Introduction
Autophagy is a cellular catabolic degradation
process which functions to optimize the bioenergetic cellular
microenvironment by degrading long-lived proteins and damaged
organelles. Autophagy uses programmed machinery that recruits a
plethora of autophagy-related genes (ATGs) which are required for
autophagosome synthesis. Autophagosomes are double membrane
structures that sequester the intended cytoplasmic portion, then
bind to the lysosome to form autophagolysosomes, where the cargo is
digested (1). Autophagy is
involved in various physiological and pathophysiological processes,
including aging, cancer and neurodegenerative diseases (2).
MicroRNAs are a short, non-coding, novel class of
gene regulators. They are synthesized in the nucleus, exported to
the cytoplasm and eventually bind to the 3′-untranslated region
(3′-UTR) of their target mRNA to repress gene expression at the
post-transcriptional level. Findings of previous studies
demonstrated the role of microRNAs in various cellular events,
including growth, apoptosis and carcinogenesis (3). MicroRNAs have recently been
characterized as modulating the process of autophagy via the
targeting of cardinal autophagy-regulating genes. miR-30a and
miR-376b have been demonstrated to target and inhibit Beclin-1
activity, thereby blocking autophagy (4,5).
Additionally, miR-101 inhibits RAB5A, which acts in the early
stages of autophagosome formation (6).
Ataxia telangiectasia mutated (ATM) is a Ser/Thr
protein kinase and a member of the phosphoinositide 3-kinase
(PI3K)-related protein kinase (PIKK) family. ATM functions to
maintain genomic stability by orchestrating the actions of several
downstream substrates involved in cell cycle arrest, apoptosis and
DNA repair in response to DNA damage-inducing agents, particularly
ionizing radiation (7). ATM has
also been identified to upregulate the autophagic response of cells
to genotoxic and oxidative stimuli (8). It has been reported that the
expression of ATM is modulated by miR-421, miR-101, miR-100 and
miR-18a in cancer cells (9–12).
miR-18a, a member of the miR-17-92 cluster, has been shown to be
significantly overexpressed in colon cancer tissues in comparison
to normal colon mucosal cells (13). Modulation of ATM expression by
miR-18a has not been exhibited in colon cancer cells. In the
present study, we explored the impact of miR-18a on the process of
autophagy and ATM expression in colon cancer cells, using the colon
cancer cell line HCT116.
Materials and methods
Cell lines and radiation
HCT116 colon cancer cells were grown in DMEM
supplemented with 10% fetal bovine serum, 2 μM glutamine, 100 IU/ml
penicillin and 100 μg/ml streptomycin sulfate, in humidified
conditions in an incubator containing 5% CO2 and at
37°C. For the X-ray irradiation procedure a 180-kVp X-ray generator
(Model XSZ-Z20/20, China) was utilized at a dose rate of 0.41
Gy/min. The study was approved by the ethics committee of Jilin
University School of Public Health. Informed consent was obtained
from patients.
RNA extraction
The total RNA of the cultured cells was extracted
with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to
the manufacturer's instructions and stored at −80°C prior to RT-PCR
analysis.
Quantitative (q) RT-PCR for measuring
miR-18a expression
For the SYBR-Green assay miRCURY LNA™ Universal RT
microRNA PCR was utilized (Exiqon, Denmark). RNA (~20 ng) was
converted to cDNA using the miRCURY LNA Universal RT microRNA PCR
with the Poly-T primer (Exiqon). Following reverse transcription,
the cDNA template was amplified using microRNA-specific and LNA
primers. qRT-PCR was performed using the Stratagene MX3000p
thermocycler according to the manufacturer's instructions. The U6
gene was used as a normalization control for all samples.
Plasmids
To construct a plasmid expressing miR-18a,
pri-miR-18a was amplified using the genomic DNA from a healthy
blood donor. PCR was performed using the rTaq enzyme
(Takara, Dalian, China). The amplified fragment was first cloned
into a PCR cloning vector (PMD-19T) and subsequently cloned into a
lentiviral vector (pCDHCMV-MCS-EF1-copGFP; System Biosciences,
Mountain View, CA, USA) at the EcoRI and BamHI sites.
Primer sequences used were: hsa-miR-18a (forward),
GCCGAATTCGTGCAGGTAGTGATATGTGC; hsa-miR-18a (reverse),
CGCGGATCCGATTTGCACAACTACATTC. The 3′-UTR of ATM (Genbank accession
no. NM_000051.3, region between 9557 and 13147 bp) carrying the
putative miR-18a binding site was amplified by PCR from the human
genomic DNA of the blood and cloned between the SacI and
HindIII sites of the pMIR-REPORT™ luciferase vector (Ambion,
Foster City, CA, USA).
The primers selected were the upstream ATM 3′-UTR
primer 5′-GCCTCTAGACTCCTGTTCTGTTCAAGTAT-3′ and the downstream ATM
3′-UTR primer 5′-GGCCCTCTAGAGCTTTTAGAATTATTTATTC-3′. PCR products
cloned into the plasmid were verified by DNA sequencing to ensure
that they were free of mutations and in the correct cloning
direction.
Luciferase reporter assays
HCT116 cells at a density of 0.5×105/well
were cultured in 24-well plates and each well was transfected with
20 ng pMIR-ATM-3′-UTR, together with 5 ng pRL-SV40 vector (Promega,
Madison, WI, USA), which contains the Renilla luciferase gene, and
50 or 100 nM of the miR-18a mimics/negative control (NC;
GenePharma, Shanghai, China) or PCDH-miR-18a/PCDH-control.
Transfection was performed using Lipofectamine™ 2000 (Invitrogen).
At 48 h post-transfection, firefly and Renilla luciferase
activities were examined using the Dual-Luciferase Reporter Assay
(Promega).
Western blot analysis
Cells were harvested and lysed in a RIPA lysis
buffer (150 mM sodium chloride, 1.0% NP-40 or Triton X-100, 0.1%
SDS, 50 mM Tris, pH 8.0, 20 mmol/l Na2PO4, pH
7.4, aprotinin, 1 mmol/l phenymethysulfonyl fluoride, 10 mg/ml
leupeptin, 100 mmol/l NaF and 2 mmol/l
Na3VO4). Total protein (100 μg) was separated
by SDS-PAGE, transferred to nitrocellulose membranes and analyzed
by immunoblotting using chemiluminescence (Santa Cruz
Biotechnology, Inc. Santa Cruz, CA, USA). The primary antibodies
used were ATM (1:300) and LC3-I/II (1:300; Cell Signaling
Technology Inc., Danvers, MA, USA), GAPDH (1:1000; Santa Cruz
Biotechnology), P62/SQSTM1 (1:500) and P70S6K and p-P70S6K (Thr389;
1:500; Abcam, Cambridge, MA, USA). The intensity of the protein
bands was quantified using Image J software and the control was set
to 1.
GFP-LC3 study
To generate the stable expression of GFP-LC3, the
HCT116 cells were transfected with the purified recombinant
plasmid, pQN-GFP-LC3, using Lipofectamin™ 2000 according to the
manufacturer's instructions. The HCT116 cells stably expressing
GFP-LC3 were plated at a density of 1×105 in a 6-well
plate with glass coverslips in the bottom and exposed to the
indicated transfections of microRNA, then irradiated. GFP-LC3
puncta were visualized under an inverted fluorescence (Olympus
XSZ-D2) microscope equipped with CCD cameras. Images were captured
for each sample at 48 h post-transfection, then analyzed
manually.
Statistical analysis
The Student's t-test was used to determine
statistical significance. P<0.01 and P<0.05 were considered
to indicate statistically significant differences.
Results
Endogenous miR-18a expression
upregulation by ionizing radiation
To determine whether a preliminary link exists
between autophagy and miR-18a expression we measured the level of
endogenous miR-18a under basal growth conditions and following
exposure to ionizing radiation (IR) in HCT116 colon cancer cells.
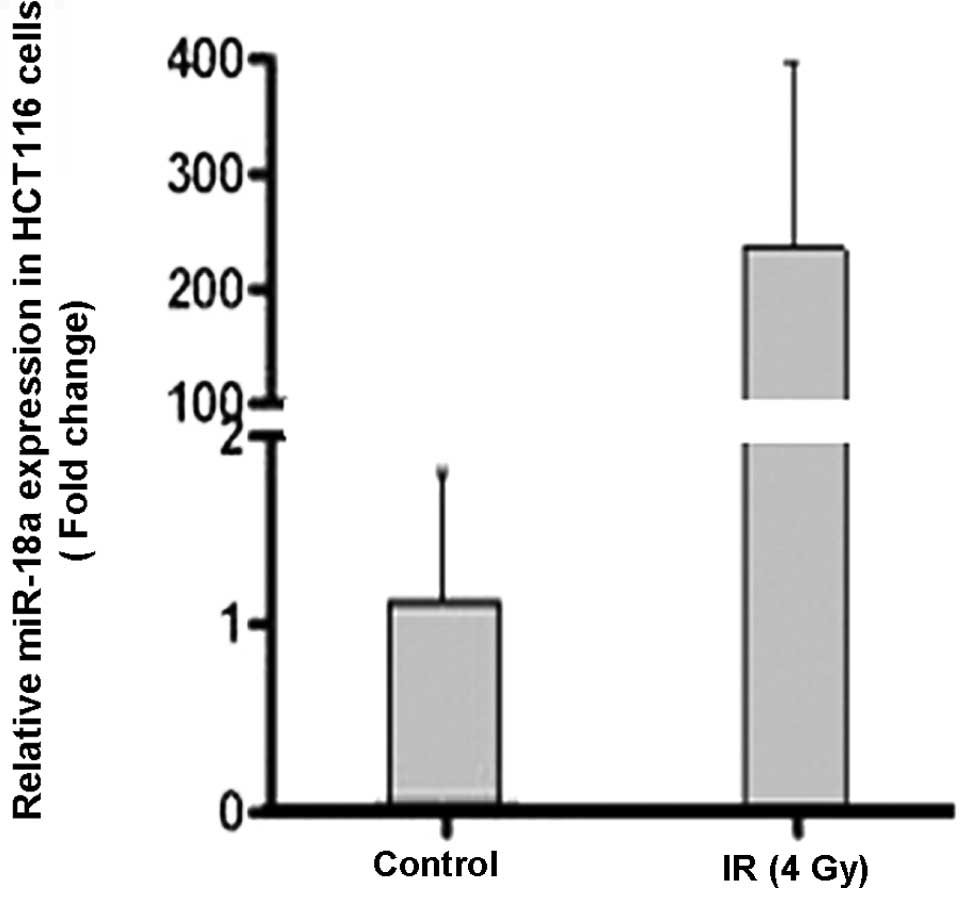
Quantitative RT-PCR was performed using the HCT116 cells collected
following exposure to 4 Gy of radiation. The expression level of
miR-18a markedly increased by >200-fold 1 h after irradiation
(Fig. 1).
Ectopic miR-18a overexpression promotes
autophagy
During autophagy, the mammalian ATG8-homolog (LC3-I)
is processed and recruited to the autophagosomes, where the
lipidated LC3-II is generated (14). Therefore, HCT116 cells stably
expressing GFP-LC3 were transfected with a miR-18a mimic, a
negative control or left without any transfection as mock cells. At
48 h post-transfection, analysis by fluorescence microscopy for the
presence of GFP-LC3 puncta was performed (Fig. 2A). miR-18a expression increased the
percentage of puncta-positive cells to a significant extent as
compared with the NC-transfected and mock cells (Fig. 2B). Notably, the percentage of
puncta-positive cells was further enhanced when miR-18a was
combined with IR, relative to cells treated with IR alone (Fig. 2C and D). Similarly, miR-18a
overexpression enhanced the LC3-II protein expression level in
non-irradiated and irradiated HCT116 cells (Fig. 2E and F). P62/SQSTM1 is a
poly-ubiquitin binding protein that was identified as being able to
bind directly to ATG8/LC3 and localize to autophagosomes to
ultimately be degraded during autophagy. Therefore, the level of
P62 reflects the autophagic turnover (15). As shown in Fig. 2G and H, in the HCT116 cells
transfected with miR-18a the expression level of the P62/SQSTM1
protein was markedly reduced in non-irradiated and irradiated cells
as compared with the NC-transfected cells. Consequently, such
findings demonstrated that miR-18a enhanced the autophagic flux.
Taken together, these results indicate that miR-18a upregulates the
process of autophagy in the HCT116 cell line and potentiates the
autophagic response of HCT116 cells to radiation.
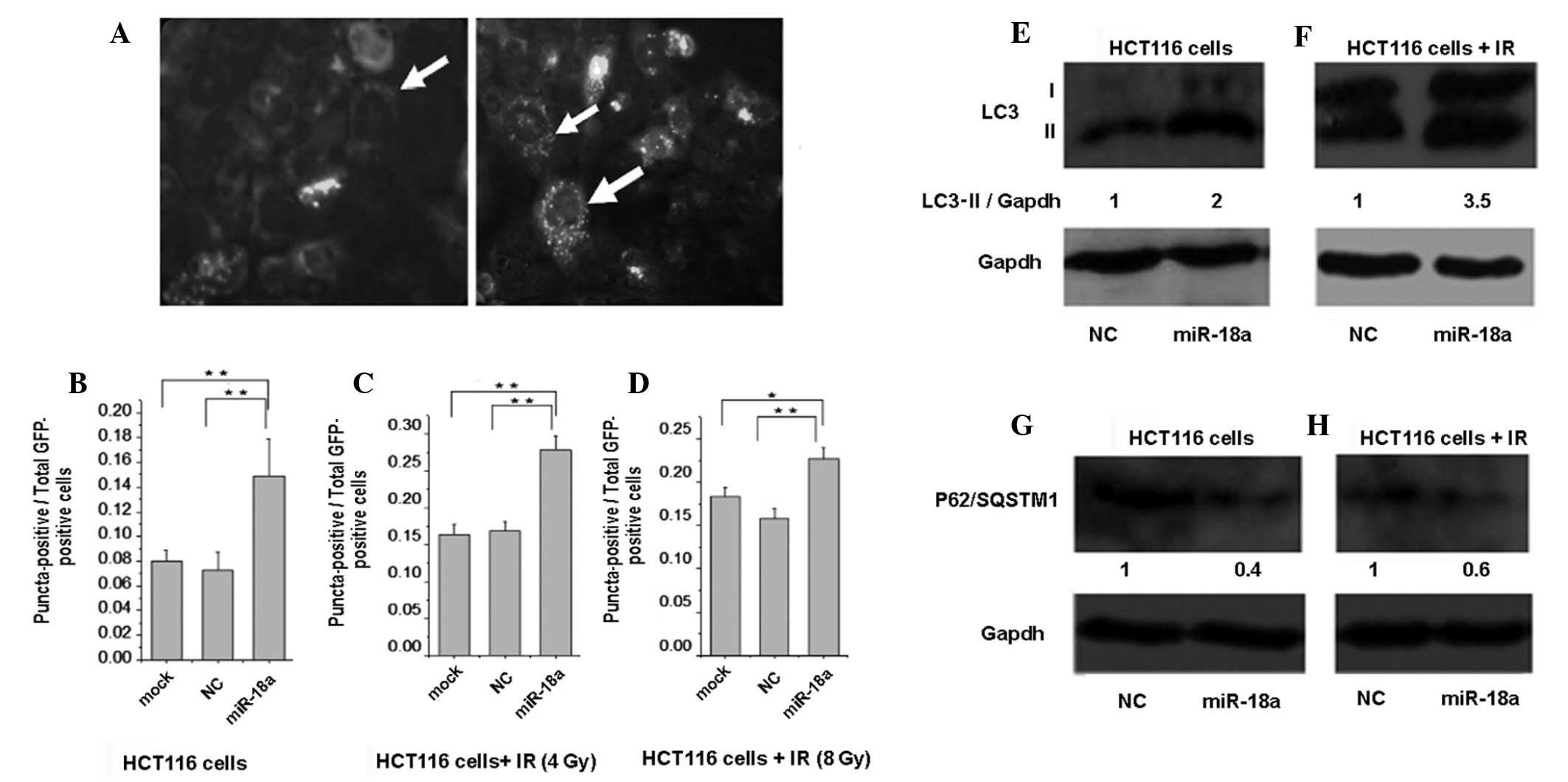 | Figure 2Ectopic miR-18a overexpression
upregulates autophagy in HCT116 cells. (A) Representative images of
HCT116 cells stably expressing GFP-LC3; the right panel with the
white arrows indicating a typical puncta-positive cell shows
induction of autophagy, while the left panel with a white arrow
indicating a GFP-LC3-positive cell shows that autophagy was not
induced. (B-D) Stably expressing GFP-LC3 HCT116 cells were
transfected with the negative control (NC) or miR-18a mimic (100
nM) or left without transfection as mock cells, then treated
accordingly with indicated doses of radiation. The graphs show the
total number of puncta-positive cells divided by the total
GFP-positive cells; *P<0.05, **P<0.01
compared to mock or NC. (E) miR-18a overexpression increases LC3-II
protein expression. HCT116 cells were transfected with NC or
miR-18a mimic (100 nM) then the cell lysate was applied to western
blot analysis 48 h post-transfection. GAPDH was used as a loading
control, n=3. (F) HCT116 cells were transfected with NC or miR-18a
mimic (100 nM) then treated with 4 Gy of IR. Western blot analysis
was conducted 48 h post-transfection (20 h post-radiation) to
measure LC3-II protein expression, n=2. (G) miR-18a overexpression
led to a decreased expression of the P62/SQSTM1 protein. HCT116
cells were transfected with NC or miR-18a mimic (100 nM), then the
cell lysate was exposed to western blot analysis 48 h
post-transfection, n=2. (H) HCT116 cells were transfected with NC
or miR-18a mimic (100 nM), then treated with 4 Gy of radiation. The
cell lysate was subjected to western blot analysis 48 h
post-transfection (20 h post-IR) to measure the P62/SQSTM1
expression level, n=2. IR, ionizing radiation. |
Ectopic miR-18a overexpression
upregulates ATM gene expression
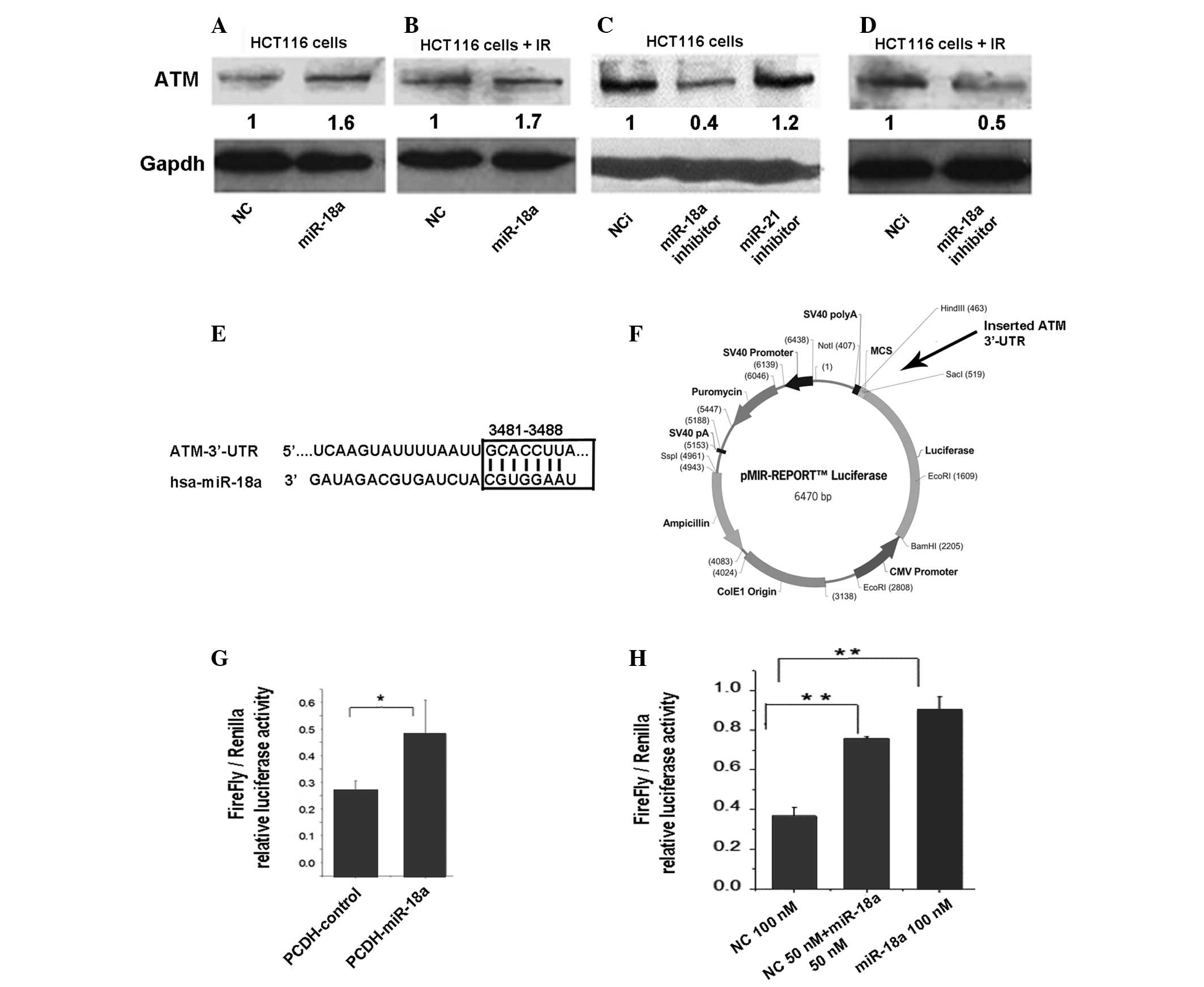
First, we explored the effect of miR-18a on
endogenous ATM protein levels in HCT116 cells by western blotting.
Using a miR-18a mimic, we identified that an ectopic increase in
miR-18a led to >50% increase in ATM protein levels in
non-irradiated and irradiated HCT116 cells relative to the negative
control (NC) transfected cells (Fig.
3A and B). Opposite trends were identified in the cells
transfected with the miR-18a inhibitor (Fig. 3C and D). According to the databases
from three popular microRNA target prediction programs (Targetscan,
miranda and miRBase), the ATM mRNA 3′-UTR contains a sequence motif
(position 3481–3488) which is complementary to the miR-18a seed
sequence and may be a putative binding site for miR-18a (Fig. 3E). Therefore, we constructed a
luciferase reporter plasmid (pMIR-ATM 3′-UTR) with the ATM 3′-UTR,
which contains a putative binding site to miR-18a, and cloned it to
a firefly luciferase reporter (Fig.
3F). PCDH vector-expressed miR-18a (PCDH-miR-18a) and synthetic
miR-18a (miR-18a mimic) were then used to evaluate the effects of
the microRNA on the reporter gene expression. At 48 h
post-transfection, the luciferase activity was assayed and
normalized to Renilla. In HCT116 cells, PCDH-miR-18a and the
miR-18a mimic significantly increased the expression of the
reporter gene with the ATM 3′-UTR tag (Fig. 3G and H). These results demonstrate
for the first time that miR-18a is able to enhance ATM gene
expression in the HCT116 colon cancer cell line, most likely by
targeting the ATM 3′-UTR.
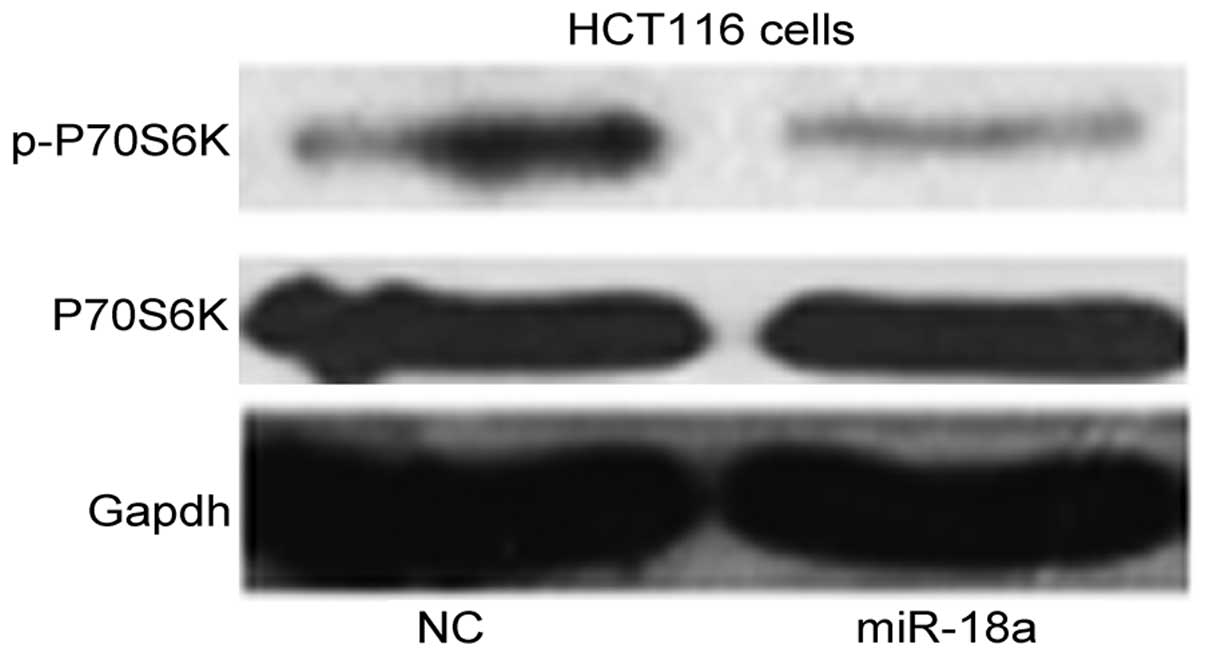
miR-18a overexpression inhibits mTORC1
activity
ATM has been demonstrated to upregulate autophagy
via inhibition of mTORC1 (8). We
hypothesized that miR-18a positively regulated ATM, inhibiting
mTORC1 and inducing autophagy. To examine whether miR-18a regulated
mTORC1, we measured P70S6K phosphorylation at Thr389 as a typical
readout of mTORC1 activity (16).
As shown in Fig. 4, miR-18a
overexpression resulted in hypophosphorylation of P70S6K (which
indicated mTORC1 activity inhibition) compared with NC-transfected
cells.
Discussion
Our knowledge concerning the molecular control of
autophagy has greatly increased during the last decade, yet
mechanisms controlling autophagic activity are not fully
understood. Abnormalities of autophagy play a role in major health
problems, including cancer and neurodegenerative diseases. The
exact role of autophagy in carcinogenesis remains elusive.
Autophagy is able to behave as a tumor suppressor or oncogene
depending on the cell context (17). A similar paradox is exhibited
during tumor therapy. Autophagy has been shown to support cancer
cell survival, as has been observed in breast cancer cells
(18). However, in other contexts,
as in HCT116 colon cancer cells, autophagy has been shown to
contribute to cell death (19,20).
Therefore, specific modulators of autophagy suitable for in
vivo use are required (21).
MicroRNAs have emerged recently as a novel class of endogenous gene
regulators, and have been implicated in various pathological and
physiological processes. Previously, a series of studies had been
working to demonstrate the role of microRNAs in the regulation of
autophagy. miR-376b overexpression was shown to attenuate
starvation- and rapamycin-induced autophagy in MCF-7 and Huh-7
cancer cells by suppressing the autophagy proteins ATG4C and BECN1
as target genes (5). Other
microRNAs, including miR-30a (via suppression of Beclin-1), miR-101
(via suppression of STMN1, RAB5A and ATG4D) and miR-204 (via
blocking the activation of LC3-II activity), have all been shown to
be potent suppressors of autophagy in cancer cells (4,6,22).
In the present study we showed that miR-18a
overexpression resulted in a significant promotion of autophagy in
HCT116 colon cancer cells, as evidenced by increased GFP-LC3 puncta
formation, increased LC3-II protein expression and a decreased
P62/SQSTM1 protein level. Moreover, miR-18a promoted
radiation-induced autophagy (Fig.
2). Additionally, the role of miR-18a in modulating autophagy
is supported by our analysis of endogenous microRNA expression,
which demonstrated a marked upregulation of miR-18a expression in
HCT116 cells following exposure to radiation (Fig. 1). ATM encodes a 370-kDa protein
with a carboxyl-terminal sequence homologous to the catalytic
domain of phosphatidylinositol 3-kinases. Its classical function is
to maintain genomic stability following the exposure of cells to
agents that induce DNA damage (double strand breaks), particularly
ionizing radiation, by phosphorylation of several downstream
substrates involved in cell cycle arrest, apoptosis and DNA repair
(23). ATM has been identified to
upregulate the autophagic response of cells to genotoxic and
oxidative stimuli. ATM stimulates downstream signaling through the
LBK/AMPK/TSC2 pathway, which in turn results in mTORC1 repression
and autophagy induction (8,24,25).
Therefore, ATM may serve as a direct link between DNA damage and
autophagy.
The 3′-UTR segment of ATM mRNA contains only a
single binding site for the seed sequence of miR-18a. We showed
that miR-18a upregulated ATM gene expression in non-irradiated and
4 Gy-irradiated HCT116 cells, as evidenced by western blotting.
Conversely, inhibition of miR-18a led to the suppression of ATM
protein expression. To explore the mechanisms we constructed
plasmids containing the binding site for miR-18a and conducted a
dual-luciferase reporter assay. Following co-transfection,
luciferase activity with the ATM 3′-UTR construct increased
significantly in the miR-18a mimic and vector expressed miR-18a
(PCDH-miR-18a)-transfected cells (Fig.
3). These results suggest that ATM expression may be elevated
by miR-18a through targeting of the 3′-UTR of ATM mRNA. However,
the theory that overexpression of miR-18a downregulates ATM
expression in breast cancer cells remains controversial (12). Similar controversial findings have
been exhibited by miR-21, which has been to shown to upregulate
Bcl-2 gene expression in pancreatic cancer cells, but also to lead
to downregulation of Bcl-2 in breast cancer cells (26,27).
Previous studies have revealed that certain microRNAs are able to
directly upregulate the expression of their target genes by binding
to the 3′-UTR (28,29). Collectively such findings suggest
that the impact of miR-18a on ATM gene expression may be cell-type
specific. Results from the present study demonstrate for the first
time that miR-18a is able to upregulate ATM target gene expression
in HCT116 cells and provides a novel clue for exploring the exact
targets and mechanisms of miR-18a in colon cancer cells. Further
comprehensive investigations are consequently required. This
provides a new challenge to further understand the mechanisms of
microRNAs. As it is well known that microRNAs target several genes
simultaneously, we hypothesize that the miR-18a induces autophagy,
at least partially, through targeting the ATM gene. This hypothesis
is supported partially by our finding that miR-18a overexpression
suppressed mTORC1 activity (Fig.
4).
Colorectal cancer (CRC) is the second most common
cause of cancer mortality in the western world. CRC develops from
an accumulation of multiple genetic and epigenetic alterations that
contribute to its diverse phenotypes. Genomic instability, which
occurs in ~5% of colorectal adenomas, constitutes a major step in
the progression to cancer (30,31).
ATM, the guardian of genomic integrity, has been shown to play a
major tumor suppressive role in colon cancer by inhibiting the
progression of pre-neoplastic lesions into neoplasia (32,33).
miR-18a belongs to the miR-17-92 cluster, located at 13q, which
encodes six microRNAs processed from a common precursor transcript.
The miR-17-92 cluster has been shown to be involved in the
pathogenesis of human cancers, including diffuse large B cell
lymphoma (34) and small cell lung
cancer (35). However, the
pathophysiological roles and targets of members of this cluster,
particularly miR-18a, are largely unknown in colon cancer (36). The results of the present study
suggest that miR-18a has the potential to modulate autophagy
through regulation of the expression of the known autophagy
activator ATM, thus providing evidence for a new role of miR-18a in
a cellular process with a significance that has increasingly been
recognized in cancer biology. An improved understanding of the
microRNA-modulated autophagic signaling networks is likely to be
crucial to the current and future cancer therapeutic strategies
(37). The ability of miR-18a to
upregulate ATM gene expression, which has potent antitumor
features, renders miR-18a a good candidate for future studies in
the field of microRNA-based epigenetic colon cancer therapy.
Acknowledgements
The authors would like to thank Dr Yin-Yuan Mo
(Department of Medical Microbiology, Immunology and Cell Biology,
Southern Illinois University School of Medicine, Springfield, IL,
USA) for providing them with a (pCDHCMV-MCS-EF1-copGFP)
lenti-vector. They are also grateful to Mr. Song Zhiheng for his
technical assistance. This study was sponsored by an NSFC grant
(30770649, 30970682) and the Research Fund for the Doctoral Program
of Higher Education of China (20100061110070).
References
|
1
|
Mizushima N: Autophagy: process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar
|
|
2
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
He L and Hannon GJ: MicroRNAs: small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X,
Liu CG and Yang JM: Regulation of autophagy by a beclin 1-targeted
microRNA, miR-30a, in cancer cells. Autophagy. 5:816–823. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Korkmaz G, le Sage C, Tekirdag KA, Agami R
and Gozuacik D: miR-376b controls starvation and mTOR
inhibition-related autophagy by targeting ATG4C and BECN1.
Autophagy. 8:165–176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Frankel LB, Wen J, Lees M, Høyer-Hansen M,
Farkas T, Krogh A, Jäättelä M and Lund AH: microRNA-101 is a potent
inhibitor of autophagy. EMBO J. 30:4628–4641. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Derheimer FA and Kastan MB: Multiple roles
of ATM in monitoring and maintaining DNA integrity. FEBS Lett.
584:3675–3681. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alexander A, Cai SL, Kim J, Nanez A, Sahin
M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, Kusewitt D,
Mills GB, Kastan MB and Walker CL: ATM signals to TSC2 in the
cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci
USA. 107:4153–4158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu H, Du L, Nagabayashi G, Seeger RC and
Gatti RA: ATM is down-regulated by N-Myc-regulated microRNA-421.
Proc Natl Acad Sci USA. 107:1506–1511. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ng WL, Yan D, Zhang X, Mo YY and Wang Y:
Over-expression of miR-100 is responsible for the low-expression of
ATM in the human glioma cell line: M059J. DNA Repair (Amst).
9:1170–1175. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yan D, Ng WL, Zhang X, Wang P, Zhang Z, Mo
YY, Mao H, Hao C, Olson JJ, Curran WJ and Wang Y: Targeting
DNA-PKcs and ATM with miR-101 sensitizes tumors to radiation. PLoS
One. 5:e113972010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Song L, Lin C, Wu Z, Gong H, Zeng Y, Wu J,
Li M and Li J: miR-18a impairs DNA damage response through
downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS
One. 6:e254542011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsuchida A, Ohno S, Wu W, Borjigin N,
Fujita K, Aoki T, Ueda S, Takanashi M and Kuroda M: Cancer miR-92
is a key oncogenic component of the miR-17-92 cluster in colon
cancer. Cancer Sci. 102:2264–2271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Klionsky DJ, Abeliovich H, Agostinis P, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy in higher eukaryotes. Autophagy. 4:151–175.
2008. View Article : Google Scholar
|
|
15
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Corradetti MN and Guan KL: Upstream of the
mammalian target of rapamycin: do all roads pass through mTOR?
Oncogene. 25:6347–6360. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu WK, Coffelt SB, Cho CH, Wang XJ, Lee
CW, Chan FK, Yu J and Sung JJ: The autophagic paradox in cancer
therapy. Oncogene. 31:939–953. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chaachouay H, Ohneseit P, Toulany M,
Kehlbach R, Multhoff G and Rodemann HP: Autophagy contributes to
resistance of tumor cells to ionizing radiation. Radiother Oncol.
99:287–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ko H, Kim YJ, Amor EC, Lee JW, Kim HC, Kim
HJ and Yang HO: Induction of autophagy by dimethyl cardamonin is
associated with proliferative arrest in human colorectal carcinoma
HCT116 and LOVO cells. J Cell Biochem. 112:2471–2479. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yuk JM, Shin DM, Song KS, Lim K, Kim KH,
Lee SH, Kim JM, Lee JS, Paik TH, Kim JS and Jo EK: Bacillus
calmette-guerin cell wall cytoskeleton enhances colon cancer
radiosensitivity through autophagy. Autophagy. 6:46–60. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Høyer-Hansen M and Jäättelä M: Autophagy:
an emerging target for cancer therapy. Autophagy. 4:574–580.
2008.
|
|
22
|
Xiao J, Zhu X, He B, Zhang Y, Kang B, Wang
Z and Ni X: MiR-204 regulate cardiomyocyte autophagy induced by
hypoxia-reoxygenation through LC3-II. Int J Cardiol. 148:110–112.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shiloh Y: ATM and related protein kinases:
safeguarding genome integrity. Nat Rev Cancer. 3:155–168. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Alexander A and Walker CL: The role of
LKB1 and AMPK in cellular responses to stress and damage. FEBS
Lett. 585:952–957. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou WJ, Deng R, Zhang XY, Feng GK, Gu LQ
and Zhu XF: G-quadruplex ligand SYUIQ-5 induces autophagy by
telomere damage and TRF2 delocalization in cancer cells. Mol Cancer
Ther. 8:3203–3213. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dong J, Zhao YP, Zhou L, Zhang TP and Chen
G: Bcl-2 upregulation induced by miR-21 via a direct interaction is
associated with apoptosis and chemoresistance in MIA PaCa-2
pancreatic cancer cells. Arch Med Res. 42:8–14. 2011. View Article : Google Scholar
|
|
27
|
Wickramasinghe NS, Manavalan TT, Dougherty
SM, Riggs KA, Li Y and Klinge CM: Estradiol downregulates miR-21
expression and increases miR-21 target gene expression in MCF-7
breast cancer cells. Nucleic Acids Res. 37:2584–2595. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vasudevan S, Tong Y and Steitz JA:
Switching from repression to activation: microRNAs can up-regulate
translation. Science. 318:1931–1934. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vasudevan S, Tong Y and Steitz JA: Cell
cycle control of microRNA-mediated translation regulation. Cell
Cycle. 7:1545–1549. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jemal A, Siegel R, Ward E, Murray T, Xu J
and Thun MJ: Cancer statistics. CA Cancer J Clin. 57:43–66.
2007.
|
|
31
|
Hermsen M, Postma C, Baak J, Weiss M,
Rapallo A, Sciutto A, Roemen G, Arends JW, Williams R, Giaretti W,
De Goeij A and Meijer G: Colorectal adenoma to carcinoma
progression follows multiple pathways of chromosomal instability.
Gastroenterology. 123:1109–1119. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bartkova J, Horejsí Z, Koed K, Krämer A,
Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C,
Ørntoft T, Lukas J and Bartek J: DNA damage response as a candidate
anti-cancer barrier in early human tumorigenesis. Nature.
434:864–870. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bartek J, Bartkova J and Lukas J: DNA
damage signalling guards against activated oncogenes and tumour
progression. Oncogene. 26:7773–7779. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ota A, Tagawa H, Karnan S, Tsuzuki S,
Karpas A, Kira S, Yoshida Y and Seto M: Identification and
characterization of a novel gene, C13orf25, as a target for
13q31–q32 amplification in malignant lymphoma. Cancer Res.
64:3087–3095. 2004.
|
|
35
|
Hayashita Y, Osada H, Tatematsu Y, Yamada
H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y and
Takahashi T: A polycistronic microRNA cluster, miR-17-92, is
overexpressed in human lung cancers and enhances cell
proliferation. Cancer Res. 65:9628–9632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van Haaften G and Agami R: Tumorigenicity
of the miR-17-92 cluster distilled. Genes Dev. 24:1–4.
2010.PubMed/NCBI
|
|
37
|
Fu LL, Wen X, Bao JK and Liu B:
MicroRNA-modulated autophagic signaling networks in cancer. Int J
Biochem Cell Biol. 44:733–736. 2012. View Article : Google Scholar : PubMed/NCBI
|