature infant-more commonthan you think. Pediatrics.
112:176–180. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dyet LE, Kennea N, Counsell SJ, Maalouf
EF, Ajayi-Obe M, Duggan PJ, Harrison M, Allsop JM, Hajnal J,
Herlihy AH, et al: Natural history of brain lesions in
extremelypreterm infants studied with serial magnetic resonance
imaging from birth and neurodevelopmental assessment. Pediatrics.
118:536–548. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hack M, Taylor HG, Drotar D, Schluchter M,
Cartar L, Wilson-Costello D, Klein N, Friedman H, Mercuri-Minich N
and Morrow M: Poor predictive validity of the bayley scales of
infant development for cognitive function of extremely low birth
weight children at school age. Pediatrics. 116:333–341. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilson-Costello D, Friedman H, Minich N,
Siner B, Taylor G, Schluchter M and Hack M: Improved
neurodevelopmental outcomes for extremely low birth weight infants
in 2000–2002. Pediatrics. 119:37–45. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leviton A, Dammann O and Durum SK: The
adaptive immune response in neonatal cerebral white matter damage.
Ann Neurol. 58:821–828. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dommergues MA, Plaisant F, Verney C and
Gressens P: Early microglial activation following neonatal
excitotoxic brain damage in mice: A potential target for
neuroprotection. Neuroscience. 121:619–628. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tahraoui SL, Marret S, Bodénant C, Leroux
P, Dommergues MA, Evrard P and Gressens P: Central role of
microglia in neonatal excitotoxic lesions of the murine
periventricular white matter. Brain Pathol. 11:56–71. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Back SA: Brain injury in the preterm
infant: New horizons for pathogenesis and prevention. Pediatr
Neurol. 53:185–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zonouzi M, Scafidi J, Li P, McEllin B,
Edwards J, Dupree JL, Harvey L, Sun D, Hübner CA, Cull-Candy SG, et
al: GABAergic regulation of cerebellar NG2 cell development is
altered in perinatal white matter injury. Nat Neurosci. 18:674–682.
2015. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sarnat HB, Philippart M, Flores-Sarnat L
and Wei XC: Timing in neural maturation: Arrest, delay,
precociousness and temporal determination of malformations.
PediatrNeurol. 52:473–486. 2015.
|
|
13
|
Andiman SE, Haynes RL, Trachtenberg FL,
Billiards SS, Folkerth RD, Volpe JJ and Kinney HC: The cerebral
cortex overlying periventricular leukomalacia: Analysis of
pyramidal neurons. Brain Pathol. 20:803–814. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Volpe JJ: Encephalopathy of prematurity
includes neuronal abnormalities. Pediatrics. 116:221–225. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheng T, Xue X and Fu J: Effect of OLIG1
on the development of oligodentrocytes and myelination in a neonal
rat PVL model induced by hypoxia-ischemia. Mol Med Rep.
11:2379–2386. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mizuno K, Hida H, Masuda T, Nishino H and
Togari H: Pretreatment with low doses of erythropoietin ameliorates
brain damage in periventricular leukomalacia by targeting late
oligodendrocyte progenitors: A rat model. Neonatology. 94:255–266.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marret S, Marchand-Martin L, Picaud JC,
Hascoët JM, Arnaud C, Rozé JC, Truffert P, Larroque B, Kaminski M
and Ancel PY; EPIPAGE Study Group, : Brain injury in very preterm
children and neurosensory and cognitive disabilities during
childhood: The EPIPAGE cohort study. PLoS One. 8:e626832013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Volpe JJ: Brain injury in premature
infants: A complex amalgam of destructive and developmental
disturbances. Lancet Neurol. 8:110–124. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang P, Chen C, Liu XB, Pleasure DE, Liu
Y and Deng W: Human iPSC-derived immature astroglia promote
oligodendrogenesis by increasing TIMP-1 Secretion. Cell Rep.
15:1303–1315. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jantzie LL, Talos DM, Jackson MC, Park HK,
Graham DA, Lechpammer M, Folkerth RD, Volpe JJ and Jensen FE:
Developmental expression of N-methyl-D-aspartate (NMDA) receptor
subunits in human white and gray matter: Potential mechanism of
increased vulnerability in the immature brain. Cereb Cortex.
25:482–495. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Haynes RL, Baud O, Li J, Kinney HC, Volpe
JJ and Folkerth DR: Oxidative and nitrative injury in
periventricular leukomalacia: A review. Brain Pathol. 15:225–233.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Back SA and Rivkees SA: Emerging concepts
in periventricular white matter injury. Semin Perinatol.
28:405–414. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Craig A, Ling Luo N, Beardsley DJ,
Wingate-Pearse N, Walker DW, Hohimer AR and Back SA: Quantitative
analysis of perinatal rodent oligodendrocyte lineage progression
and its correlation with human. Exp Neurol. 181:231–240. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Emery B: Transcriptional and
post-transcriptional control of CNS myelination. Curr Opin
Neurobiol. 20:601–607. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu Y, Silverstein FS, Skoff R and Barks
JD: Hypoxic-ischemic oligodendroglial injury in neonatal rat brain.
Pediatr Res. 51:25–33. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Segovia KN, McClure M, Moravec M, Luo NL,
Wan Y, Gong X, Riddle A, Craig A, Struve J, Sherman LS and Back SA:
Arrested oligodendrocyte lineage maturation in chronicperinatal
white matter injury. Ann Neurol. 63:520–530. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Manning SM, Boll G, Fitzgerald E, Selip
DB, Volpe JJ and Jensen FE: The clinically available NMDA receptor
antagonist, memantine, exhibits relative safety in the developing
rat brain. Int J Dev Neurosci. 29:767–773. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xapelli S, Bernardino L, Ferreira R, Grade
S, Silva AP, Salgado JR, Cavadas C, Grouzmann E, Poulsen FR,
Jakobsen B, et al: Interaction between neuropeptide Y (NPY) and
brain-derived neurotrophic factor in NPY-mediated neuroprotection
against excitotoxicity: A role for microglia. Eur J Neurosci.
27:2089–2102. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Domercq M, Sánchez-Gómez MV, Sherwin C,
Etxebarria E, Fern R and Matute C: System xc- and glutamate
transporter inhibition mediates microglial toxicity to
oligodendrocytes. J Immunol. 178:6549–6556. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Leviton A and Gressens P: Neuronal damage
accompanies perinatal white-matter damage. Trends Neurosci.
30:473–478. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Haynes RL, Xu G, Folkerth RD, Trachtenberg
FL, Volpe JJ and Kinney HC: Potential neuronal repair in cerebral
white matter injury in the human neonate. Pediatr Res. 69:62–67.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Talos DM, Fishman RE, Park H, Folkerth RD,
Follett PL, Volpe JJ and Jensen FE: Developmental regulation of
alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor
subunit expression in forebrain and relationship to regional
susceptibility to hypoxic/ischemic injury. I. Rodent cerebral white
matter and cortex. J Comp Neurol. 497:42–60. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zubiaurre-Elorza L, Soria-Pastor S, Junque
C, Segarra D, Bargalló N, Mayolas N, Romano-Berindoague C and
Macaya A: Gray matter volume decrements in preterm children with
periventricular leukomalacia. Pediatr Res. 69:554–560. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lockshin RA and Zakeri Z: Apoptosis,
autophagy, and more. Int J Biochem Cell Biol. 36:2405–2019. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Natoli G, Costanzo A, Guido F, Moretti F
and Levrero M: Apoptotic, non-apoptotic, and anti-apoptotic
pathways of tumor necrosis factor signalling. Biochem Pharmacol.
56:915–920. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Budd RC: Activation-induced cell death.
Curr Opin Immunol. 13:356–362. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Balduini W, Carloni S and Buonocore G:
Autophagy in hypoxia-ischemia induced brain injury. J Matern Fetal
Neonatal Med. 25 Suppl 1:S30–S34. 2012. View Article : Google Scholar
|
|
39
|
Barteczek P, Li L, Ernst AS, Böhler LI,
Marti HH and Kunze R: Neuronal HIF-1α and HIF-2α deficiency
improves neuronal survival and sensorimotor function in the early
acute phase after ischemic stroke. J Cereb Blood Flow Metab.
37:291–306. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xiang Q, Zhou WY, Hu WX, Wen Z, He D, Wu
XM, Wei HP, Wang WD and Hu GZ: Neuroprotective effects of Rhizoma
Dioscoreae polysaccharides against neuronal apoptosis induced by in
vitro hypoxia. ExpTher Med. 10:2063–2070. 2015. View Article : Google Scholar
|
|
41
|
Olgun Y, Kırkım G, Kolatan E, Kıray M,
Bağrıyanık A, Şerbetçioğlu B, Yılmaz O, Gökmen N, Ellidokuz H,
Kumral A and Sütay S: Otoprotective effect of recombinant
erythropoietin in a model of newborn hypoxic-ischemic
encephalopathy. Int J Pediatr Otorhinolaryngol. 77:739–746. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pimentel-Coelho PM, Magalhães ES, Lopes
LM, deAzevedoL C, Santiago MF and Mendez-Otero R: Human cord blood
transplantation in a neonatal rat model of hypoxic-ischemic brain
damage: Functional outcome related to neuroprotection in the
striatum. Stem Cells Dev. 19:351–358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Reggiori F: Membrane origin for autophagy.
Curr Top Dev Biol. 74:1–30. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tsujimoto Y and Shimizu S: Another way to
die: Autophagic programmed cell death. Cell Death Differ. 12 Suppl
2:S1528–S1534. 2005. View Article : Google Scholar
|
|
45
|
Tang JF, Wen Q, Sun J, Zhang WM and Zhu
HL: Advances in the researches on the biological activities and
inhibitors of phosphatidylinositol 3-kinase. Anticancer Agents Med
Chem. 14:673–687. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gallagher LE, Williamson LE and Chan EY:
Advances in autophagy regulatory mechanisms. Cells. 5:E242016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Carloni S, Girelli S, Scopa C, Buonocore
G, Longini M and Balduini W: Activation of autophagy and Akt/CREB
signaling play an equivalent role in the neuroprotective efect of
rapamycin in neonatal hypoxia-ischemia. Autophagy. 6:366–377. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shi R, Weng J, Zhao L, Li XM, Gao TM and
Kong J: Excessive autophagy contributes to neuron death in cerebral
ischemia. CNS Neurosci Ther. 18:250–260. 2012. View Article : Google Scholar : PubMed/NCBI
|