Introduction
Epstein-Barr virus (EBV) is a member of the
γ-herpesvirus family. EBV-associated lymphoid malignancies include
a subset of Burkitt lymphoma (BL), acquired immune deficiency
syndrome lymphoma, Hodgkin’s lymphoma, post-transplant lymphoma,
age-associated B-cell lymphoma, and peripheral T- and natural
killer-cell lymphomas (1–3). The EBV life cycle includes distinct
latent and lytic genetic programs (4–6).
Several therapeutic strategies requiring the activation of EBV
lytic genes for tumor cell killing have been described (7,8). The
switch from latent to lytic EBV infection is mediated by the
expression of two EBV immediate-early (IE) viral proteins, BZLF1
and BRLF1. Lytic EBV replication damages the cancer cells and
triggers host immune responses against EBV and the infected cells
(9). During lytic infection, EBV
encodes viral kinases that phosphorylate the antiviral nucleoside
analogue ganciclovir (GCV) to produce its active cytotoxic activity
(10). Taken together, these
findings suggest that EBV-targeted cancer therapy warrants
investigation.
Nuclear factor-κB (NF-κB) is a significant
transcriptional factor involved in the regulation of cell
apoptosis, cell cycle progression and carcinogenic transformation.
Constitutive NF-κB activity has been observed in lymphoma (11). Recent studies have demonstrated
that the transforming EBV-encoded latent membrane protein 1 (LMP1)
induces constitutive NF-κB activity (12) and that elevated NF-κB levels
promote the survival and proliferation of infected cells and
inhibit lytic gene promoter activation, lytic protein synthesis and
lytic replication (13–16). These findings suggest that
inhibiting NF-κB is an effective and novel method for treating
EBV-positive malignancies.
Parthenolide (PN) is a sesquiterpene lactone
(17) found in medicinal plants,
particularly in feverfew (Tanacetum parthenium). The
nucleophilic nature of its methylene-γ-lactone ring and epoxide
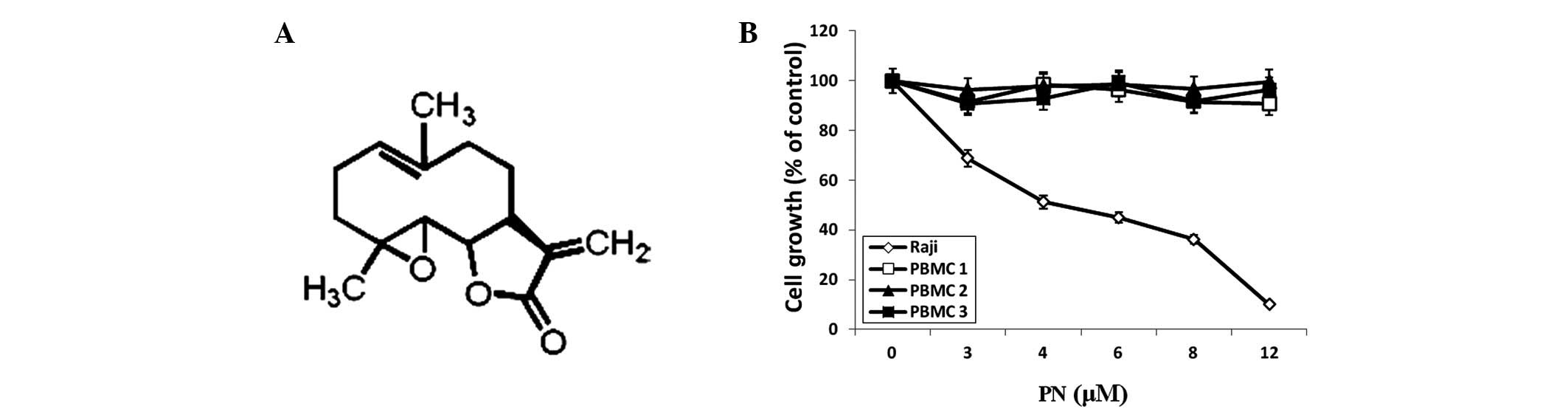
group enables rapid interactions with biological sites (Fig. 1A). PN is a herbal medicine that has
been used to treat migraines and rheumatoid arthritis for
centuries. Recently, PN has been identified to have several other
properties, including antitumor activity, inhibition of DNA
synthesis and inhibition of cell proliferation, in various cancer
cell lines (18). In addition, PN
sensitizes cancer cells to other antitumor agents (19,20).
Accumulating evidence has shown that PN is capable of inhibiting
the activity of the NF-κB subunit RelA/p65 by inhibiting the IκB
kinase-mediated phosphorylation of IκB (21), suggesting that PN is a novel
therapeutic agent for treating EBV-positive lymphoma.
In this study, we analyzed the effect of PN on the
survival of Raji EBV-positive lymphoma cells. We confirmed that PN
inhibited RelA/p65 activity and showed that it induced EBV lytic
replication in Raji cells, resulting in EBV-positive cell death
in vitro. When PN was used in combination with GCV, the
anticancer potency of PN was enhanced. These findings indicate that
PN may be a novel agent for targeting EBV-associated Burkitt
lymphoma.
Materials and methods
Cell culture
The EBV-positive BL cell line, Raji, was obtained
from ATCC (Manassas, VA, USA). The cells were cultured in RPMI-1640
supplemented with 10% fetal bovine serum, 100 U/ml penicillin and
100 μg/ml streptomycin at 37°C and 5% CO2. Peripheral
blood mononuclear cells (PBMCs) were purified from the blood
donors’ buffy coats by Ficoll gradient centrifugation. This study
was conducted in accordance with the Helsinki protocol and approved
by the Xiamen University Institutional Review Board.
Reagents and antibodies
PN was purchased from Sigma-Aldrich (St. Louis, MO,
USA). PN was dissolved in dimethyl sulfoxide (DMSO; Sigma Chemical
Co., St. Louis, MO, USA) as a stock solution of 1 μmol/ml and
diluted in the culture medium immediately prior to use. The final
concentration of DMSO in all experiments was <0.01%. GCV was
purchased from North China Pharmaceutical Group Corporation
(Shijiazhuang, Hebei, China) and
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT)
was purchased from Sigma. An annexin V fluorescein conjugate
(FITC) and propidium iodide (PI) apoptosis detection kit
was purchased from Invitrogen (Grand Island, NY, USA). An NF-κB
(RelA/p65) transcription factor assay kit was purchased from Cayman
Chemical (Ann Arbor, MI, USA). Antibodies against RelA/p65
(1:1000), poly (ADP-ribose) polymerase (PARP; 1:1000), caspase-8
(1:1000), caspase-9 (1:1000), Oct-1 (1:1000) and horseradish
peroxidase-conjugated anti-rabbit immunoglobulin (1:3000) were
purchased from Cell Signaling Technology (Beverly, MA, USA).
Anti-β-actin antibody (1:3000) was obtained from Epitomics
(Burlingame, CA, USA).
Cell proliferation assay
Raji cells and PBMCs in 180 μl RPMI-1640 were
inoculated onto 96-well plates. Four parallel rows of wells were
set up for each group. The MTT assay was performed 48 h following
treatment with PN at different concentrations in the presence or
absence of GCV. The absorbance rate [optical density (OD)] of each
well was measured at 570 nm using an enzyme-linked immunosorbent
detector (DG3022A). Growth inhibition was determined as a
percentage of the control.
Apoptosis detection assays and cell cycle
analysis
Raji cells were treated with 0, 4 or 6 μmol/l PN for
48 h. Raji cells were washed and stained with annexin
VFITC and PI according to the manufacturer’s
instructions. Apoptosis was quantified using a FACSort flow
cytometer and CellQuest (BD Biosciences, Mountain View, CA, USA).
Apoptosis was also detected via 4′,6-diamidino-2-phenylindole
(DAPI) staining. Briefly, Raji cells were washed with
phosphate-buffered saline and stained with 1 mg/ml DAPI for 30 min
at 37°C. Slides were then washed with phosphate-buffered saline,
air dried and covered with coverslips for analysis via fluorescence
microscopy using a Leica DM2500 microscope (Buffalo Grove, IL,
USA). For cell cycle analysis, the Raji cells were fixed with cold
methanol overnight and then treated with PI (2 μg/ml) and RNase
prior to flow cytometry.
Caspase-3 assays
Caspase-3 activity was measured in the control and
PN-treated Raji cells. Caspase-3 activity assays were performed
using a colorimetric substrate according to the manufacturer’s
instructions (EMD Chemicals, Gibbstown, NJ, USA). Briefly, Raji
cells were lysed, and 50 μg of the resulting cell lysates were
added to an assay buffer to a total volume of 90 μl and incubated
at 37°C for 10 min. A colorimetric substrate for caspase-3 (final
concentration, 200 μmol/l) was then added to the mixture.
Absorbance values at 405 nm were recorded for each sample following
incubation at 37°C for 2 h.
NF-κB (RelA/p65) activity
Raji cells were incubated with 4 or 6 μmol/l PN for
6, 12 or 24 h. The cells were collected and nuclear proteins were
extracted. NF-κB (RelA/p65) activity was detected using a NF-κB
(RelA/p65) transcription factor assay kit. The OD of each well was
measured at 450 nm using a Model 680 spectrophotometer (Bio-Rad,
Philadelphia, PA, USA).
Western blot analysis
Raji cells were incubated with 4 μmol/l PN for 24 h.
The nuclear and cytoplasmic extracts were then purified to detect
RelA/p65. Preparation of nuclear and cytoplasmic fractions was
performed using the NE-PER extraction reagent kit (Pierce
Biotechnology, Rockford, IL, USA) according to the manufacturer’s
instructions. Briefly, cells were suspended in cytoplasmic
extraction reagent I containing 1% protease inhibitor cocktail
(Sigma) and 1% phosphatase inhibitor cocktail (Sigma) and then
incubated on ice for 10 min. Cytoplasmic extraction reagent II (11
μl) was added to the mixture, vortexed and incubated on ice for 1
min. The sample was centrifuged at maximum speed (13,000 × g) for
10 min at 4°C. The supernatant (cytoplasmic fraction) was
transferred to a new tube and stored at −80°C. The pellet was
resuspended in 100 μl of ice-cold nuclear extraction reagent and
vortexed for 15 sec every 10 min for a total of 40 min. The sample
was then centrifuged (13,000 × g) for 10 min at 4°C. The
supernatant (nuclear fraction) was transferred to a new tube and
stored at −80°C.
Raji cells were treated with 0, 4 or 6 μmol/l PN for
48 h. Whole-cell extracts were purified to detect PARP, caspase-8
and caspase-9. To prepare the whole-cell lysates, the cells were
lysed in a lysis buffer (Cell Signaling). Cell lysates were kept on
ice for 30 min and centrifuged at 13,000 × g for 10 min at 4°C.
The protein concentration was determined by Bradford
assay (Bio-Rad). Sample proteins (50 μg) were solubilized in 1%
sodium dodecyl sulfate (SDS) sample buffer and subjected to
SDS-polyacrylamide gel electrophoresis (PAGE) on a 4–15% gel
(Bio-Rad). Proteins were transferred from the gel onto a
polyvinylidene difluoride membrane and immunoblotted with various
specific primary antibodies and appropriate horseradish
peroxidase-conjugated secondary antibodies. Proteins were
visualized using an enhanced chemiluminescence western blotting
detection system (Amersham, Sweden). Densitometric digital analysis
of protein bands was performed to quantify each protein band using
Quantity One 1-D analysis software version 4.1.0 (Bio-Rad). Each
protein was normalized by the intensity of the β-actin or Oct-1
housekeeping gene in each sample.
Detection of BZLF1 and BRLF1 mRNA
expression in Raji cells using reverse transcriptase polymerase
chain reaction (RT-PCR)
Raji cells were collected at 6, 12 and 24 h after
incubation with PN 4 μmol/l. BZLF1 and BRLF1 mRNA
expression was detected using RT-PCR. The sequences of primers used
were GGGACAAGCAAACACCAC (sense) and TTTACACCTGACCCATACC
(anti-sense) for BZLF1; CCATACAGGACACAACACCTCA (sense) and
ACTCCC GGCTGTAAATTCCT (anti-sense) for BRLF1; GTGGGG
CGCCCCAGGCACCA (sense) and CTCCTTAATGTC ACGCACGATTTC (anti-sense)
for β-actin. The PCR conditions for BZLF1 were
denaturation at 95°C for 2 min; annealing for 30 cycles each at
94°C for 30 sec, 58°C for 30 sec, and 72°C for 60 sec; and an
extension at 72°C for 10 min. The PCR conditions for BRLF1 were
denaturation at 95°C for 2 min; annealing for 30 cycles each at
94°C for 30 sec, 58°C for 30 sec, and 72°C for 90 sec; and an
extension at 72°C for 10 min. The PCR products were subjected to
PAGE using a 1.5% agarose gel and visualized using Goldview
staining. The fragments were analyzed using the Quantity One 4.52
analysis system (Bio-Rad).
Statistical analysis
Experiments were conducted in triplicate. The
results were presented as the means ± standard deviation. Data were
processed using SPSS 13.0 software (SPSS Inc., Chicago, IL, USA),
and statistical differences were determined using analysis of
variance followed by a q test. P<0.05 was considered to indicate
a statistically significant difference.
Results
PN inhibited Raji cell growth
Results of the MTT assay analysis showed that PN
inhibited Raji cell growth in a dose-dependent manner (P<0.05
compared with the control group), with a half maximal inhibitory
concentration value of 5.07 μmol/l, but had no effect on normal
PBMCs (Fig. 1B).
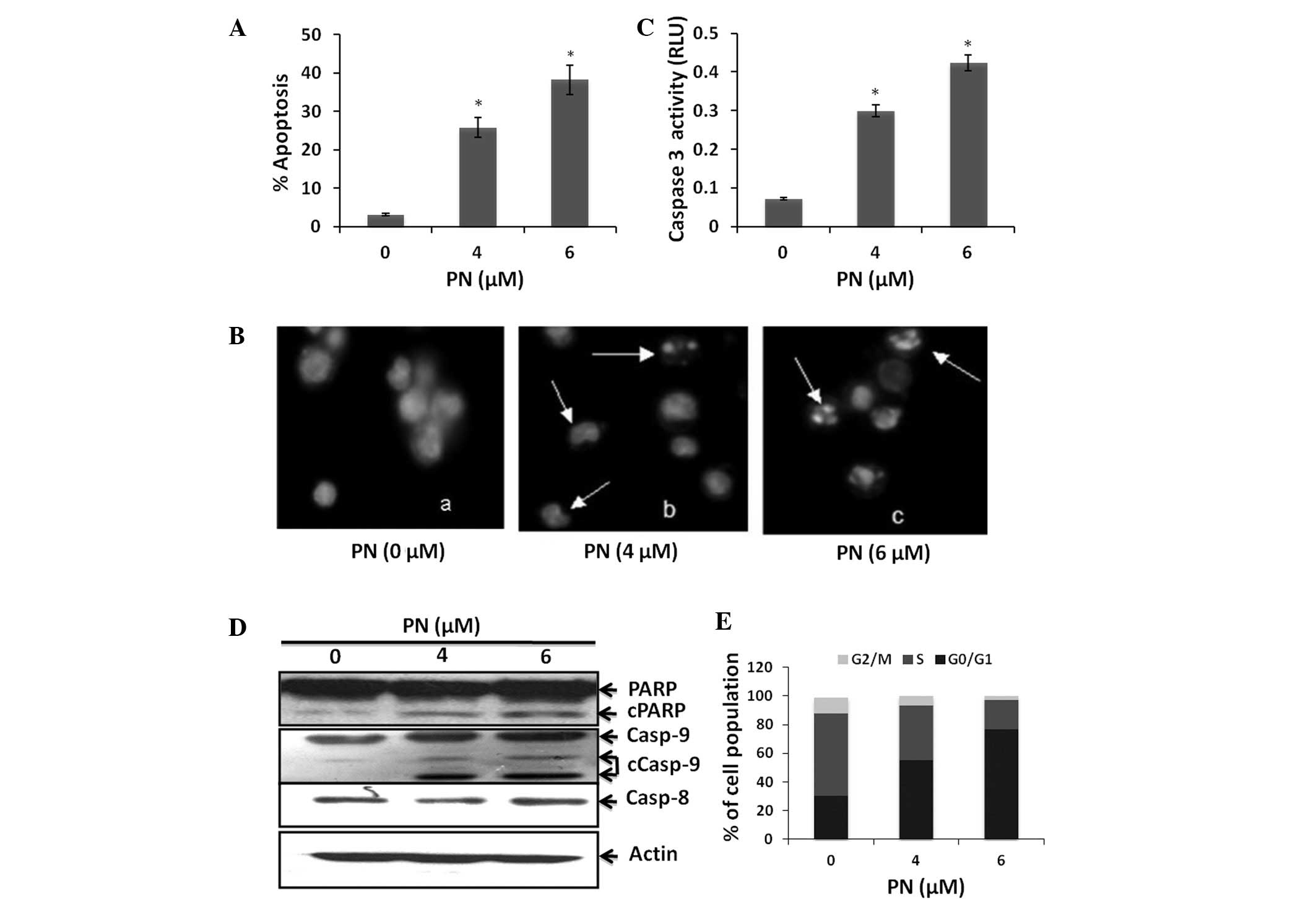
PN induced apoptosis and affects the cell
cycle in Raji cells
To investigate whether PN is capable of inhibiting
Raji cell growth by inducing cell death, we treated Raji cells with
0, 4 or 6 μmol/l PN for 48 h. Induction of cell apoptosis was
examined using both an annexin VFITC and PI assay and
DAPI staining. Our results revealed that PN induced Raji cell
apoptosis in a dose-dependent manner (P<0.05 compared with the
control group; Fig. 2A and B). To
verify that these cells underwent apoptosis, we measured the
generation of caspase-3 activity. Raji cells treated with PN had
increased caspase-3 activity (Fig.
2C). In addition, a known caspase substrate, poly- (ADP-ribose)
polymerase, was cleaved after PN treatment (Fig. 2D). Furthermore, we found that PN
activated caspase-9 but not caspase-8 (Fig. 2D). These results suggest that PN
induced Raji cell apoptosis via the mitochondrial pathway.
To determine whether the inhibition of Raji cell
proliferation induced by PN was associated with changes in cell
cycle progression, a cell cycle analysis was performed on Raji
cells treated with PN. Treatment with 4 and 6 μmol/l PN
significantly increased the cell population in the G0/G1 phase by
24.9 and 46.5%, respectively, and decreased the cell population in
the S phase by 19.2 and 36.7%, respectively (P<0.05 compared
with the control group; Fig.
2E).
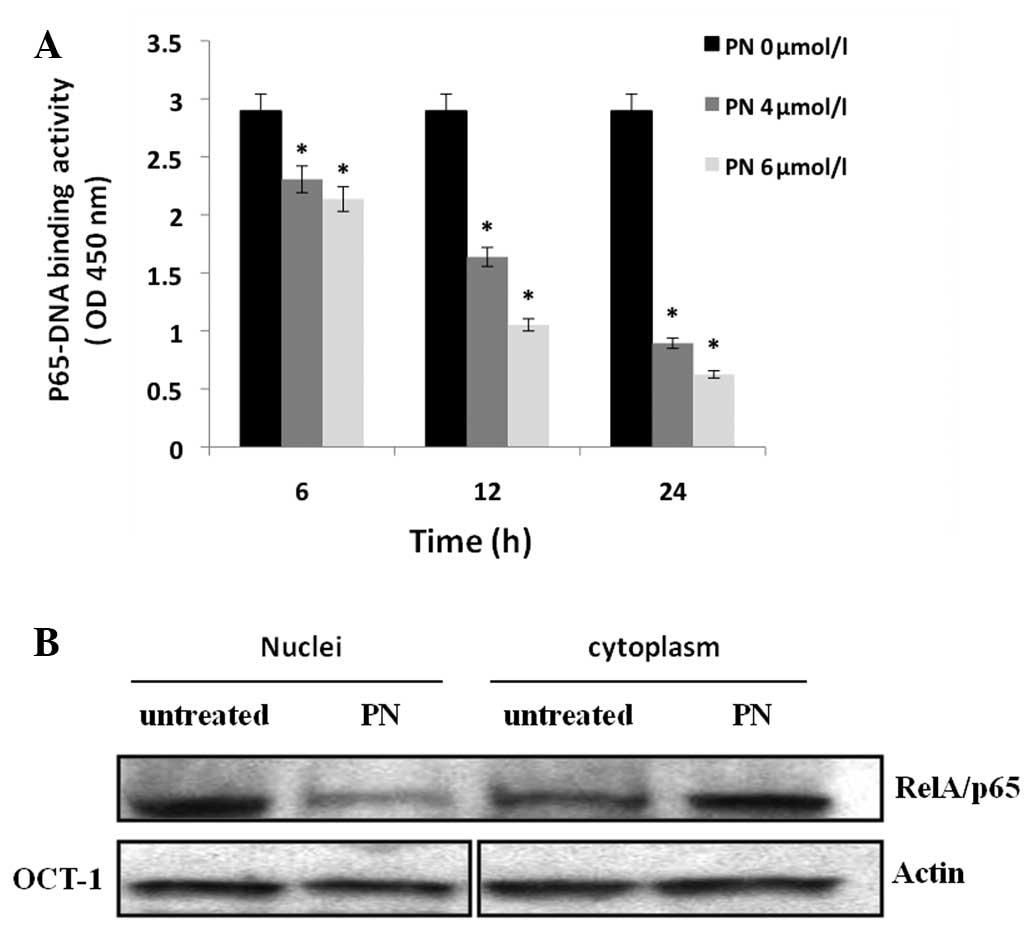
PN inhibited NF-κB activity in Raji
cells
To determine whether PN has an effect on NF-κB
activity in Raji cells, we used a NF-κB transcription factor assay
kit to detect NF-κB DNA-binding activity. Our results indicated
that the RelA/p65-DNA binding activity gradually decreased between
6 and 24 h after the addition of PN (Fig. 3A). We also investigated the
expression of RelA/p65 in the cytoplasm and the nucleus separately
using western blot analysis. As shown in Fig. 3B, RelA/p65 expression in the
nucleus of these cells was reduced following incubation with
PN.
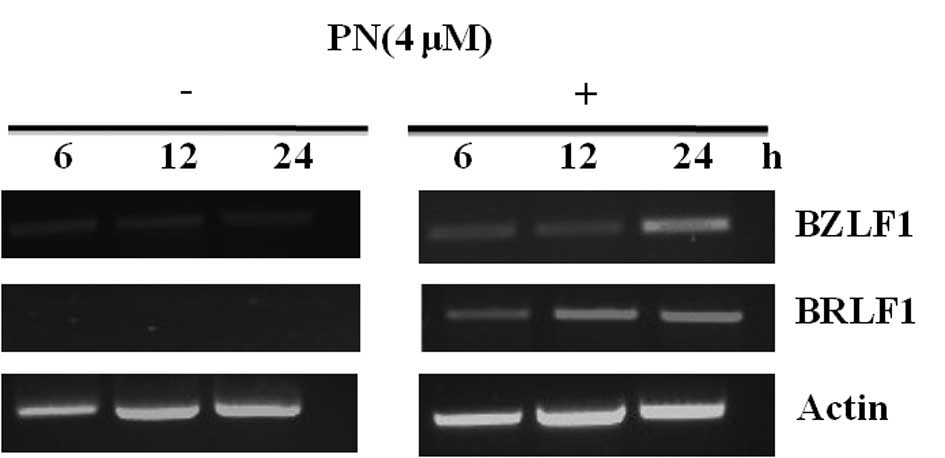
PN reactivated EBV in Raji cells
Expression of BZLF1 and BRLF1 mRNA was
increased in Raji cells treated with 4 or 6 μmol/l of PN (P<0.05
compared with the control group), indicating that PN induced EBV
lytic replication in the Raji cells (Fig. 4).
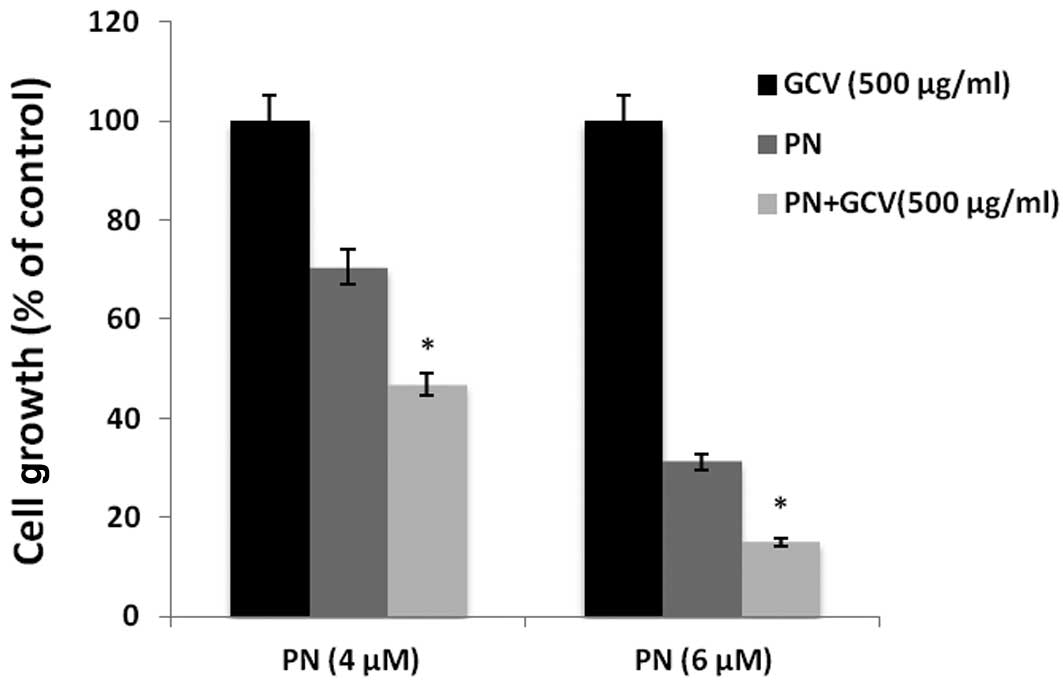
GCV amplified cytotoxicity induced by PN
in Raji cells
Raji cells were incubated with the indicated
concentrations of PN, in the presence or absence of GCV for 48 h.
Findings of the MTT assay showed that Raji cell viability was
further diminished by GCV combined with PN in comparison to PN
alone (P<0.05; Fig. 5). These
findings suggest that the NF-κB inhibitor PN reactivated EBV lytic
replication, allowing GCV to produce its active cytotoxic form,
thereby inducing the cytotoxic activity of GCV.
Discussion
Our findings have shown that PN inhibited Raji cell
growth in a dose-dependent manner and increased the cell population
in the G0/G1 phase while decreasing the cell population in the S
phase. These results suggest that PN inhibits Raji cell
proliferation by inducing cell cycle arrest at the G0/G1 phase. We
also observed that PN induced Raji cell growth arrest by inducing
Raji cell apoptosis via the mitochondrial pathway.
BL development has been reported to be markedly
associated with EBV. The fact that EBV is generally present in
cancer cells but rarely found in healthy cells represents an
opportunity for targeted cancer therapy (22). One approach is to activate the
lytic replication cycle of latent EBV, when viral proteins are
expressed at a high level and progeny viruses are produced. Lytic
EBV replication damages the cancer cells and triggers host immune
responses against EBV and the infected cells (23–26).
Entry into the viral lytic cycle is initiated by the expression of
two IE EBV proteins, Z Epstein-Barr virus replication activator,
encoded by BZLF1 (27), and Rta,
encoded by BRLF1 (28). The two IE
proteins activate the viral early genes, resulting in a cascade of
events that lead to progeny virion. Activation of NF-κB is a
feature of numerous viral infections (29). NF-κB activation during viral
infection is believed to be a protective response of the host to
the viral pathogen. Therefore, a number of viruses have evolved
distinct strategies to control NF-κB activity to evade the immune
response (30,31). Additionally, viruses may modulate
the NF-κB pathway to enhance viral replication or prevent
virus-induced apoptosis. Overexpression of NF-κB inhibits the
activation of lytic promoters from EBV, suggesting that NF-κB is a
novel target for the disruption of virus latency and therefore for
the treatment of EBV-related malignancies (32).
Although the mechanisms mediating the various
effects of PN in different diseases are not entirely clear, several
studies have shown that a significant aspect of the antitumor
activity of this compound appears to be associated with its
activity in inhibiting the NF-κB signal pathway by preventing the
degradation of IκBα (33). Our
results showed that PN suppressed NF-κB activity in Raji cells by
restricting the location of NF-κB from the cytoplasm to the
nucleus, which is in agreement with previous reports that showed
that PN is an inhibitor of NF-κB activity (34–36),
and acted as one mechanism for killing EBV-positive Raji cells.
However, our finding that PN induced a fully lytic form of EBV
suggests that PN has a second mechanism for killing tumor cells. We
found that treatment with PN increased BZLF1 and
BRLF1 mRNA expression, indicating that PN was capable of
inducing EBV lytic replication in Raji cells. In this study, GCV
amplified the cytotoxicity induced by PN, indicating a synergistic
cytotoxicity. Our results also suggest lytic virus replication in
PN-treated Raji cells, as GCV functions only in the lytic phase of
EBV infection and our findings showed that GCV treatment alone had
no cytotoxic effect on Raji cells. Moreover, the inhibition of
NF-κB activity specifically causes lytic cytotoxicity in
EBV-positive Raji cells.
In conclusion, findings of this study have shown the
ability of PN to induce EBV lytic replication and produce apparent
anticancer effects in EBV-positive carcinoma cells through the
inhibition of NF-κB activity. When GCV was added to PN, the
cytotoxic effect was even more potent. However, more studies are
required to test the effect of PN in EBV-positive malignancies in
animal models.
Acknowledgements
This study was supported by grants from the Fujian
Provincial Department of Science and Technology (2009-CXB-57 and
2011J01252) and Bureau of Science and Technology of Xiamen, China
(3502Z20094012). We thank Dr Lan V Pham from MD Anderson Cancer
Center for valuable comments. We appreciate Dr Markeda Wade from MD
Anderson Cancer Center for revising our manuscript.
References
|
1
|
Yang L, Parkin DM, Whelan S, Zhang S, Chen
Y, Lu F and Li L: Statistics on cancer in China: cancer
registration in 2002. Eur J Cancer Prev. 14:329–335. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parkin DM: The global health burden of
infection-associated cancers in the year 2002. Int J Cancer.
118:3030–3044. 2006.PubMed/NCBI
|
|
3
|
Cohen JI, Kimura H, Nakamura S, Ko YH and
Jaffe ES: Epstein-Barr virus-associated lymphoproliferative disease
in non-immunocompromised hosts: a status report and summary of an
international meeting, 8–9 September 2008. Ann Oncol. 20:1472–1482.
2009.PubMed/NCBI
|
|
4
|
Thorley-Lawson DA and Gross A: Persistence
of the Epstein-Barr virus and the origins of associated lymphomas.
N Engl J Med. 350:1328–1337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thorley-Lawson DA and Allday MJ: The
curious case of the tumour virus: 50 years of Burkitt’s lymphoma.
Nat Rev Microbiol. 6:913–924. 2008.PubMed/NCBI
|
|
6
|
Cohen JI, Bollard CM, Khanna R and
Pittaluga S: Current understanding of the role of Epstein-Barr
virus in lymphomagenesis and therapeutic approaches to
EBV-associated lymphomas. Leuk Lymphoma. 49(Suppl 1): 27–34. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Perrine SP, Hermine O, Small T, et al: A
phase 1/2 trial of arginine butyrate and ganciclovir in patients
with Epstein-Barr virus-associated lymphoid malignancies. Blood.
109:2571–2578. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fu DX, Tanhehco Y, Chen J, et al:
Bortezomib-induced enzyme-targeted radiation therapy in
herpesvirus-associated tumors. Nat Med. 14:1118–1122. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Landais E, Saulquin X, Scotet E, et al:
Direct killing of Epstein-Barr virus (EBV)-infected B cells by CD4
T cells directed against the EBV lytic protein BHRF1. Blood.
103:1408–1416. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feng WH, Israel B, Raab-Traub N, Busson P
and Kenney SC: Chemotherapy induces lytic EBV replication and
confers ganciclovir susceptibility to EBV-positive epithelial cell
tumors. Cancer Res. 62:1920–1926. 2002.PubMed/NCBI
|
|
11
|
Hailfinger S, Nogai H, Pelzer C, et al:
Malt1-dependent RelB cleavage promotes canonical NF-kappaB
activation in lymphocytes and lymphoma cell lines. Proc Natl Acad
Sci USA. 108:14596–14601. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Song YJ and Kang MS: Roles of TRAF2 and
TRAF3 in Epstein-Barr virus latent membrane protein 1-induced
alternative NF-kappaB activation. Virus Genes. 41:174–180. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cahir-McFarland ED, Davidson DM, Schauer
SL, Duong J and Kieff E: NF-kappa B inhibition causes spontaneous
apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells.
Proc Natl Acad Sci USA. 97:6055–6060. 2000. View Article : Google Scholar
|
|
14
|
Chaudhary PM, Jasmin A, Eby MT and Hood L:
Modulation of the NF-kappa B pathway by virally encoded death
effector domains-containing proteins. Oncogene. 18:5738–5746. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Izumi KM and Kieff ED: The Epstein-Barr
virus oncogene product latent membrane protein 1 engages the tumor
necrosis factor receptor-associated death domain protein to mediate
B lymphocyte growth transformation and activate NF-kappaB. Proc
Natl Acad Sci USA. 94:12592–12597. 1997. View Article : Google Scholar
|
|
16
|
Keller SA, Schattner EJ and Cesarman E:
Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary
effusion lymphoma cells. Blood. 96:2537–2542. 2000.PubMed/NCBI
|
|
17
|
Smolinski AT and Pestka JJ: Comparative
effects of the herbal constituent parthenolide (Feverfew) on
lipopolysaccharide-induced inflammatory gene expression in murine
spleen and liver. J Inflamm (Lond). 2:62005. View Article : Google Scholar
|
|
18
|
Pareek A, Suthar M, Rathore GS and Bansal
V: Feverfew (Tanacetum parthenium L.): A systematic review.
Pharmacogn Rev. 5:103–110. 2011.
|
|
19
|
deGraffenried LA, Chandrasekar B,
Friedrichs WE, et al: NF-kappa B inhibition markedly enhances
sensitivity of resistant breast cancer tumor cells to tamoxifen.
Ann Oncol. 15:885–890. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakshatri H, Rice SE and Bhat-Nakshatri P:
Antitumor agent parthenolide reverses resistance of breast cancer
cells to tumor necrosis factor-related apoptosis-inducing ligand
through sustained activation of c-Jun N-terminal kinase. Oncogene.
23:7330–7344. 2004. View Article : Google Scholar
|
|
21
|
Dai Y, Guzman ML, Chen S, et al: The NF
(Nuclear factor)-kappaB inhibitor parthenolide interacts with
histone deacetylase inhibitors to induce MKK7/JNK1-dependent
apoptosis in human acute myeloid leukaemia cells. Br J Haematol.
151:70–83. 2010. View Article : Google Scholar
|
|
22
|
Israel BF and Kenney SC: Virally targeted
therapies for EBV-associated malignancies. Oncogene. 22:5122–5130.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Flemington EK: Herpesvirus lytic
replication and the cell cycle: arresting new developments. J
Virol. 75:4475–4481. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang H, Zhao Y, Zeng L, Tang M, El-Deeb A,
Li JJ and Cao Y: BZLF1 controlled by family repeat domain induces
lytic cytotoxicity in Epstein-Barr virus-positive tumor cells.
Anticancer Res. 24:67–74. 2004.PubMed/NCBI
|
|
25
|
Chen YL, Law PY and Loh HH: Inhibition of
PI3K/Akt signaling: an emerging paradigm for targeted cancer
therapy. Curr Med Chem Anticancer Agents. 5:575–589. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Feng WH and Kenney SC: Valproic acid
enhances the efficacy of chemotherapy in EBV-positive tumors by
increasing lytic viral gene expression. Cancer Res. 66:8762–8769.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Urier G, Buisson M, Chambard P and
Sergeant A: The Epstein-Barr virus early protein EB1 activates
transcription from different responsive elements including AP-1
binding sites. EMBO J. 8:1447–1453. 1989.PubMed/NCBI
|
|
28
|
Zalani S, Holley-Guthrie E and Kenney S:
Epstein-Barr viral latency is disrupted by the immediate-early
BRLF1 protein through a cell-specific mechanism. Proc Natl Acad Sci
USA. 93:9194–9199. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mogensen TH and Paludan SR: Molecular
pathways in virus-induced cytokine production. Microbiol Mol Biol
Rev. 65:131–150. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hiscott J, Kwon H and Genin P: Hostile
takeovers: viral appropriation of the NF-kappaB pathway. J Clin
Invest. 107:143–151. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Santoro MG, Rossi A and Amici C: NF-kappaB
and virus infection: who controls whom. EMBO J. 22:2552–2560. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brown HJ, Song MJ, Deng H, Wu TT, Cheng G
and Sun R: NF-kappaB inhibits gammaherpesvirus lytic replication. J
Virol. 77:8532–8540. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kwok BH, Koh B, Ndubuisi MI, Elofsson M
and Crews CM: The anti-inflammatory natural product parthenolide
from the medicinal herb Feverfew directly binds to and inhibits
IkappaB kinase. Chem Biol. 8:759–766. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hayashi S, Koshiba K, Hatashita M, et al:
Thermosensitization and induction of apoptosis or cell-cycle arrest
human prostate cancer androgen-independent cell lines. Int J Mol
Med. 28:1033–1042. 2011.PubMed/NCBI
|
|
35
|
Gunn EJ, Williams JT, Huynh DT, et al: The
natural products parthenolide and andrographolide exhibit
anti-cancer stem cell activity in multiple myeloma. Leuk Lymphoma.
52:1085–1097. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ghashghaeinia M, Toulany M, Saki M, et al:
The NFkB pathway inhibitors Bay 11-7082 and parthenolide induce
programmed cell death in anucleated Erythrocytes. Cell Physiol
Biochem. 27:45–54. 2011. View Article : Google Scholar : PubMed/NCBI
|