Introduction
Duchenne muscular dystrophy (DMD) and Becker
muscular dystrophy (BMD) are the most common childhood muscular
dystrophies, and are caused by mutations in the dystrophin gene,
which encodes dystrophin protein at the Xp-21 locus. DMD and BMD
occur with a birth prevalence rate of 15.9–19.5 cases per 100,000
live male births and 1 case per 6,000–8,000 live male births,
respectively (1,2). Approximately 60–65% of mutations are
deletions, 5–15% are duplications and 30% are point mutations
(3). There are two hotspots in the
dystrophin gene. The most important hotspots are located in exon
45–55 and exon 2–19 (4). However,
DMD and BMD are X-linked recessive diseases, and thus females are
the carriers of the mutation.
The majority of DMD/BMD carriers remain
asymptomatic, while only 2.5–10% of carriers are symptomatic and
classified as manifesting carriers (MCs) (5). The symptoms of MCs range from mild
muscle weakness to severe abnormal gait with frequent falls, and
difficulty in rising from the floor or walking on tiptoes. Certain
patients even have rapidly progressive muscular dystrophy similar
to male dystrophinopathy. Diagnosis of DMD/BMD carriers is based on
clinical symptoms, family history, biochemistry markers,
echocardiography, pathology, molecular genetic analysis and
linkage-testing. Identification of DMD/BMD carriers may present
challenges, particularly in the absence of a family history of
DMD/BMD. Genetic analysis of the dystrophin gene is typically
required for diagnosis, particularly for genetic consulting and
prenatal diagnosis.
In the present study, clinical data were collected
and analyzed from 154 probable female carriers. The creatine kinase
(CK) level of the subjects, and its correlation with alanine
transaminase (ALT) and aspartate transaminase (AST) levels were
analyzed. Multiplex ligation-dependent probe amplification (MLPA)
for exons of the dystrophin gene, combined with a muscle disease
panel based on next-generation sequencing (NGS), was applied to
detect the MCs. Two of the MCs were confirmed by muscle
pathological examination. The status of probable carriers was
determined by MLPA and Sanger sequencing according to the mutations
of probands. The present study presents a non-invasive, easy
screening for MCs of DMD/BMD, and describes an effective, accurate
method for the detection of deletions/duplications and small
mutations in DMD/BMD carriers.
Materials and methods
Participants
The study participants were selected between January
2013 and October 2017 at the First Affiliated Hospital of Guangxi
Medical University (Nanning, China). The subjects included male DMD
patients (proband), symptomatic females and their relatives,
including mothers, maternal grandmothers, maternal aunts and
sisters of probands with confirmed DMD dystrophin gene mutations.
The present study was conducted in strict accordance with the
recommendations in the Guidelines of the Centers for Disease
Control and Prevention (1). The
study was approved by the Ethics Committee of the First Affiliated
Hospital of Guangxi Medical University. Informed consent was
obtained from all participants included in the study.
Inclusion and exclusion criteria
Subjects included in the present study fulfilled one
of the following criteria: i) Exhibited clinical manifestations,
such as muscle weakness, walking with frequent falling, abnormal
gait, and difficulty running, jumping and going upstairs; ii) were
maternal female relatives of probands with confirmed diagnosis of
DMD/BMD; and iii) exhibited a persistent CK level that was >2
times greater than the upper limit of the normal range (CK, <178
U/l). Symptomatic females who were confirmed by genetic testing to
suffer from another muscular dystrophy disease, such as limb-girdle
muscular dystrophy (LGMD), or females with incomplete clinical
information were excluded from the present study.
Clinical analysis
The clinical data (including the chief complaint,
history of present illness, growth and development history, past
medical history and family history) of all subjects were collected
and reviewed retrospectively. Blood samples were collected and
levels of serum enzymes CK, liver function and cardiac enzymes
concentrations were measured using a Hitachi 7600-020 analyzer
(Hitachi, Ltd., Tokyo, Japan). All assays were conducted according
to the manufacturer's instructions using the same batch reagent.
Echocardiography findings and muscle biopsies were analyzed.
Genetic analysis
All probands and MCs underwent MLPA for dystrophin
gene exons. If no mutation was identified by MLPA, NGS-based muscle
disease panel tests (containing 169 known muscle disease-associated
genes, including for LGMD and facioscapulohumeral muscular
dystrophy) was performed. When large deletions/duplications were
observed by MLPA analysis, MLPA was also used to classify the
female relatives. If a point mutation was identified by NGS in
probands, The mutation site was amplified by PCR (the primers were
designed by the website Primer Z(http://genepipe.ncgm.sinica.edu.tw/primerz/primerz4.do),
and then sequenced by Sanger sequencing for probands and their
female relatives.
MLPA analysis
DNA samples were extracted from peripheral blood
obtained from the subjects according to standard procedures
[FlexiGene DNA kit (cat. no. 51206; Qiagen GmbH, Hilden, Germany)].
All 79 exons of the dystrophin gene were screened by MLPA.
Two sets of reagents (SALSA MLPA probe sets P034 and P035) were
used to perform the MLPA reaction according to the manufacturer's
instructions (MRC-Holland BV, Amsterdam, The Netherlands). Genomic
DNA was denatured, hybridized, ligated, and amplified. Amplified
products were analyzed on an ABI model 3500XL capillary sequencer
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). The initial
data were analyzed using GeneMapper software (version 140701.0000),
and the peak area of each fragment was compared with that of 3
control samples.
Target enrichment of genomic DNA and
sequencing
A minimum of 3 µg DNA was used to construct the
indexed Illumina libraries according to the manufacturer's
instructions. A final library size of 200–300 bp, including adapter
sequences, was selected. All exons of the dystrophin gene
were target-enriched using the dystrophin Exon Enrichment
kit (MyGenostics, Inc., Baltimore, MD, USA). The muscle diseases
panel was a complete kit designed by the Zhongguancun Huakang Gene
Institute (Beijing, China) and synthesized using the Agilent
SureSelect Target Enrichment technique (Agilent Technologies, Inc.,
Santa Clara, CA, USA). The capture experiment was conducted
according to the manufacturer's instructions. The enrichment
libraries were sequenced on an Illumina HiSeq 2000 sequencer
(Illumina, Inc., San Diego, CA, USA) for paired-read 100 bp
sequencing.
Bioinformatics analysis
Following HiSeq 2000 sequencing (Illumina, Inc.),
high-quality reads were retrieved from raw reads by filtering out
the low-quality reads and adaptor sequences using the Solexa QA
package (sourceforge.net/projects/solexaqa/files/) and the
cutadapt program (hpc.nih.gov/apps/cutadapt.html), respectively. The
SOAPaligner program (soap.genomics.org.cn/) was then used to align the
clean read sequences to the human reference genome (hg19). To
detect exon duplications and deletions, the coverage of each
position was plotted by base position. Higher coverage of a region
indicated duplication, whereas regions that were not covered
corresponded to deletions. Identical sequences were produced by
polymerase chain reaction (PCR) duplication to obtain cluster
formation (human genomic DNA template). After the PCR duplicates
were removed using Picard software (http://broadinstitute.github.io/picard/; version
2.6.0-SNAPSHOT), single-nucleotide polymorphisms (SNPs) were
identified using the SOAPsnp program (http://soap.genomics.org.cn/soapsnp.html).
Subsequently, the reads were realigned to the reference genome
using BWA, and insertions or deletions (InDels) were identified
using the GATK program (www.broadinstitute.org/gsa/wiki/index.php/Home_Page).
The identified SNPs and InDels were annotated using the
Exome-assistant program (http://122.228.158.106/exomeassistant).
Muscle pathology
Biopsy of the right gastrocnemius was performed in 2
female patients (both MCs) and 1 male DMD patient, and non-muscular
dystrophy muscle tissue was utilized as a control. Each patient
underwent open muscle biopsy from the right gastrocnemius under
local anesthesia. Fresh specimens were fixed in 10% neutral
buffered formalin and further processed into paraffin-embedded
blocks. The morphology was observed under a microscope following
hematoxylin and eosin staining. In addition, immunohistochemistry
was used to evaluate dystrophin protein expression. Briefly,
sections were incubated at 4°C overnight with a rabbit polyclonal
antibody targeting dystrophin (cat. no. RB-9024; 1:100; Thermo
Fisher Scientific, Inc.), and then incubated at room temperature
for 30 min with Supervision TM Universal (6) (Anti-Mouse/Rabbit) Detection Reagent
(HRP) (cat. no. D-3004; Lab Vision Corporation, Fremont, CA, USA)
conjugated to peroxidase in Tris-HCI buffer containing carrier
protein and anti-microbial agent. The experiment was performed
according to the manufacturer's protocol.
Statistical analysis
Statistical analysis was conducted using SPSS
software (version 16.0; SPSS, Inc., Chicago, IL, USA).
Independent-samples t-test was used to compare the difference in CK
level between Group 1 (MCs) and Group 2 (asymptomatic female
carriers with high CK levels). Pearson's correlation coefficient
(r) was used to identify the probable correlation of CK level with
the AST and ALT levels in female carriers. Receiver operating
characteristic (ROC) curve analysis was used to distinguish the
predicted value of CK in DMD/BMD carriers. P<0.05 was considered
to denote differences that were statistically significant.
Results
Participant characteristics
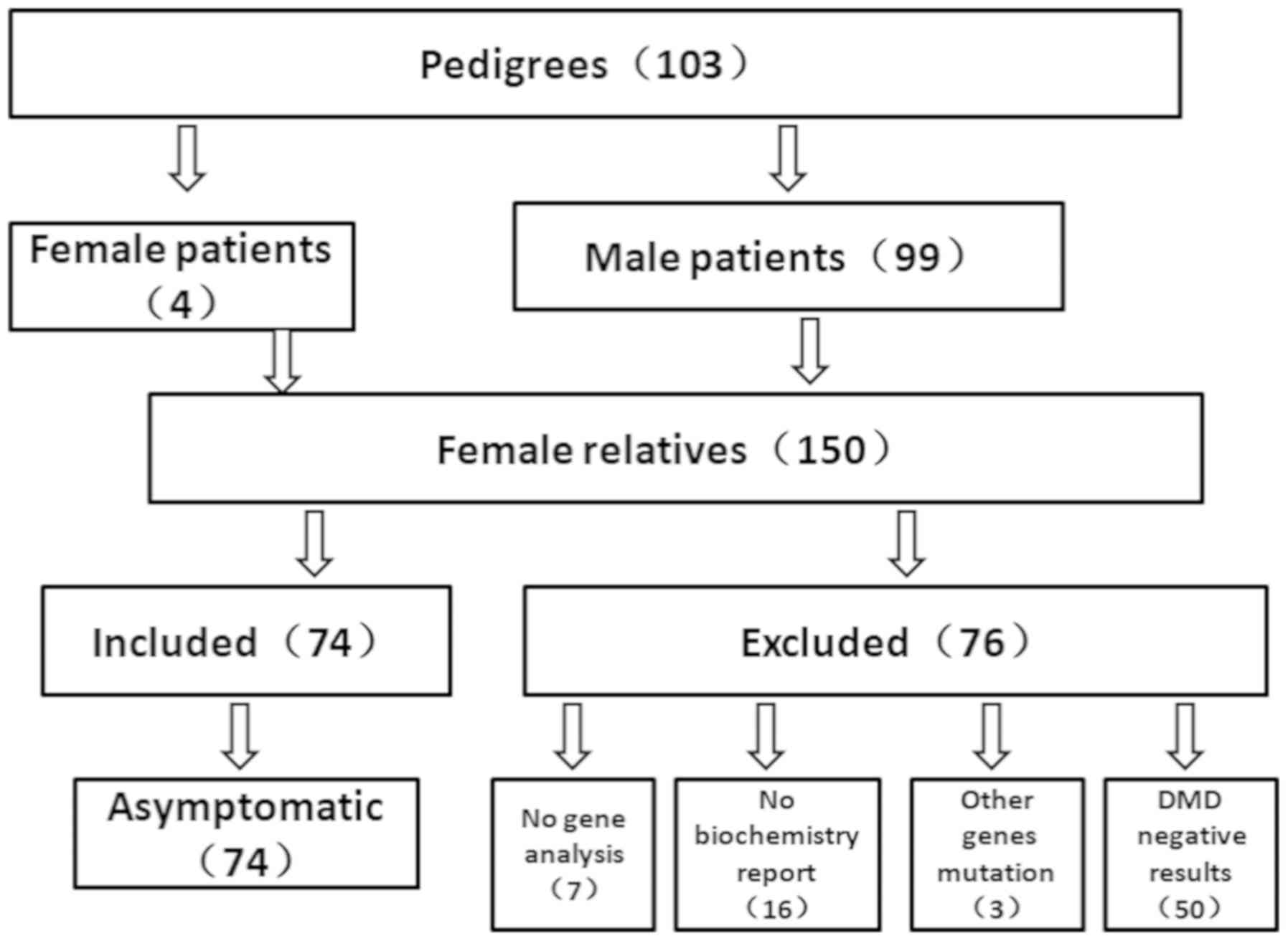
A total of 103 families from the south of China were
recruited in the present study, including 4 MCs and 99 male DMD
patients. Furthermore, 150 female relatives were collected. Of
these 150 participants, 23 participants were excluded due to
incomplete clinical information, and 3 participants were excluded
due to testing positive for a mutation in other genes that resulted
in muscular dystrophy disease, confirmed as LGMD by muscle disease
panel analysis. A homozygous mutation c.583G>A in the SGCA gene
(LGMD2D), compound heterozygous mutations c.620T>C and
c.823C>T in the FKRP gene (LGMD2I), and compound heterozygous
mutations c.77848C>T and c.97099C>T in the TTN gene (LGMD2J)
were detected in these 3 cases. A total of 50 participants were
also excluded as their DMD genetic analysis results were negative.
Ultimately, 78 females were enrolled into the present study. Among
these, 4 cases were MCs and 74 cases were asymptomatic female
carriers, including 2 cases who were carriers of suspected germline
mosaicism with no mutation in the dystrophin gene (Fig. 1).
Clinical symptoms
The present study included 4 symptomatic female
carriers. Two of these MCs were admitted to the hospital due to
elevated CK levels. All exhibited a variety of symptoms, including
falls, waddling gait and tiredness (Table I).
 | Table I.Clinical and genetic characteristic of
4 MCs. |
Table I.
Clinical and genetic characteristic of
4 MCs.
| MC patient | Age at onset | Family history | Severity | Clinical
symptoms | CK (U/l) | AST (U/l) | ALT (U/l) | Biopsy | Mutation
position |
|---|
| 1 | 2 years | Positive | DMD-like | Mild motor
developmental delay, easy fatigue, slow gait, Gowers sign (+), calf
pseudohypertrophy, muscle weakness of upper limbs, lower limbs
4(+) | 6,162 | 107 | 154 | Small amount of the
proliferation of connective tissue in the muscle fibers, few
stromal small vessels with small amount of granulocytes
Immunohistochemical examination for dystrophin antibody exhibited a
mosaic reduction of the protein expression | Duplication at exons
45–55 |
| 2 | 3 years | Negative | DMD-like | Easy fatigue, Gowers
sign (+) | 5,306 | 171 | 97 | Not performed | Duplication at exons
9–10 and 69–70 |
| 3 | Early childhood | Positive | Mild BMD-like | Slow gait, myalgia,
easy fatigue, muscle weakness of lower and upper limb 4 (+) | 4,713 | 83 | 101 | Not performed | Duplication at exons
45–55 |
| 4 | 4 years | Unknown
(adopted) | Mild, BMD-like | Myalgia, muscle
weakness of lower and upper limb 5 | 2,158 | 91 | 60 | Some muscle atrophy,
fuzzy muscle fibers Immunohistochemical examination for dystrophin
antibody exhibited a mosaic reduction of the protein
expression | Duplication at exons
49–52 |
Laboratory examination
Variation of CK level
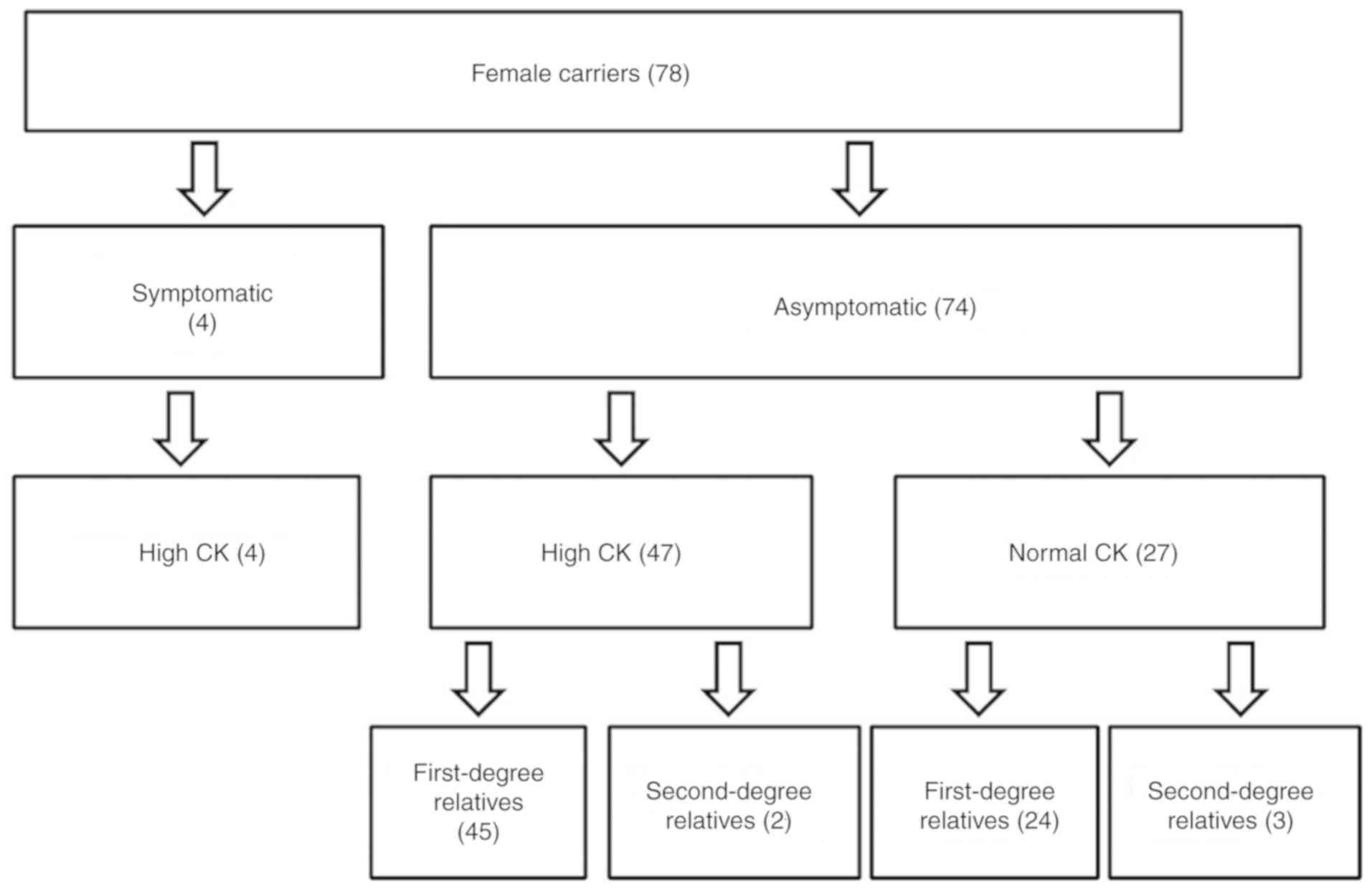
The 4 MCs exhibited an increased CK level (100%),
and the mean CK level was 4,584.75 U/l. Among the 74 asymptomatic
carriers, 47 cases had high CK levels, including 45 first-degree
relatives and 2 sec-degree relatives of DMD/BMD probands (Fig. 2), with a mean CK level of 987.8 U/l
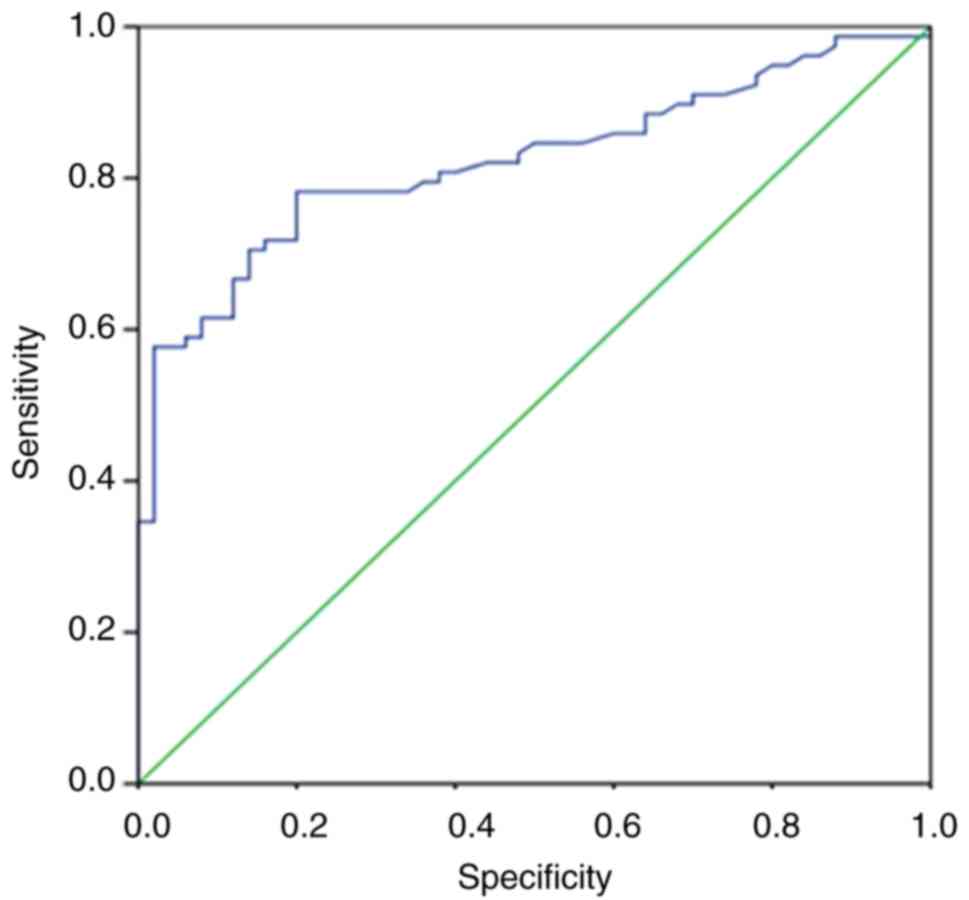
(range, 180–7,012 U/l). The ROC curve analysis revealed that the CK
level was an excellent predictor for distinguishing DMD/BMD
carriers. The area under the ROC curve was 0.822 (P<0.001), the
sensitivity was 65.38% and the specificity was 92.1% (Fig. 3). Furthermore, a total of 27
asymptomatic female carriers had normal CK levels, accounting for
24 first-degree relatives and 3 sec-degree relatives (Fig. 2). The mean CK level of these
subjects was 103.59 U/l (range, 24–174 U/l; Table II). Female carriers were then
divided into 3 groups according to their CK level: Group 1, MCs;
Group 2, asymptomatic female carriers with high CK levels; and
Group 3, asymptomatic female carriers with normal CK levels
(Table II). Independent-samples
t-test was used to compare the difference in CK level between Group
1 and Group 2, indicating that the CK level of MCs was significant
higher in comparison with that of asymptomatic female carriers with
high CK (P<0.001).
| Table II.CK level of all female carriers
included in the study. |
Table II.
CK level of all female carriers
included in the study.
| Group | No. (n=78) | CK level (U/l) |
|---|
| Group 1 | 4 | 6162, 5306, 4713,
2158 |
| Group 2 | 47 | 7012, 4142, 2724,
2078, 2065, 1647, 1516, 1454, 1419, 1390, 1233, 1183, 1169, 1095,
967, 944, 920, 855, 794, 759, 758, 655, 633, 582, 550, 525, 515,
500, 494, 484, 460, 434, 431, 415, 372, 355, 335, 325, 322, 322,
318, 288, 242, 197, 186, 183, 180 |
| Group 3 | 27 | 174, 167, 157, 154,
147, 135, 128, 128, 128, 118, 110, 106, 102, 97, 96, 90, 84, 83,
81, 76, 73, 72, 71, 69, 66, 61, 24 |
Correlation of CK with AST and ALT
levels
Among the 4 symptomatic and 74 asymptomatic female
carriers, 61 cases exhibited normal ALT and AST levels, whereas 17
cases had elevated ALT and AST levels. The mean ALT level was 33.56
U/l, with a minimum of 9 U/l and a maximum of 154 U/l. Similarly,
the mean AST level was 38.20 U/l, with a minimum of 14 U/l and a
maximum of 171 U/l. Further analysis indicated that the variation
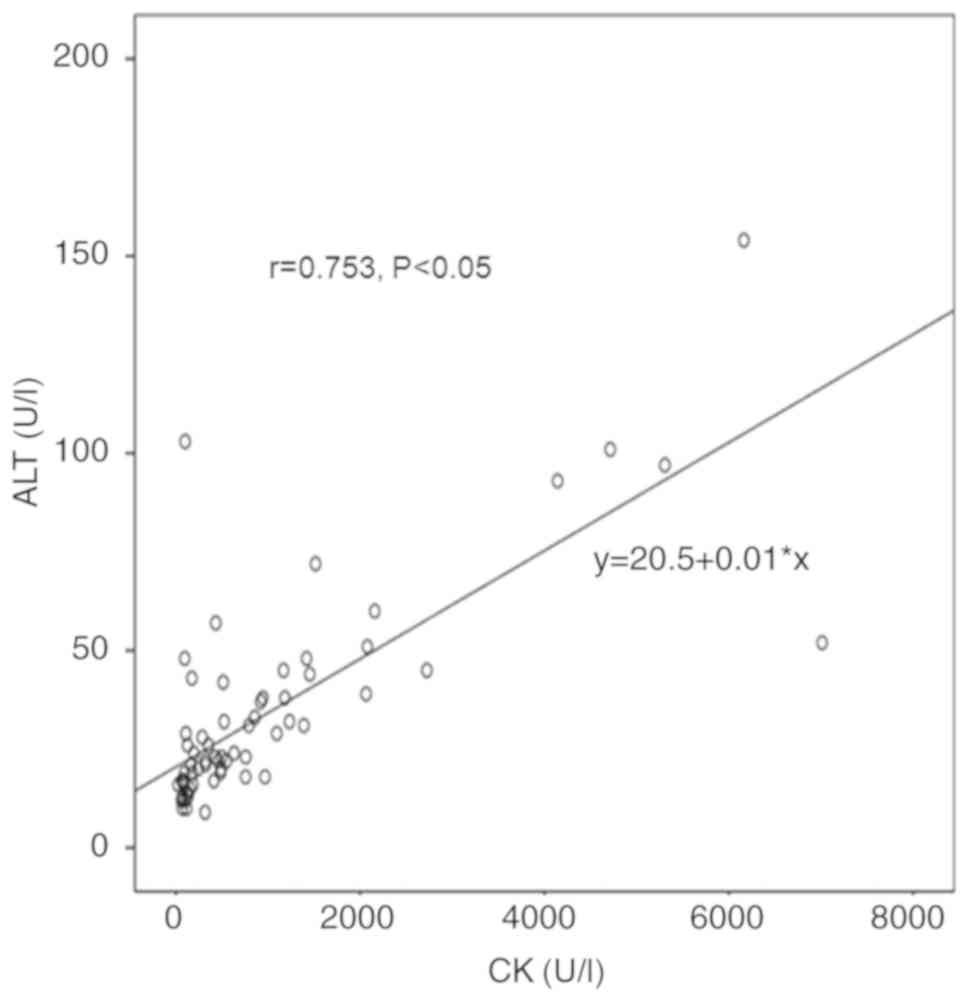
in ALT and CK levels exhibited a positive correlation (r=0.753,
P<0.05; Fig. 4), while a
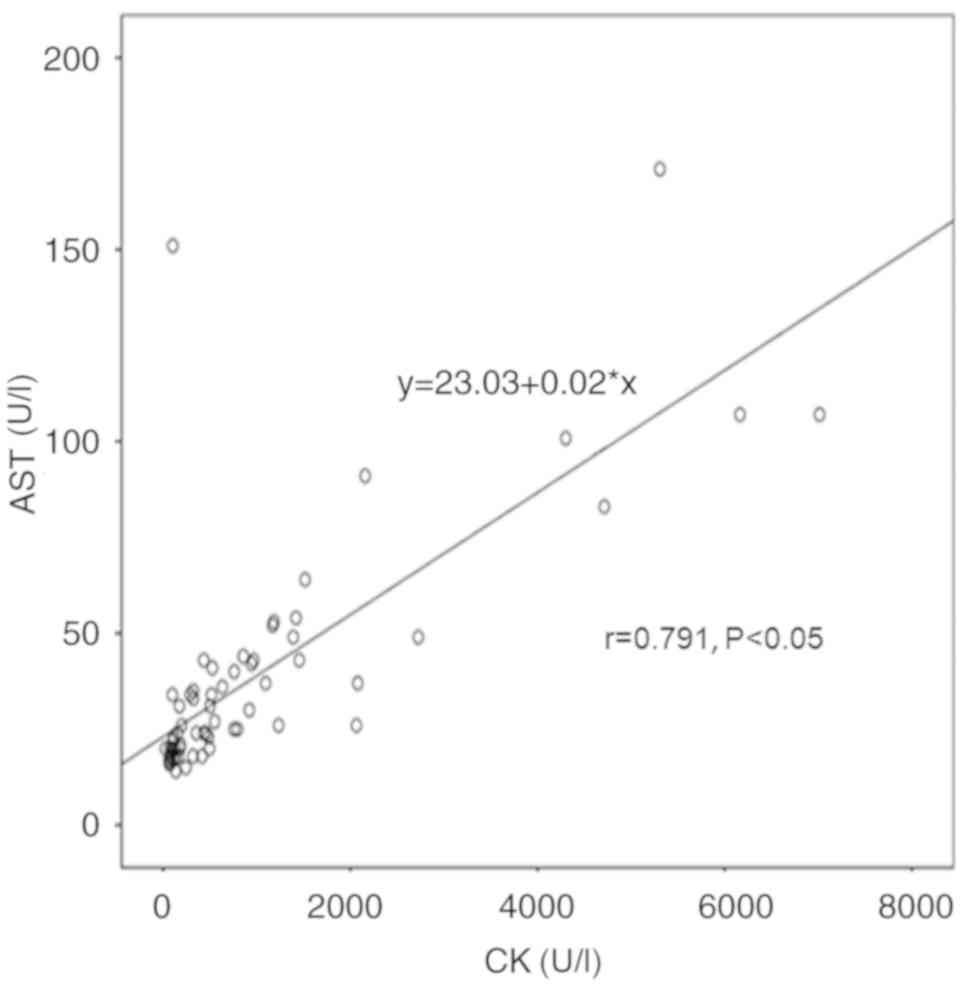
positive correlation was also observed between AST and CK levels
(r=0.791, P<0.05; Fig. 5). AST
exhibited a stronger correlation with CK levels as compared with
that of ALT and CK levels.
Molecular characteristics of female
carriers
In the present study, 4 MCs were detected by MLPA
analysis of the dystrophin gene exons. All of the MCs exhibited
duplication mutations, and duplications of exons 9–10, 69–70, 49–52
and 45–55 were identified. The other 74 asymptomatic carriers were
analyzed by MLPA combined with Sanger sequencing. Among these, 31
(41.89%) cases carried deletion mutations, 33 (44.59%) cases
exhibited point mutations, and only 10 cases (13.51%) exhibited
duplication mutations. In addition, 2 of the asymptomatic females
were carriers of suspected germline mosaicism without mutations in
dystrophin gene, although their daughters had the same mutations as
the probands. Overall, 34 different mutations were characterized in
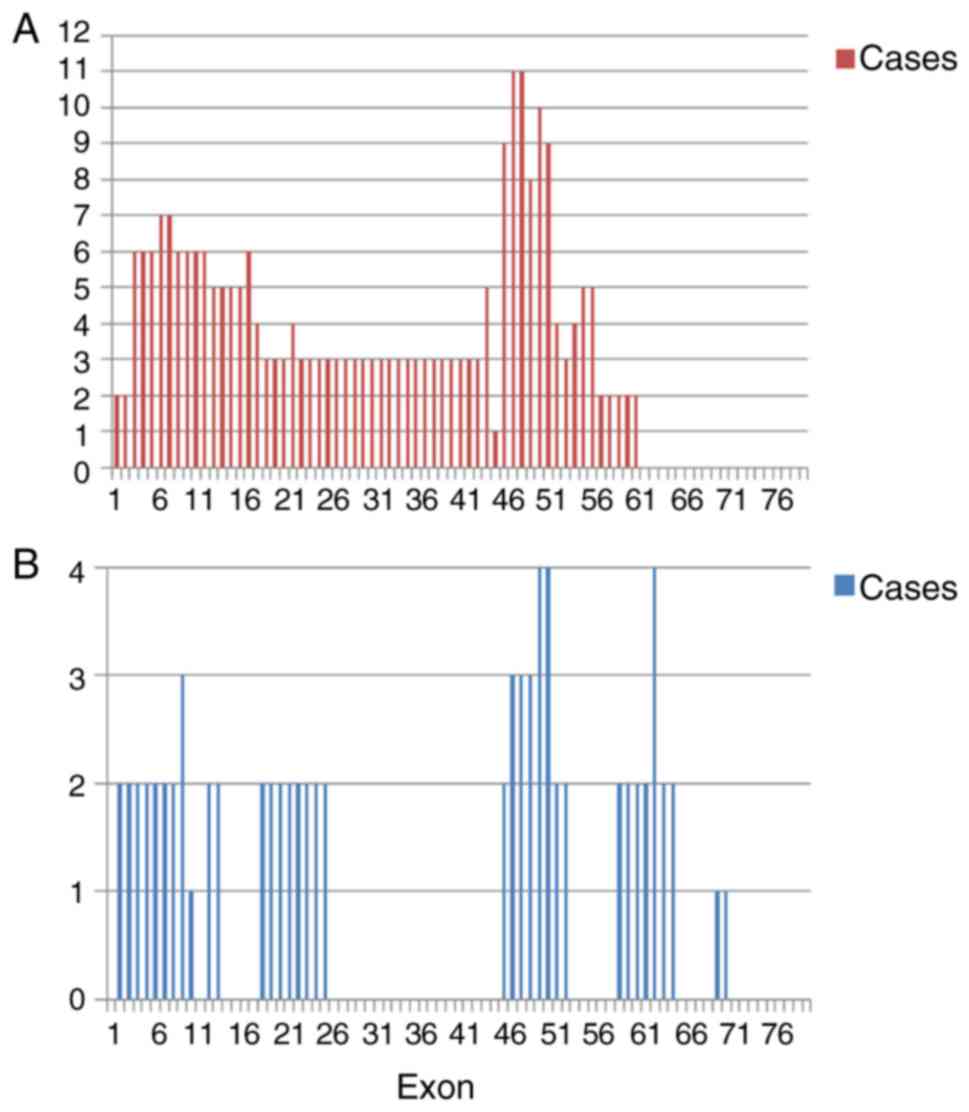
the present study. The deletion breakpoints were mainly clustered
at exons 45–55, followed by exons 3–16 (Fig. 6A), whereas duplication breakpoints
were clustered at the 3′ end of the dystrophin gene (Fig. 6B). The point mutation detection
rate and distribution of mutations according to type, including 22
nonsense, 4 frame shift and 7 splice site mutations, are shown in
Table III.
| Table III.Point mutations of female carriers,
analyzed by Sanger sequencing. |
Table III.
Point mutations of female carriers,
analyzed by Sanger sequencing.
| Patient | Base change | Effect | Exon ID | Mutation type |
|---|
| 1 | c.2605C>T | P.Gln869Ter | Exon 20 | Nonsense |
| 2 | c.6318G>A | p.Trp2106Ter | Exon 44 | Nonsense |
| 3 |
c.1860_1861delGT |
p.Leu620LeufsTer14 | Exon 16 | Frameshift |
| 4 | c.8027+1G>A | – | Intron 54 | Splicing |
| 5 | c.8087 delT |
p.Leu2696ArgfsTer30 | Exon 55 | Frameshift |
| 6 | c.6292C>T | p.Arg2098Ter | Exon 44 | Nonsense |
| 7 | c.5488A>T | p.Arg1830Ter | Exon 39 | Nonsense |
| 8 | c.1332-9A>G | – | Intron 11 | Splicing |
| 9 | c.7657C>T | p.Arg2553Ter | Exon 52 | Nonsense |
| 10 | c.133C>T | p.Gln45Ter | Exon 3 | Nonsense |
| 11 | c.8010G>A | p.Trp2670Ter | Exon 54 | Nonsense |
| 12 |
c.10498_10499delAG | p.Ser3500Ter | Exon 74 | Frameshift |
| 13 | c.10108C>T | p.Arg3370Ter | Exon 70 | Nonsense |
| 14 | c.3151C>T | p.Arg1051Ter | Exon 23 | Nonsense |
| 15 |
c.10223+1G>A | – | Intron 69 | Splicing |
| 16 | c.5488A>T | pArg1830Ter | Exon 39 | Nonsense |
| 17 | c.3982C>T | p.Gln1328Ter | Exon 29 | Nonsense |
| 18 | c.490A>T | p.K164X | Exon 6 | Nonsense |
| 19 | c.10108C>T | p.Arg3370Ter | Exon 70 | Nonsense |
| 20 | c.3982C>T | p.Gln1328Ter | Exon 29 | Nonsense |
| 21 | c.3721G>T | p.Glu1241Ter | Exon 27 | Nonsense |
| 22 | c.10171C>T | p.Arg3391Ter | Exon 70 | Nonsense |
| 23 | c.10171C>T | p.Arg3391Ter | Exon 70 | Nonsense |
| 24 | c.1332-8A>G | – | Intron 11 | Splicing |
| 25 | c.3603+1G>A | – | Intron 26 | Splicing |
| 26 | c.1800-1G>A | – | Intron 14 | Splicing |
| 27 | c.462+1G>T | – | Intron 6 | Splicing |
| 28 | c.133C>T | p.Gln45Ter | Exon 3 | Nonsense |
| 29 | c.691T>A | p.Tyr231Asn | Exon 8 | Frameshift |
| 30 | c.5488A>T | pArg1830Ter | Exon 39 | Nonsense |
| 31 | c.5488A>T | pArg1830Ter | Exon 39 | Nonsense |
| 32 | c.10171C>T | p.Arg3391Ter | Exon 70 | Nonsense |
| 33 | c.3151C>T | p.Arg1051Ter | Exon 23 | Nonsense |
In addition, it was observed that 31 (36%) mothers
were negative for dystrophin gene analysis, and the associated
probands had possible de novo mutations. A total of 6 female
relatives exhibited negative genetic results by MLPA and NGS,
however, their CK levels were abnormally high (Table IV). These cases were first degree
relatives of the probands, and a muscle disease diagnosis could not
be made by muscle panel analysis. Therefore, the possibility that
these subjects were DMD carriers cannot be excluded, particularly
given the possibility of somatic mosaicism. Certainly, other
factors that would cause high CK levels should be considered.
| Table IV.CK level of female relatives with
high CK level and negative genetic results. |
Table IV.
CK level of female relatives with
high CK level and negative genetic results.
| Female
relative | CK (U/l) | AST (U/l) | ALT (U/l) | Mutation of
proband |
|---|
| 1 | 297 | 24 | 18 | DEL EX46-59 |
| 2 | 194 | 14 | 19 | DEL EX46-48 |
| 3 | 632 | 19 | 18 | DEL EX45-55 |
| 4 | 257 | 20 | 16 | DEL EX50-54 |
| 5 | 191 | 13 | 19 | DEL EX04-11 |
| 6 | 311 | 38 | 43 | DEL EX51 |
Muscle biopsy results
Among the 4 symptomatic carriers, 2 patients were
subjected to muscle biopsy (Fig. 7A
and B). The proliferation of connective tissue in the muscle
fibers was low, and individual interstitial small vessels cut
through only a few granulocytes and lymphocytes. ‘Muscle fiber
necrosis was observed and regeneration was not evident, except when
combined with clinical myositis. The immune positivity of spectrin
in all biopsies indicated well-preserved sarcolemmal integrity,
ensuring that false negative results for other sarcolemmal
membrane-associated proteins could be ruled out. Furthermore,
immunohistochemical examination with monoclonal antibodies against
dystrophin indicated a mosaic reduction of the protein expression
in these MCs. The control biopsy presented a normal expression of
dystrophin, with a consistent and uniform brown muscle fiber
membrane. By contrast, in male DMD patients, almost no dystrophin
expression was observed in the muscle fiber membrane (Fig. 7C-E).
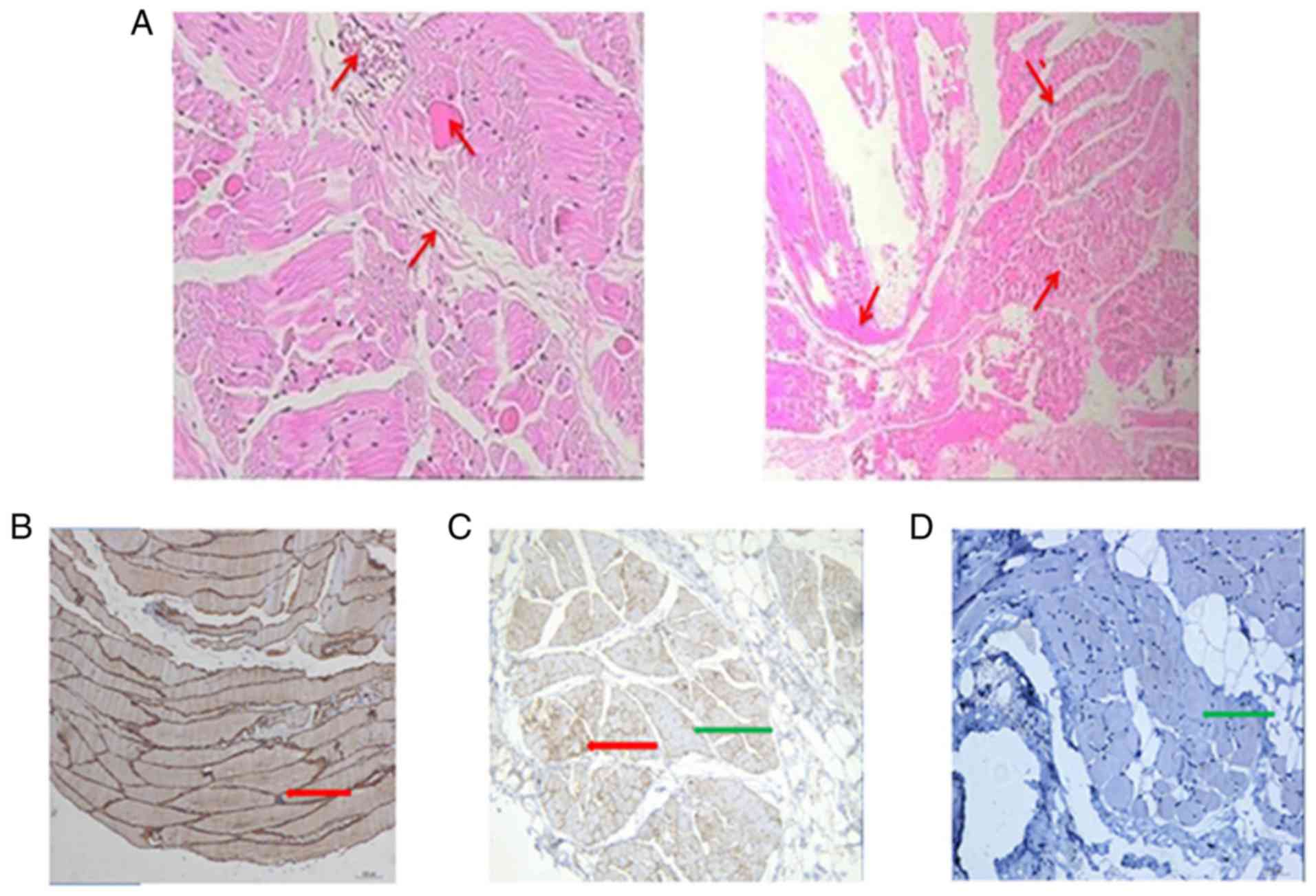 | Figure 7.Sections with HE staining and
immunolabeling with dystrophin antibodies of patient 1
(magnification, ×100). (A) HE staining of muscle biopsies of
patient 1, exhibiting a small amount of connective tissue
proliferation in the muscle fibers, few stromal small vessels with
a small amount of granulocytes and lymphocytes infiltration.
Dystrophin expression in the biopsies obtained from a (B) control,
(C) female MC (patient 1) and (D) male DMD patient. The control
section presented a normal expression of dystrophin, with a
consistent and uniform brown muscle fiber membrane. The MC (patient
1) exhibited random presence of normal and dystrophin-deficient
fibers, indicating a mosaic expression of dystrophin. By contrast,
the male DMD patient exhibited almost no expression of dystrophin
in the muscle fiber membrane. The red arrow indicates expression of
dystrophin in brown muscle fiber membrane, and the green arrow
indicates absence of dystrophin expression. HE, hematoxylin-eosin;
MC, manifesting carrier; DMD, Duchenne muscular dystrophy. |
Discussion
Dystrophinopathy is the most common muscular
dystrophy and involves X-linked recessive inheritance, while MCs
are rare. In the present study, 4 MC females (4/78 participants;
4.49%) were reported, who presented with muscle weakness and
elevated CK levels. Duplication mutations in dystrophin gene were
detected in all of the MCs. It has previously been reported that
2.5–10% carriers experience symptoms ranging from mild muscle
weakness to rapidly progressive DMD-like muscular dystrophy
(5), which is consistent with the
results of the present study. Several disease-causing mechanisms
have been implicated in DMD/BMD MCs; however, the most frequently
reported mechanism to provoke symptoms in DMD/BMD carriers is
skewed X-inactivation (7). In
X-linked dominant diseases, X inactivation can influence the
severity of the phenotype or the survival of females with
heterozygous lethal mutations. Our next step will be relevant to
the X inactivation analysis of these carriers to clarify its
pathogenesis.
The phenotypes of MCs of DMD/BMD are similar to LGMD
and are, therefore, likely to be misdiagnosed with LGMD. MCs with
motor weaknesses should be distinguished from other myodystrophies.
As many as 17% of dystrophinopathies are misdiagnosed as LGMD
(8). In the present study, 3 cases
among the 78 cases exhibited progressive motor weaknesses similar
to DMD and elevated CK levels. A subsequent muscle disease panel
analysis revealed their diagnosis as LGMD, which was confirmed by
muscle biopsy and immunostaining. In addition to weakness symptoms,
an MC with motor developmental delay was detected, but without any
other intellectual or cognitive disorders or mental illnesses.
These findings are in accordance with those reported by Seeman
et al (9) in 2010. More
recently, the incidence of mental retardation in MCs was found to
be as high as 7% (10). However,
all 4 female symptomatic carriers in the present study did not
exhibit evidence of mental retardation.
As CK is a component of the muscle fiber cytosol,
its serum activity is considered to be positively correlated with
progressing muscle damage. An elevated CK level is one of the
characteristic features of DMD/BMD, particularly when screening
neonatal carriers. To the best of our knowledge, the present study
is the first to use a ROC curve to analyze the diagnostic
sensitivity of CK level in DMD/BMD, and the results suggested that
the CK level has high specificity and sensitivity for the diagnosis
of female carriers. For female relatives with negative genetic
tests, particularly those with CK values up to 1.5–3 times the
normal upper limit, muscle biopsy and immunohistochemical analyses
are recommended to evaluate dystrophin protein expression to
confirm the status of probable carriers. As previous studies have
suggested, chronic persistent CK elevation indicates the
possibility of neuromuscular disease (11). Wang et al (12) reported that among a group of 201
female relatives, 97 of the 98 first-degree relatives with high
levels of CK were DMD or BMD carriers, while 31 of the remaining
103 subjects exhibiting normal CK levels were diagnosed as
carriers. Therefore, the possibility that female relatives with
normal levels of CK could be carriers cannot be excluded. In the
present study, it was also observed that CK levels were positively
correlated with the levels of ALT and AST. Elevated ALT/AST may be
the presenting sign of muscle disease in children, which provides
an opportunity for early diagnosis. For patients with unexplained
long-lasting hypertransaminasemia, serum CK activity should be
measured and muscle biopsy should be conducted at an early stage
for correct diagnosis of muscular dystrophy. The sensitivity of ALT
and AST was observed to be 21.79% in the present study, which is
significantly lower compared with that for the CK level. Therefore,
the CK level is a better predictor of DMD/BMD carriers, and female
relatives with elevated ALT/AST should also be screened for serum
CK activity to identify potential carriers. While a total of 6
female relatives in the present study had a high CK level, they did
not present any mutations in the dystrophin gene. Since
these patients did not agree to undergo muscle biopsy, they cannot
be excluded as possible carriers.
The limitations of MLPA and NGS analyses can lead to
false negatives, while germline mosaicism carriers would also show
a negative result in genetic tests. Luce et al (13) reported three recombination events
and suggested that germline mosaicism had occurred in these
families. In addition, high CK levels may be due to other
non-neuromuscular factors, such as physical activity or muscle
trauma (14), and further study
would help to exclude the possibility of myopathy. Currently,
muscle biopsy immunostaining has become important for the diagnosis
of myopathy. In the present study, the muscle biopsy immunostaining
results of two symptomatic carriers exhibited mosaic distribution.
Approximately 20% of carriers had abnormal dystrophin
immunostaining on muscle biopsy, with a mosaic pattern of
dystrophin-positive and dystrophin-negative fibers present
(15). When the gene mutation
analysis is negative, muscle biopsy with immunohistochemical
staining would be useful to assist in the further diagnosis of
carriers.
In the present study, 74 asymptomatic carriers were
detected by MLPA combined with Sanger sequencing. Among these,
41.89% of cases involved deletion mutations, 44.59% of cases
exhibited point mutations in exons and 13.51% exhibited duplication
mutations. These results are similar to those of a previous study
(16). Additionally, all of 4 MCs
carried frameshift and duplication mutations. By contrast, point
mutations and deletions were more common in female asymptomatic
carriers, as previously reported in the literature (17). More frameshift and duplication
mutations were observed in MCs in the present study, which may be
due to the small sample size. Therefore, larger samples should be
used in future studies. Large segmental deletions and duplications
in female carriers of dystrophin gene mutations involved the
majority of the exons. Deletion breakpoints were mainly clustered
at exons 45–55, followed by exons 3–16, which was similar to the
results previously reported in the literature (4). However, in the present study,
duplications were concentrated in the 3′ end of the DMD gene, in
contrast with previously reported DMD gene mutation hot spots at
the 5′ end (17). An earlier study
analyzed 92 suspected DMD/BMD male patients, and reported that the
deletion and duplication hot spots in the dystrophin gene were the
same as those of female carriers in the present study (18). The discrepancy between the results
of the present and previous studies may be due to the small sample
we collected.
According to the literature, approximately
two-thirds of the genetic mutations in male DMD/BMD patients were
inherited from their carrier mothers, while more than one-third
were de novo mutations (19). In the present study, 31 (36%)
mothers did not exhibit a mutation in the dystrophin gene, as
determinedby MLPA and Sanger sequencing analysis. This indicated
that these probands may present de novo mutations.
Furthermore, 2 cases of germline mutation female carriers who were
the mothers of the 2 probands were identified They did not exhibit
a mutation in the dystrophin gene by MLPA and NGS analyses, and had
a normal CK level. However, their daughters presented the same
mutation as the probands, although this mutation was absent in the
fathers of these cases. This evidence suggests that these mothers
are highly likely to have germline mosaicism. Thus, mothers with a
DMD/BMD child should be aware of the possibility that they are
DMD/BMD carriers, particularly those who show an elevated CK level,
even when no mutation in the dystrophin gene is detected by genetic
analysis. A number of researchers recommend that mothers who have a
DMD child should obtain prenatal diagnosis again when pregnant. In
order to prevent DMD/BMD, attention must be paid to
dystrophinopathy carrier screening, genetic counseling and prenatal
diagnosis.
In conclusion, measurement of the CK level is a
non-invasive, easy and excellent screening method for DMD/BMD
carriers, and this level was observed to be strongly correlated
with ALT and AST. The present study reported that MLPA for exons of
the dystrophin gene, along with a muscle disease panel analysis,
was effective for the diagnosis of MCs, while MLPA combined with
Sanger sequencing determined carrier status. However, a number of
carriers remained that could not be confirmed by this strategy.
DMD/BMD female relatives with negative genetic testing results,
particularly those with high CK levels, are recommended to undergo
prenatal diagnosis. Furthermore, female relatives with negative
genetic tests and persistent high CK levels should be recommended
for muscle biopsy and immunohistochemical analysis to evaluate
dystrophin protein expression in order to confirm the status of
probable carriers.
Acknowledgements
Not applicable.
Funding
This study was supported by the Chinese Natural
Science Foundation (grant no. 81760215), the First Affiliated
Hospital of Guangxi Medical University starting fund for study
abroad returnees (grant no. 2010001), and the Natural Science
Foundation of Guangxi Province (grant no. 03201216025D).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DL conducted the study and analyzed the genetic test
results. JZho and YX summarized the clinical information, analyzed
the genetic test results and drafted the manuscript. VB helped to
summarize the clinical information and analyze the genetic test
results. GC analyzed the pathological biopsy results. YD performed
the pathological biopsy. HL and JZha obtained the clinical
information. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All procedures performed in this study involving
human participants were in accordance with the ethical standards of
the institutional and national research committee. Informed consent
was obtained from all participants included in the study.
Patient consent for publication
Consent for publication was obtained from all
participants included in the study.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ALT
|
alanine transaminase
|
|
AST
|
aspartate transaminase
|
|
BMD
|
Becker muscular dystrophy
|
|
CK
|
creatine kinase
|
|
DMD
|
Duchenne muscular dystrophy
|
|
InDels
|
insertions or deletions
|
|
LGMD
|
limb-girdle muscular dystrophy
|
|
NGS
|
next-generation sequencing
|
|
MCs
|
manifesting carriers
|
|
MLPA
|
multiplex ligation-dependent probe
amplification
|
|
ROC
|
receiver operating characteristic
|
|
SNP
|
single-nucleotide polymorphism
|
References
|
1
|
Bushby K, Finkel R, Birnkrant DJ, Case LE,
Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, et
al: Diagnosis and management of Duchenne muscular dystrophy, part
1: Diagnosis, and pharmacological and psychosocial management.
Lancet Neurol. 9:77–93. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ryder S, Leadley RM, Armstrong N, Westwood
M, de Kock S, Butt T, Jain M and Kleijnen J: The burden,
epidemiology, costs and treatment for Duchenne muscular dystrophy:
An evidence review. Orphanet J Rare Dis. 12:792017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Magri F, Govoni A, D'Angelo MG, Del Bo R,
Ghezzi S, Sandra G, Turconi AC, Sciacco M, Ciscato P, Bordoni A, et
al: Genotype and phenotype characterization in a large
dystrophinopathic cohort with extended follow-up. J Neurol.
258:1610–1623. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lopez-Hernandez LB, Gomez-Diaz B,
Luna-Angulo AB, Anaya-Segura M, Bunyan DJ, Zuniga-Guzman C,
Escobar-Cedillo RE, Roque-Ramirez B, Ruano-Calderon LA,
Rangel-Villalobos H, et al: Comparison of mutation profiles in the
Duchenne muscular dystrophy gene among populations: Implications
for potential molecular therapies. Int J Mol Sci. 16:5334–5346.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Taylor PJ, Maroulis S, Mullan GL, Pedersen
RL, Baumli A, Elakis G, Piras S, Walsh C, Prosper-Gutierrez B, De
La Puente-Alonso F, et al: Measurement of the clinical utility of a
combined mutation detection protocol in carriers of Duchenne and
Becker muscular dystrophy. J Med Genet. 44:368–372. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang P, Du CW, Kwan M, Liang SX and Zhang
GJ: The impact of p53 in predicting clinical outcome of breast
cancer patients with visceral metastasis. Sci Rep. 3:22462013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Juan-Mateu J, Rodriguez MJ, Nascimento A,
Jiménez-Mallebrera C, González-Quereda L, Rivas E, Paradas C,
Madruga M, Sanchez-Ayaso P, Jou C, et al: Prognostic value of
X-chromosome inactivation in symptomatic female carriers of
dystrophinopathy. Orphanet J Rare Dis. 7:822012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arikawa E, Hoffman EP, Kaido M, Nonaka I,
Sugita H and Arahata K: The frequency of patients with dystrophin
abnormalities in a limb-girdle patient population. Neurology.
41:1491–1496. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Seemann N, Campbell C, Hammond R and
Prasad C: 9 year old girl with progressive weakness. Brain Pathol.
20:255–256. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mercier S, Toutain A, Toussaint A, Raynaud
M, de Barace C, Marcorelles P, Pasquier L, Blayau M, Espil C,
Parent P, et al: Genetic and clinical specificity of 26 symptomatic
carriers for dystrophinopathies at pediatric age. Eur J Hum Genet.
21:855–863. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dabby R, Sadeh M, Herman O, Berger E,
Watemberg N, Hayek S, Jossiphov J and Nevo Y: Asymptomatic or
minimally symptomatic hyperCKemia: Histopathologic correlates. Isr
Med Assoc J. 8:110–113. 2006.PubMed/NCBI
|
|
12
|
Wang Q, Yang X, Yan Y, Song N, Lin C and
Jin C: Duchenne or Becker muscular dystrophy: A clinical, genetic
and immunohistochemical study in China. Neurol India. 59:797–802.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luce LN, Ottaviani D, Ferrer M, Szijan I,
Cotignola J and Giliberto F: Molecular diagnosis of
dystrophinopathies using a multi-technique analysis algorithm.
Muscle Nerve. 49:249–256. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Voermans NC, de Visser M, Wokke JH and
Brusse E: Increased CK activity in serum without symptoms: Further
investigations often unnecessary. Ned Tijdschr Geneeskd.
157:A63152013.(In Dutch). PubMed/NCBI
|
|
15
|
Hoogerwaard EM, Ginjaar IB, Bakker E and
de Visser M: Dystrophin analysis in carriers of Duchenne and Becker
muscular dystrophy. Neurology. 65:1984–1986. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Soltanzadeh P, Friez MJ, Dunn D, von
Niederhausern A, Gurvich OL, Swoboda KJ, Sampson JB, Pestronk A,
Connolly AM, Florence JM, et al: Clinical and genetic
characterization of manifesting carriers of DMD mutations.
Neuromuscul Disord. 20:499–504. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee SH, Lee JH, Lee KA and Choi YC:
Clinical and genetic characterization of female dystrophinopathy. J
Clin Neurol. 11:248–251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhong J, Xu T, Chen G, Liao H, Zhang J and
Lan D: Genetic analysis of the dystrophin gene in children with
Duchenne and Becker muscular dystrophies. Muscle Nerve. 56:117–121.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mukherjee M, Chaturvedi LS, Srivastava S,
Mittal RD and Mittal B: De novo mutations in sporadic deletional
Duchenne muscular dystrophy (DMD) cases. Exp Mol Med. 35:113–117.
2003. View Article : Google Scholar : PubMed/NCBI
|