Introduction
There is evidence that oxidative stress is a major
stimulus in the pathogenesis of cardiovascular diseases, including
atherosclerosis, hypertension, myocardial infarction and heart
failure (1,2). Hypoxia and ischemia may increase the
production of intracellular reactive oxygen species (ROS) and
consequently induce cardiomyocyte necrosis and apoptosis (3,4).
Therefore, the development of an effective antioxidant strategy to
reduce oxidative stress, which has attracted considerable interest
from investigators, may provide insights into delaying or
preventing cardiomyocyte death in ischemic heart diseases (5).
The mammalian target of rapamycin (mTOR) is the
central controlling mechanism of cellular growth and metabolism.
mTOR stimulates protein synthesis and cell growth through the
inhibition of eukaryotic initiation factor 4E-binding protein-1
(4EBP1) and the activation of ribosomal protein S6 kinase (S6K)
(6–8). The Akt-mediated activation of the
mTOR pathway is a major regulator of the cellular response to
hypoxia and other microenvironmental stresses (9). Several proteins are involved in the
regulation of mTORC1 activity, including proline-rich Akt substrate
of 40 kDa (PRAS40), DEP domain-containing mTOR-interacting protein
(DEPTOR) or regulated in development and DNA damage responses-1
(REDD1) (10–12).
REDD1, as a hypoxia-regulated HIF-1 target gene, is
important in the TSC1/TSC2-mediated inhibition of mTOR (13). REDD1 is markedly induced by
numerous stress stimuli, including hypoxia, oxidative stress and
energy depletion. More recent studies have suggested that REDD1
functions as a direct regulator of mitochondrial metabolism;
however, the molecular mechanisms by which cell stress and hypoxia
induce REDD1 expression have yet to be determined (14,15).
Salidroside (Fig.
1), as an adaptogen purified from Rhodiola rosea (R.
rosea), has been reported to exert antioxidative, anti-aging,
neuroprotective and cardioprotective effects, implying that
salidroside may play a central role in the alleviation of
mitochondrial-generated ROS and modulation of mitochondrial-related
apoptosis signaling in multiple types of cells (16). A previous study has demonstrated
that pretreatment with salidroside upregulated the HIF-1α protein
and induced its translocation in order to protect cardiomyocytes
against hypoxia-induced injury (17). Nevertheless, the mechanism
underlying its antioxidative effects remains unclear and requires
further investigation.
In the present study, we investigated the potential
anti-apoptosis activity of salidroside using human umbilical vein
endothelial cells (HUVECs). Our results indicated that salidroside
markedly inhibits hydrogen peroxide
(H2O2)-induced HUVEC apoptosis and ROS
production. Furthermore, we demonstrated that the anti-apoptosis
effect of salidroside may be mediated by activation of the
PI3K/Akt/mTOR pathway. Furthermore, the antioxidative effect of
salidroside is dependent on REDD1 activity and requires the
transcription factor HIF-1α.
In conclusion, our results demonstrated that the
activation of REDD1 by salidroside affects cell viability and ROS
production, and may be a promising candidate for antioxidant
therapy in cardiovascular disease.
Materials and methods
Materials
Salidroside, an active component of the medicinal
plant R. rosea, with 99% purity confirmed by HPLC, was
purchased from the National Institute for the Control of
Pharmaceutical and Biological Products (Beijing, China);
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and
Lipofectamine™ 2000 were purchased from Invitrogen Life
Technologies (Carlsbad, CA, USA); FITC-conjugated annexin-V was
purchased from BD Biosciences (San Jose, CA, USA); and LY294002 was
purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture and treatment
HUVECs (ATCC, Teddington, UK) were cultured in F-12K
medium with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM
L-glutamine, 30 μg/ml endothelial cell growth supplement and 10%
FBS at 37°C in 5% CO2 and 95% humidity. For
H2O2 treatment, cells were first screened
with increasing doses of H2O2 and the optimal
concentration (200 μmol) was selected for further experiments. For
antioxidant treatment, the optimal concentration (100 μg/ml) of
salidroside was selected for further experiments. Cells were
cultured with salidroside or a saline vehicle for 24 h, followed by
H2O2 (200 μmol) treatment. For LY294002
treatment, cells were cultured with salidroside followed by
LY294002 and H2O2 treatment.
Plasmid construction, lentiviral
packaging and infection
To silence REDD1 expression, we constructed a
vector-based REDD1-shRNA to interfere with the expression of REDD1.
The oligonucleotides used were
5′-CCGGTGATGCCTAGCCAGTTGGTAACTCGAGTTACCAACTGGCTAGGCATCATTTTTG-3′
and
5′-AATTCAAAAATGATGCCTAGCCAGTTGGTAACTCGAGTTACCAACTGGCTAGGCATCA-3′,
and were cloned into the AgeI and EcoRI sites of the
pLKO.1-puro vector (Sigma Aldrich). Lentiviruses were created by
co-transfecting HEK 293T cells with a packaging vector. Lentiviral
supernatants were collected 48 h post-transfection and target cells
were infected with 3 ml virus supernatant containing 8 μg/ml
polybrene for 24 h. The media was replaced with complete medium.
Puromycin (3 μg/ml) was added to the cell culture media two days
after infection. The knockdown efficiency was monitored by western
blot analysis. The formation of REDD1-shRNA was confirmed using DNA
sequencing.
Cell viability assay
Cells were seeded in 96-well culture plates at
104 cells/well and allowed to attach to the plates. The
culture medium was removed and renewed with fresh medium containing
H2O2 at various concentrations, ranging from
0–400 μM and incubated for 6 h. For the salidroside assays, cells
were pretreated with salidroside for 24 h prior to the addition of
H2O2. Cells receiving saline served as a
vehicle control and were equivalent to no treatment. The Cell
Counting kit-8 (CCK-8) reagent (Sigma Aldrich) was added and
incubated at 37°C for 2 h according to the manufacturer’s
instructions. Optical density (OD) values at 450 nm were read by a
microplate reader. Each experiment was performed three times
independently.
Flow cytometric assays
To evaluate apoptosis in HUVECs, flow cytometric
analysis was performed using annexin-V. Cells were treated with 100
μg/ml salidroside or saline for 24 h, then 200 μmol
H2O2 was added. Cells were detached by
exposure to trypsin 6 h later. Apoptosis was detected in cells
washed with PBS by staining with annexin-V and propidium iodide
according to the manufacturer’s instructions (BD Biosciences).
Stained cells were analyzed using flow cytometry with CellQuest
(Becton-Dickinson, Franklin Lakes, NJ, USA) software.
The cells, which had been treated or remained
untreated with salidroside or H2O2, were
incubated with a chloromethyl derivative of H2DCFDA
(CM-H2DCFDA; Invitrogen Life Technologies) in the dark
for 30 min at 37°C. Following washing, the ROS level of cells was
immediately analyzed by flow cytometry.
Protein extraction and western blot
analysis
Cells were washed three times with ice-cold PBS and
subsequently lysed in RIPA buffer containing 1 mM PMSF. The
supernatant was collected and the protein concentration of each
sample was determined using a BCA Protein Assay kit (Pierce
Biotechnology, Inc., Rockford, IL, USA) according to the
manufacturer’s instructions. Total proteins (50 μg) were loaded
onto 10% sodium dodecyl sulfate-polyacrylamide gels for
electrophoresis and transferred onto 0.22-μm PVDF membranes
(Millipore, Billerica, MA, USA), which were blocked with 8% non-fat
milk. The primary antibodies used to probe the membranes included
anti-HIF-1α [Cell Signaling Technology, Inc. (CST), Beverley, MA,
USA; #3716], anti-REDD1 (ProteinTech Group, Chicago IL, USA;
10638-1-AP), anti-Akt (CST, #9272), anti-p-Akt (CST, #9271),
anti-p-p70S6K (CST, #6198), anti-p-mTOR (CST, #2971) and anti-GAPDH
(ProteinTech Group, 60004-1-Ig) at 4°C overnight. Following
washing, the membranes were incubated for 1 h at room temperature
with horseradish peroxidase-conjugated secondary antibody. Proteins
were detected using an advanced enhanced chemiluminescence (ECL)
system (GE Healthcare, Chalfont St. Giles, UK).
Statistical analysis
The results of all experiments are expressed as the
means ± SD from individual experiments. Data were compared using
the Student’s t-test using GraphPad Prism 5.0 software (GraphPad
Software Inc., San Diego, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Salidroside attenuates
H2O2-induced HUVEC cytotoxicity and excessive
generation of ROS
In order to investigate the effect of salidroside on
the growth of endothelial cells, we evaluated the effect of
salidroside on the viability of HUVECs using the CCK-8 assay.
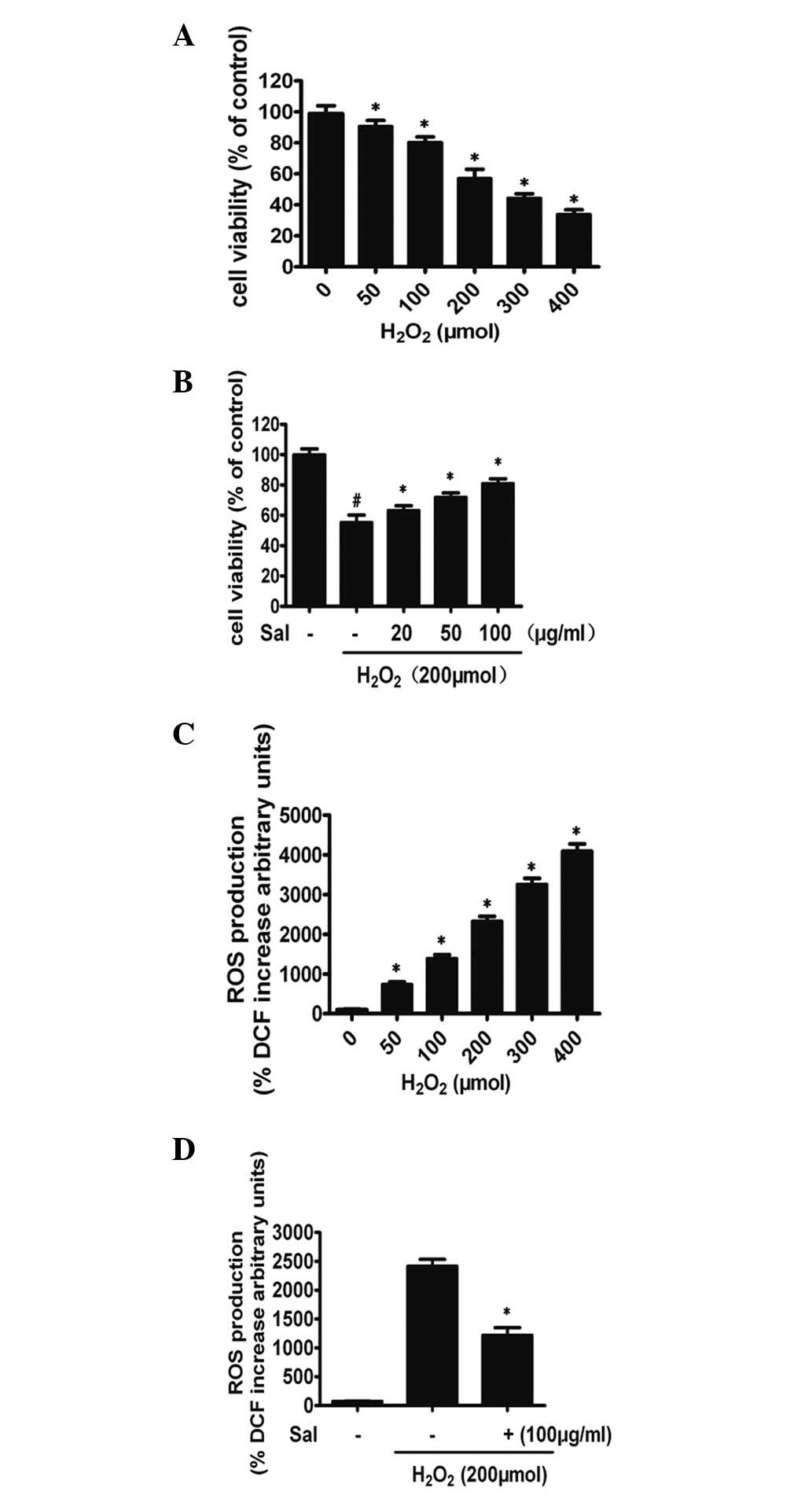
H2O2 (50–400 μmol) significantly decreased
the cell viability of HUVECs in a concentration-dependent manner
(P<0.01; Fig. 2A). When exposed
to 200 μmol H2O2 for 6 h, HUVEC viability
(61.9±4.1%, n=3) decreased by 38.1% compared with the control group
(P<0.01, both n=3). Therefore, 200 μmol of
H2O2 was used as the optimal concentration in
the subsequent experiments. To examine the concentration-dependent
effect of salidroside, cells were pretreated with 20, 50 or 100
μg/ml salidroside or saline vehicle for 24 h prior to being exposed
to 200 μmol H2O2 for 6 h. The decrease in
HUVEC viability induced by H2O2 was improved
significantly by treatment with salidroside in a
concentration-dependent manner. A significant increase in cell
viability was evident following pretreatment with 100 μg/ml of
salidroside (Fig. 2B). The optimal
concentration (100 μg/ml) of salidroside was selected for further
experiments. In addition, salidroside at each concentration applied
alone did not cause any apparent cytotoxicity (data not shown). The
dichlorofluorescein (DCFH) assay results indicated that salidroside
reduced the levels of ROS in HUVECs under
H2O2. When HUVECs were subjected to
H2O2 for 6 h, the concentration of
intracellular ROS increased, compared with the control group
(P<0.01; Fig. 2C). However,
preincubation with 100 μg/ml salidroside for 24 h followed by
coincubation with 200 μmol H2O2 for a further
6 h markedly decreased the H2O2-induced ROS
level. ROS production decreased by 22.9% compared with cells
treated with H2O2 alone (P<0.01; Fig. 2D).
Salidroside inhibits
H2O2-induced HUVEC apoptosis
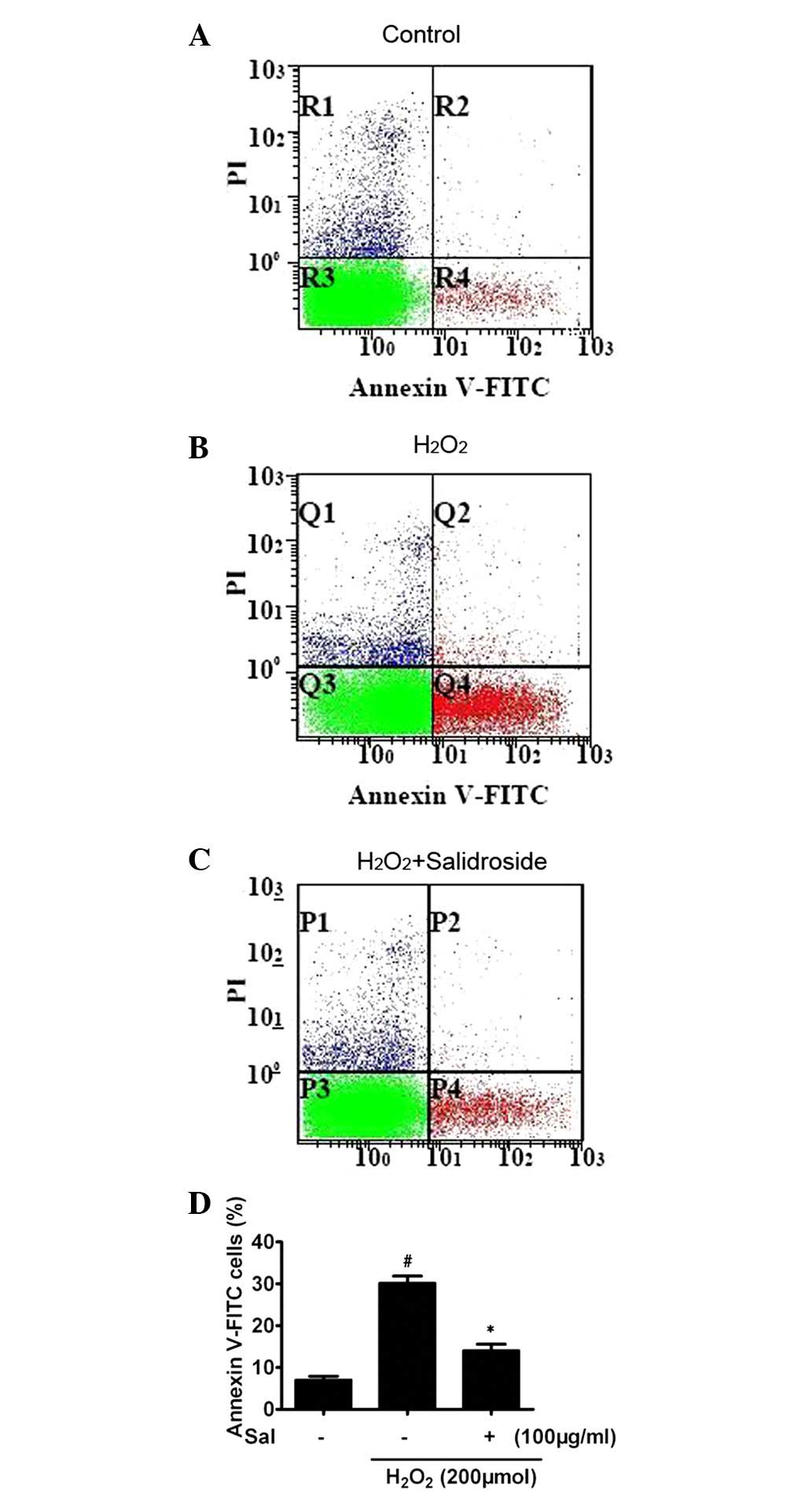
CCK-8 does not discriminate between necrosis and
apoptosis. We evaluated the possible anti-apoptotic effect of
salidroside on HUVECs subjected to H2O2 using
annexin-V/PI double staining and flow cytometry analysis. In the
control group, the majority of cells were viable (93.7%).
H2O2 increased the percentage of apoptotic
cells. Compared with the control group, 6 h stimulation of
H2O2-induced apoptosis produced a 5.3-fold
increase in apoptotic cells (6.5±0.8%, P<0.01, n=3; Fig. 3A and B). Pretreatment with
salidroside for 24 h and cotreatment with
H2O2 for a further 6 h significantly
attenuated the apoptosis induced by H2O2,
with 51.5% fewer apoptotic cells compared with the cells treated
with H2O2 alone (P<0.01, n=4; Fig. 3C and D).
Salidroside increases the expression of
REDD1 protein and activates the Akt/mTOR pathway
HIF-1α reduces the production of ROS under hypoxic
conditions, which mediate the cells adaptive response to hypoxia
and ischemic. It is doubtful as to whether REDD1 is also associated
with the process of salidroside protection against
H2O2-induced ROS. Therefore, we investigated
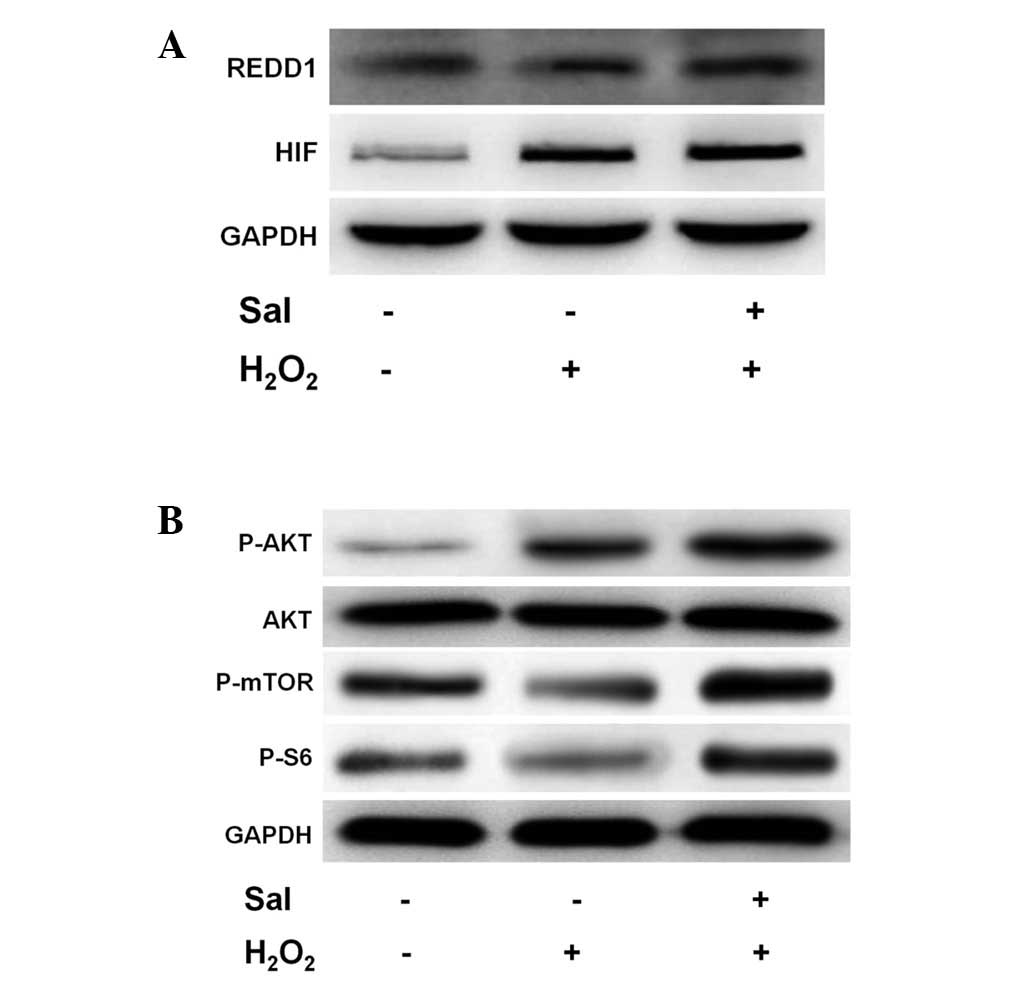
the levels of REDD1 and consequently, the expression of HIF-1α in
response to H2O2 and salidroside. HUVECs were
stimulated with salidroside for 24 h and subsequently with
H2O2 for a further 6 h; following this, the
REDD1 and HIF-1α protein levels were analyzed. Incubation of HUVECs
in H2O2 stimulated REDD1 expression (Fig. 4A). Notably, this expression was
enhanced when the HUVECs were incubated with salidroside under
H2O2. This result indicates that REDD1 may be
activated under H2O2 conditions and
salidroside. Similarly, H2O2 and salidroside
conditions led to an increase in the levels of HIF-1α protein,
consistent with previous findings.
In order to fully elucidate the potential signaling
pathway involved in anti-apoptotic induction by salidroside under
oxidative stress conditions in HUVECs, we examined the implication
of protein kinases activated in response to salidroside. The mTOR
pathway has been reported to enhance translation and transcription,
enabling cell growth. Therefore, we investigated whether
salidroside activated mTOR in the HUVECs treated with
H2O2, and if so, whether Akt modulates this
activation. Therefore, HUVECs were pretreated with salidroside and
subjected to 6 h of treatment with H2O2. We
performed western blot analysis in order to detect the effect of
salidroside on the phosphorylation of Akt and mTOR in HUVECs. We
observed that H2O2 and salidroside
pretreatment increased the expression of phospho-Akt (serine 473;
Fig. 4B). However, mTOR and S6
phosphorylation decreased significantly in HUVECs treated with
H2O2 alone. Notably, phosphorylation levels
were significantly elevated following pretreatment with salidroside
(Fig. 4B).
Antioxidative effect of salidroside is
dependent on REDD1 activation
To determine whether salidroside-induced
antioxidatives require the function of REDD1, we examined the
effect of transfecting HUVECs with siRNAs targeting REDD1.
Initially, we examined the effect of siRNAs targeting REDD1.
Western blot analysis revealed that siRNAs were highly effective at
decreasing REDD1 protein levels (Fig.
5A). Furthermore, REDD1 siRNAs significantly reduced the
salidroside-induced antioxidative effect (Fig. 5B). Thus, salidroside protects
against oxidative stress-induced cell death and this protective
effect is dependent upon REDD1 activation. These data also confirm
that activation of REDD1 is protective against oxidative
stress.
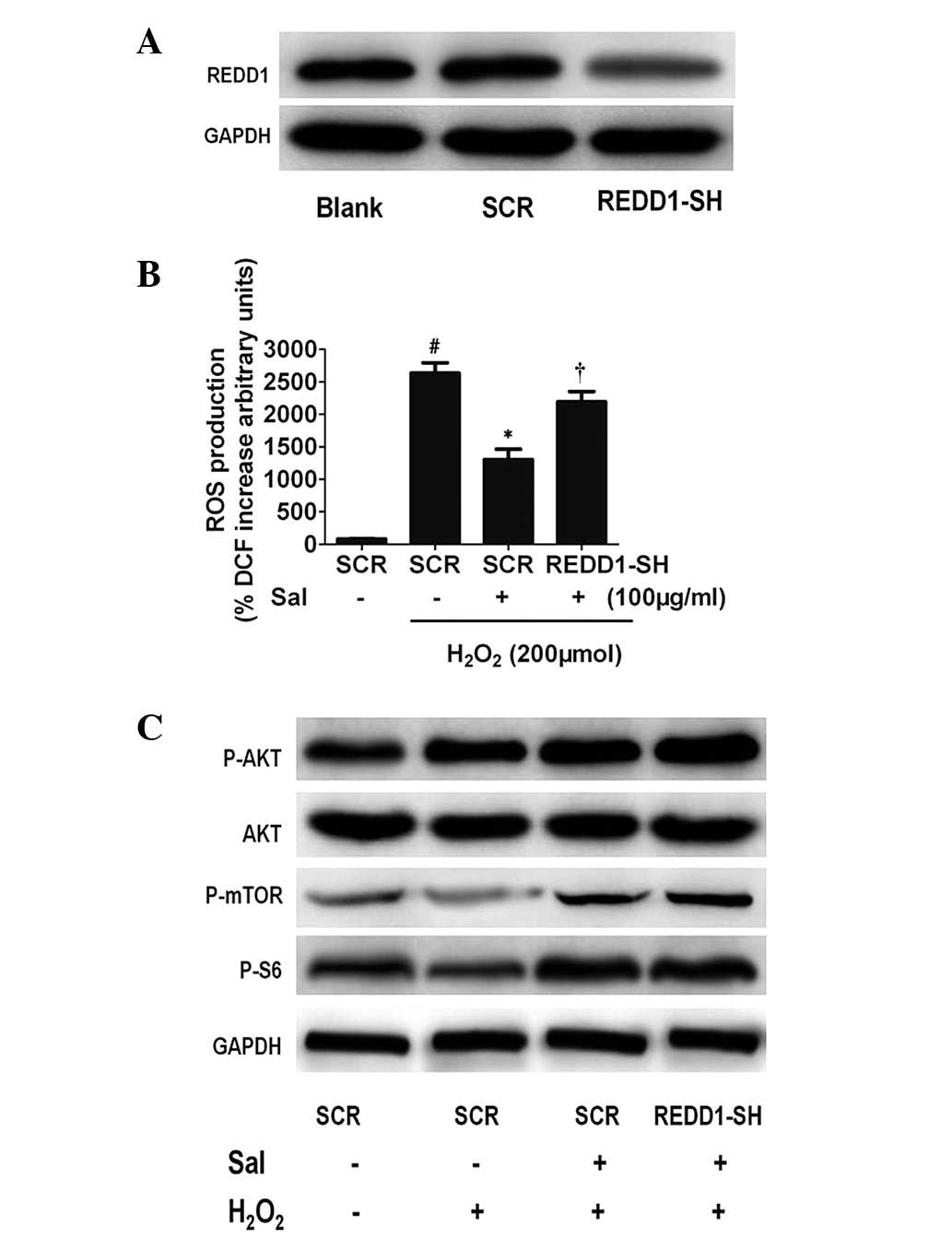 | Figure 5The antioxidative effect of Sal is
dependent on REDD1 activation. (A) Knockdown of REDD1 in HUVECs by
lentiviral REDD1-directed shRNA (shREDD1) or the control group,
assessed by western blot analysis. GAPDH served as a loading
control. (B) REDD1 knockdown decreases the antioxidative effect of
Sal under H2O2. Equal numbers of the
indicated REDD1−/− HUVECs were treated under
H2O2 with or without Sal, and increased ROS
production in whole REDD1−/− versus wild-type
cells was measured using DCFH. #P<0.01, compared with
the control. *P<0.01, compared with the
H2O2 treatment group. †P<0.01,
compared with the SCR group. (C) Western blot analysis shows that
Akt/mTOR pathway expression remained upregulated in REDD1-depleted
HUVECs as well as untransfected and control siRNA HUVECs subjected
to hypoxic stress. REDD1, regulated in development and DNA damage
responses-1; HUVECs, human umbilical vein endothelial cells; ROS,
reactive oxygen species; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase, DCFH, dichlorofluorescein; Sal, salidroside. |
PI3K participates in the regulation of
mTOR expression by salidroside
To determine whether PI3K activation is involved in
the induction of mTOR expression by salidroside, we treated HUVECs
with the PI3K inhibitor LY294002. We subjected HUVECs to
salidroside for 24 h under H2O2 conditions in
the absence or presence of inhibitors, and mTOR expression was
detected by immunoblotting. Salidroside and
H2O2 stimulate Akt expression, and the
combination of the treatments led to an additive effect. However,
LY294002 blocked the effect of salidroside on the expression of
mTOR. Furthermore, LY294002 did not prevent the effect of ROS
production (Fig. 6). These results
suggested that in HUVECs, salidroside stimulates mTOR expression
through PI3K/mTOR signaling pathways. These data demonstrate that
Akt may be a central regulator of the mTOR pathway for the
anti-apoptotic effect of salidroside.
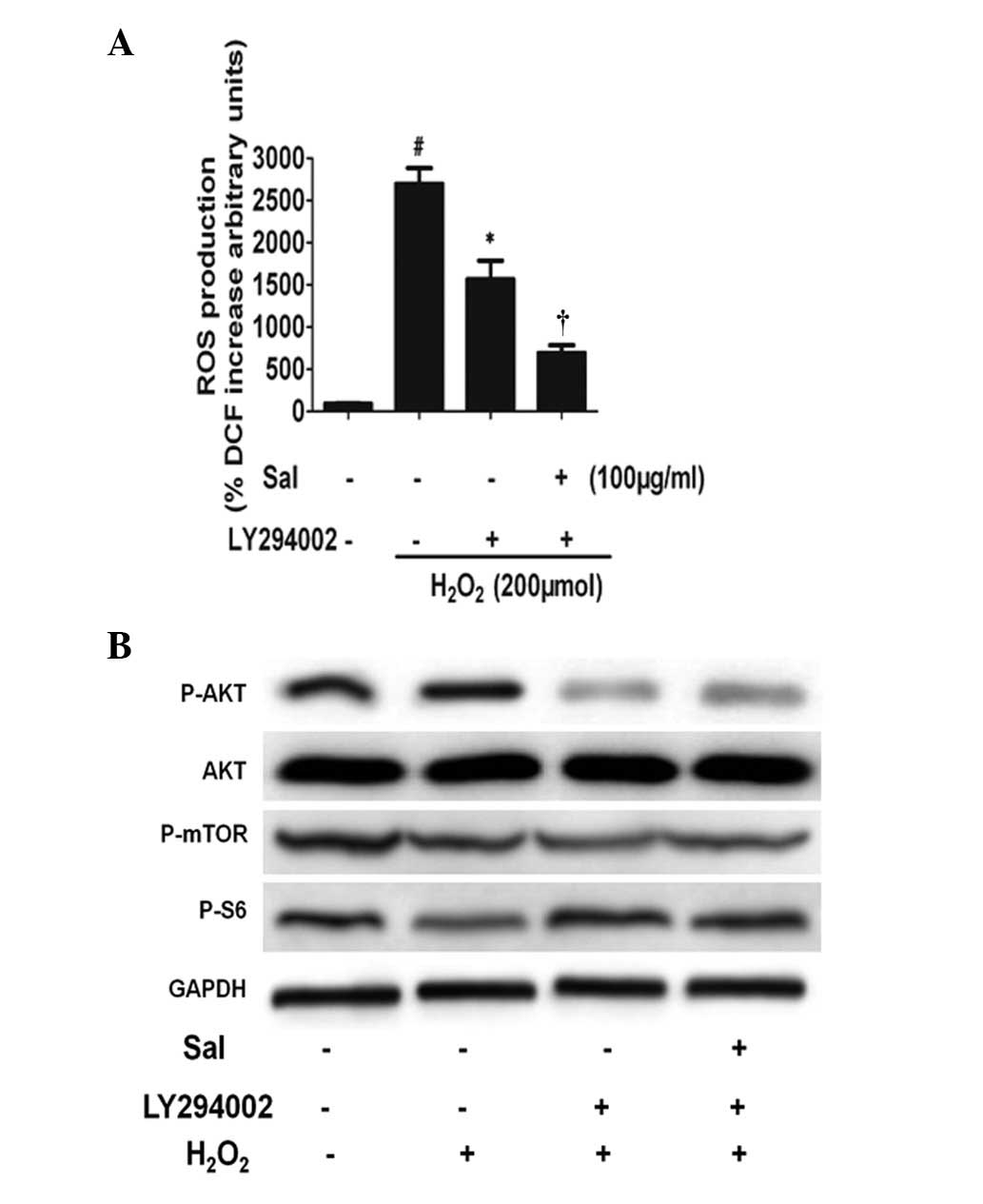 | Figure 6The PI3K pathway participates in the
regulation of mTOR expression by Sal. (A) Changes in ROS production
induced by either Sal or H2O2 treatment alone
or in combination in the absence and presence of LY294002,
respectively. HUVECs were preincubated with Sal, and then
coincubated with H2O2 for a further 6 h, and
LY294002 was added 30 min prior to treatment with Sal.
#P<0.01, compared with the control.
*P<0.01, compared with the H2O2
treatment group. †P<0.01, compared with the
H2O2 + LY294002 treatment group. (B)
Activation of the mTOR pathway by Sal is dependent on PI3K/Akt.
PI3K/Akt acted in parallel to activate the mTOR pathway following
treatment with Sal. The PI3K inhibitor LY294002 blocks the increase
in mTOR and PS6 protein levels induced by Sal under
H2O2 conditions. Levels of phosphorylated Akt
(Ser473), mTOR and p-p70S6K were determined by immunoblotting.
Total Akt and GAPDH expression are shown as sample loading
controls. A representative experiment of three independent
experiments is shown. HUVECs, human umbilical vein endothelial
cells; ROS, reactive oxygen species; GAPDH, glyceraldehyde
3-phosphate dehydrogenase; Sal, salidroside. |
Discussion
In the present study, we demonstrated that increased
REDD1 expression in response to salidroside may participate in the
mechanism by which salidroside inhibited
H2O2-induced HUVEC oxidative stress, and
salidroside-induced mTOR expression occurs through the PI3K/mTOR
signaling pathway to protect HUVECs against apoptosis induced by
H2O2.
The necrotic and apoptotic processes induced by
oxidative stress are involved in the pathogenesis of cardiac
ischemic-reperfusion injury, which may ultimately lead to heart
failure (1,18). Our present study confirmed that
H2O2 significantly decreases cell viability,
as evidenced by CCK-8, whereas pretreatment with various
concentrations (10, 50 and 100 μg/ml) of salidroside attenuated the
H2O2-decreased cell viability in a
dose-dependent manner. These results indicated that salidroside
exhibited an apparent protective effect against
H2O2-induced cytotoxicity and inhibited ROS
production in response to H2O2 in HUVECs.
Recently, there has been increased interest in the
role of the mTOR pathway in the heart. While the evidence is
mounting that the activation of mTOR may be an important cellular
signaling event for preventing apoptotic processes, the role of
mTOR in regulating cardioprotection is less clear in that it has
been examined in a number of settings with what appear to be
disparate conclusions (19,20).
Recent studies have examined the role of Akt in the protective
effect of salidroside, and the conclusions reached were that
salidroside prevented apoptosis in neuronal and cardiac cells,
through the activation of the PI3K/Akt pathway (21,22).
These findings indicated the regulation of Akt by salidroside,
which aroused our interest in the role of mTOR and the downstream
target, p70S6K, in the heart. In addition, examination of the
regulation of salidroside may be crucial in identifying the
molecular mechanisms that regulate salidroside in the heart under
conditions of oxidative stress. Thus, we aimed to further
characterize the regulation of mTOR by salidroside.
Therefore, we determined whether salidroside
affected the phosphorylation of the mTOR pathway in HUVECs. In this
study, we identified one key anti-apoptotic role for salidroside
under H2O2: The activation of mTOR. The
activation of mTOR pathway members was analyzed by western blot
analysis using the specific antibody against the phosphorylated
form of mTOR and SP6. We observed that treatment of HUVECs with
salidroside increased the level of mTOR phosphorylation and
SP6.
Previous studies have reported that salidroside
induced the anti-apoptotic effect, so we also examined the cell
apoptosis of HUVECs exposed to salidroside. As shown in Fig. 3, following the treatment of the
cells with an increased concentration of salidroside for 24 h, a
significant decrease in apoptosis occurred in HUVECs, which may be
correlated to the salidroside-induced upregulation of mTOR.
We demonstrated that oxidative stress inactivates
mTOR. Numerous studies have demonstrated that this inactivation is
dependent on HIF activity and HIF-inducible REDD1 (12,13,23,24).
REDD1, an important HIF-1 effector, is important in the TSC1
(hamartin)/TSC2 (tuberin)-mediated inhibition of mTOR (13). However, Hernández et al
discovered that REDD-1 is downregulated by ROS by inactivating TSC2
and activating mTOR (25).
However, our results indicate that stimulation with
H2O2 increased REDD1 expression, which
coincided with the upregulated expression of HIF-1α in HUVECs.
Furthermore, we observed that pretreatment with salidroside
followed by coincubation with H2O2, increased
REDD1 and HIF-1α levels compared with those treated by
H2O2 alone. The synergy between salidroside
and H2O2 is due to the fact that they
regulate REDD1 through distinct mechanisms. Although our data
support a regulatory role for REDD1 under oxidative stress
conditions, we observed the upregulation of mTOR following
treatment with salidroside, which varies from the mTOR inhibition
of oxidative stress occurring independently of REDD1. We propose
that mTOR regulation involves multiple pathways, including
mediation by the AMPK/TSC2/Rheb pathway and inhibition by the
HIF/REDD1/2 pathway or the other HIF-independent pathways affected
by mTOR, including PI3K/Akt. In this regard, the elucidation of the
basic mechanisms affecting this network of signaling molecules will
offer essential information to improve our understanding of
salidroside as well as supporting the pharmacological
mechanism-based application of salidroside.
Akt is an important survival kinase in cell
proliferation and survival, which is activated downstream of PI3K
(26). Akt-mediated protection
inhibits cardiomyocyte necrotic and apoptotic death induced by
deleterious stimuli (27). The
existence of several possible routes by which Akt modulates the
mTOR pathway suggests that this may be a central mechanism of mTOR
regulation. This study aimed to identify whether PI3K signaling
pathways regulated mTOR activity in HUVECs exposed to salidroside
under oxidative stress conditions. Herein, we delineate a novel
signaling pathway regulated by salidroside-activated Akt that leads
to the activation of mTOR. This conclusion is supported by the
following results: i) Under oxidative stress conditions,
salidroside induces markedly higher phosphorylation levels of Akt
compared with those treated by H2O2 alone,
which is similar to the result reported by Zhu et
al(22); ii) LY294002
efficiently suppressed the activation of Akt and the induction of
mTOR expression by salidroside under oxidative stress conditions;
iii) LY294002 inhibited the phosphorylation of mTOR; iv) the
downregulation of mTOR inhibited the activation of SP6; and v) mTOR
expression was unaffected by siRNAs targeting REDD1. We delineated
the pathway upstream of mTOR activation in response to salidroside
under oxidative stress, defining Akt as an essential activator of
the mTOR pathway in this situation.
Due to the fact that REDD1 is essential in limiting
the generation of ROS by an unidentified mechanism and in ROS
attenuation by salidroside, we hypothesized that the mechanism by
which salidroside inhibits H2O2-induced
oxidative stress in HUVECs lies in the upregulation of the REDD1
protein. To more clearly define the role of REDD1, we examined the
production of ROS in REDD1-knockdown HUVECs. The
REDD1−/− cells demonstrated a substantial
elevation in ROS production, and treatment with salidroside was
insufficient to normalize the ROS levels to wild-type levels
(Fig. 5).
In conclusion, we demonstrated that salidroside
protects HUVECs against oxidative injury induced by
H2O2 and that the potential mechanisms may
involve increasing REDD1 expression to prevent the generation of
ROS, modulating the expression of HIF-1α and regulating the
activation of the PI3K/Akt pathway followed by activating the
downstream molecules of mTOR and SP6 to reduce
H2O2-induced apoptosis. These combined
effects may explain the marked protective effect of
salidroside.
Acknowledgements
This study was financially supported by the funding
of the Natural Science Foundation of China (no. 81001561).
References
|
1
|
Hirooka Y, Sagara Y, Kishi T and Sunagawa
K: Oxidative stress and central cardiovascular regulation.
Pathogenesis of hypertension and therapeutic aspects. Circ J.
74:827–835. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stocker R and Keaney JF Jr: Role of
oxidative modifications in atherosclerosis. Physiol Rev.
84:1381–1478. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Suzuki YJ, Jain V, Park AM and Day RM:
Oxidative stress and oxidant signaling in obstructive sleep apnea
and associated cardiovascular diseases. Free Radic Biol and Med.
40:1683–1692. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kumar D and Jugdutt BI: Apoptosis and
oxidants in the heart. J Lab Clin Med. 142:288–297. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu YZ, Huang SH, Tan BK, Sun J, Whiteman
M and Zhu YC: Antioxidants in Chinese herbal medicines: a
biochemical perspective. Nat Prod Rep. 21:478–489. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fingar DC and Blenis J: Target of
rapamycin (TOR): an integrator of nutrient and growth factor
signals and coordinator of cell growth and cell cycle progression.
Oncogene. 23:3151–3171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Proud CG: The multifaceted role of mTOR in
cellular stress responses. DNA Repair (Amst). 3:927–934. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hornberger TA, Stuppard R, Conley KE, et
al: Mechanical stimuli regulate rapamycin-sensitive signalling by a
phosphoinositide 3-kinase-, protein kinase B- and growth
factor-independent mechanism. Biochem J. 380:795–804. 2004.
View Article : Google Scholar
|
|
9
|
Reiling JH and Sabatini DM: Stress and
mTORture signaling. Oncogene. 25:6373–6383. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peterson TR, Laplante M, Thoreen CC, et
al: DEPTOR is an mTOR inhibitor frequently overexpressed in
multiple myeloma cells and required for their survival. Cell.
137:873–886. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vander Haar E, Lee S, Bandhakavi S,
Griffin TJ and Kim DH: Insulin signalling to mTOR mediated by the
Akt/PKB substrate PRAS40. Nat Cell Biol. 9:316–323. 2007.PubMed/NCBI
|
|
12
|
Sofer A, Lei K, Johannessen CM and Ellisen
LW: Regulation of mTOR and cell growth in response to energy stress
by REDD1. Mol Cell Biol. 25:5834–5845. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brugarolas J, Lei K, Hurley RL, et al:
Regulation of mTOR function in response to hypoxia by REDD1 and the
TSC1/TSC2 tumor suppressor complex. Genes Dev. 18:2893–2904. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shoshani T, Faerman A, Mett I, et al:
Identification of a novel hypoxia-inducible factor 1-responsive
gene, RTP801, involved in apoptosis. Mol Cell Biol. 22:2283–2293.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ellisen LW, Ramsayer KD, Johannessen CM,
et al: REDD1, a developmentally regulated transcriptional target of
p63 and p53, links p63 to regulation of reactive oxygen species.
Mol Cell. 10:995–1005. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu S, Liu M, Gu X and Ding F:
Neuroprotective effects of salidroside in the PC12 cell model
exposed to hypoglycemia and serum limitation. Cell Mol Neurobiol.
28:1067–1078. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang J, Liu A, Hou R, et al: Salidroside
protects cardiomyocyte against hypoxia-induced death: a HIF-1
alpha-activated and VEGF-mediated pathway. Eur J Pharmacol.
607:6–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakayama H, Chen X, Baines CP, et al:
Ca2+- and mitochondrial-dependent cardiomyocyte necrosis
as a primary mediator of heart failure. J Clin Invest.
117:2431–2444. 2007.
|
|
19
|
Buss SJ, Muenz S, Riffel JH, et al:
Beneficial effects of mammalian target of rapamycin inhibition on
left ventricular remodeling after myocardial infarction. J Am Coll
Cardiol. 54:2435–2446. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lajoie C, El-Helou V, Proulx C, Clément R,
Gosselin H and Calderone A: Infarct size is increased in female
post-MI rats treated with rapamycin. Can J Physiol Pharmacol.
87:460–470. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang L, Ding W, Sun H, et al: Salidroside
protects PC12 cells from MPP+-induced apoptosis via
activation of the PI3K/Akt pathway. Food Chem Toxicol.
50:2591–2597. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu Y, Shi YP, Wu D, et al: Salidroside
protects against hydrogen peroxide-induced injury in cardiac H9c2
cells via PI3K-Akt dependent pathway. DNA Cell Biol. 30:809–819.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
DeYoung MP, Horak P, Sofer A, Sgroi D and
Ellisen LW: Hypoxia regulates TSC1/2-mTOR signaling and tumor
suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev.
22:239–251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu L, Cash TP, Jones RG, Keith B,
Thompson CB and Simon MC: Hypoxia-induced energy stress regulates
mRNA translation and cell growth. Mol Cell. 21:521–531. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hernández G, Lal H, Fidalgo M, et al: A
novel cardioprotective p38-MAPK/mTOR pathway. Exp Cell Res.
317:2938–2949. 2011.PubMed/NCBI
|
|
26
|
Wang BH, Shravah J, Luo HL, Raedschelders
K, Chen DD and Ansley DM: Propofol protects against hydrogen
peroxide-induced injury in cardiac H9c2 cells via Akt activation
and Bcl-2 up-regulation. Biochem Biophys Res Commun. 389:105–111.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miyamoto S, Murphy AN and Brown JH: Akt
mediated mitochondrial protection in the heart: metabolic and
survival pathways to the rescue. J Bioenerg Biomembr. 41:169–180.
2009. View Article : Google Scholar : PubMed/NCBI
|