Introduction
Thrombocytopenia-absent radius (TAR) syndrome (MIM
274000) is a rare condition (0.5:100,000 in Spain) (1), characterized by absence of the radii
with the presence of thumbs and thrombocytopenia (2). Numerous studies have identified the
presence of a minimally deleted 200-kb region at chromosome band
1q21.1 in patients with TAR, but it is not sufficient to cause the
phenotype (3,4). A study identified two rare single
nucleotide polymorphisms (SNPs) in the regulatory region of the
RBM8A gene that are involved in TAR syndrome through the reduction
of the expression of the RBM8A-encoded Y14 protein (4). The first allele (rs139428292 G>A),
which is located in the 5′ untranslated region (UTR) region of the
gene, was demonstrated to have a minor allele frequency (MAF) of
3.05%, and the second allele (rs201779890 G>C), located in the
first intron of the gene, exhibited a MAF of 0.42%, in 7,504
healthy individuals from Cambridge BioResource (Cambridge, UK)
(4,5).
Prenatal detection of the disease may be possible
for pregnancies known to be at risk and for pregnancies in which
radial anomalies are identified on routine ultrasound examination,
which has traditionally been used to identify TAR syndrome, using a
variety of molecular genetics methods.
The present study describes the clinical, molecular
and molecular cytogenetic studies of a prenatally diagnosed fetus
with TAR syndrome with compound inheritance of a small (334 kb)
deletion, as detected by array-comparative genomic hybridization
(CGH), and of a 5′UTR low frequency allele (rs139428292) in gene
RBM8A, as detected by Sanger sequencing.
Case report
Case presentation
This case report is presented with the consent of
the patient’s family. A 29-year-old female, gravida 3, para 1, with
a 3.5-year-old healthy child, presented in the first trimester of
pregnancy. Previous family history revealed a pregnancy with
prenatal ultrasound findings consistent with TAR syndrome at 12
weeks. Increased nuchal translucency (NT; 4.9 mm), omphalocele with
intestinal contents and a single umbilical artery were observed.
The hands were in ulnar flexion and there was bilateral absence of
one of the long bones of the forearm (either the radius or ulna),
which was not possible to clearly define at that stage. The
adjusted risk for trisomy 21 was 1 in 54, whereas the adjusted risk
for trisomies 18 and 13 was 1 in 260. Following genetic counseling,
the parents opted for invasive prenatal diagnosis, and chorionic
villus sampling (CVS) was performed. Chromosome analysis revealed a
normal female karyotype (46, XX). No further genetic analysis was
performed. The karyotypes of the parents were normal. Pregnancy was
terminated by choice at 13 weeks. There was no pathological
examination of the fetus due to the lack of parental consent.
In the presented pregnancy, ultrasound examination
at 13 weeks of gestation revealed an increased NT of 8.1 mm. The
fetal crown-rump length was 53.1 mm and the fetal heart rate was
171 beats per minute. The upper limbs were shorter than normal, but
no other structural defects were identified during the first
trimester ultrasound examination. Following genetic counseling, the
parents opted for invasive prenatal diagnosis.
Cytogenetics analysis
CVS culture was according to the standard procedures
(6). CVS was performed at 13 weeks
and fetal DNA was extracted directly using InstaGene matrix resin
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The chorionic
villi were cultured and GTG banding (300–400 bands) of chromosomes
revealed a normal male karyotype (46, XY). Due to their previous
history, the parents decided to further investigate with molecular
cytogenetic testing.
Microarray analysis
Array-CGH analysis was conducted on DNA from CVS
using a SurePrint G3 Human GE 8×60k, Oligo Microarray kit that has
a backbone resolution of ~200 kb (Agilent Technologies, Santa
Clara, CA, USA), as described in a previous study (7). The statistical analysis, using
aberration detection method-2 [aberration filter/threshold set at 6
and annotation genomic build NCBI37 (NCBI, Bethesda, MD, USA)],
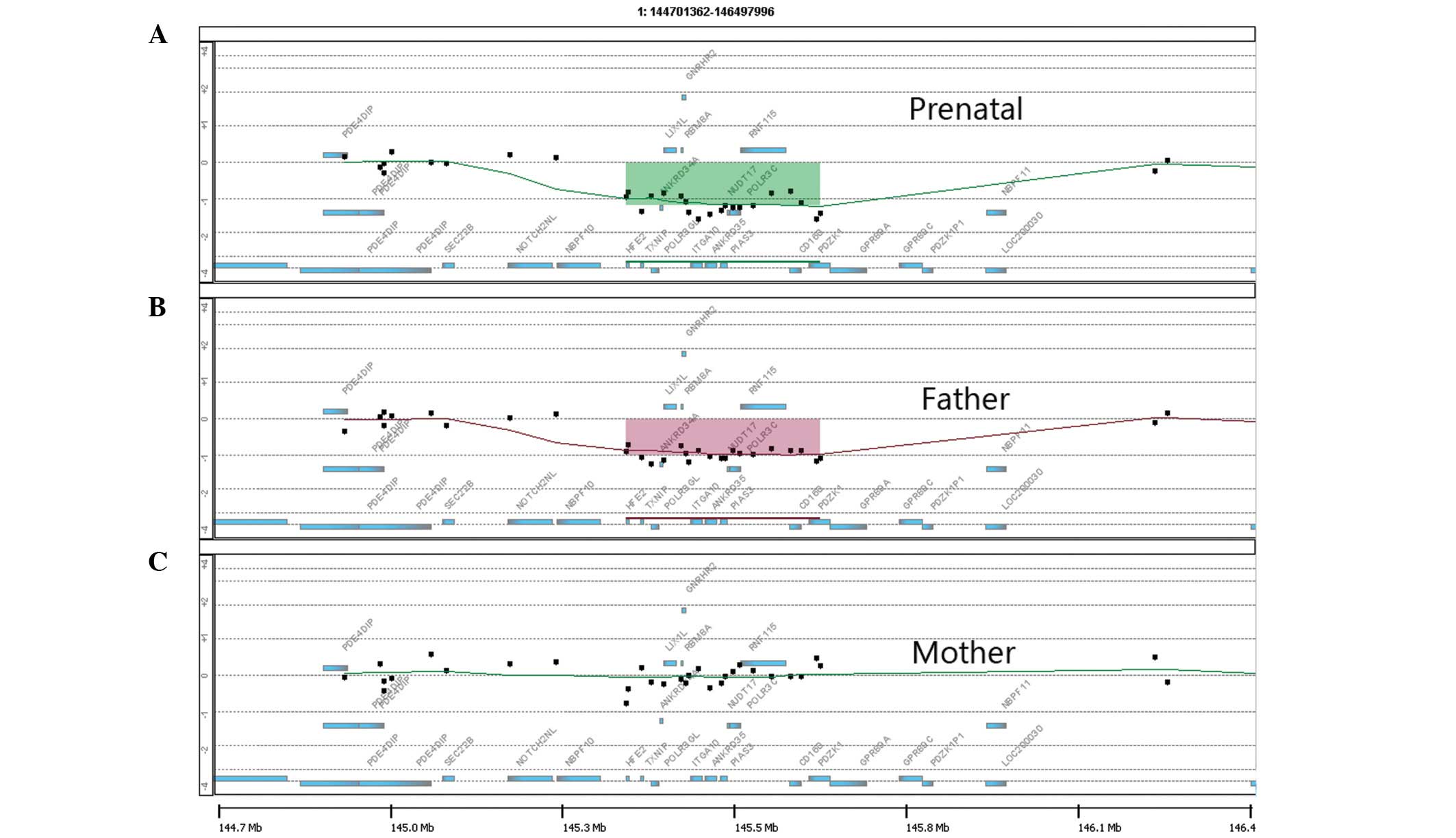
revealed a 334-kb deletion in the long arm of chromosome 1, located
within the 1q21.1 region (chr1:145,413,388-145,747,269) of the
fetus (Fig. 1A). The same
chromosomal abnormality was identified in the father (Fig. 1B) and no abnormal copy number
variations were identified in the mother (Fig. 1C).
qPCR
The array-CGH results were confirmed by copy number
profiling using a qPCR method, as described previously (8). Two genes, POLR3GL
(chr1:145,456,236-145,470,388) and CD160
(chr1:145,695,798-145,715,614), which map to the ends of the
putatively deleted region, were amplified with specific primers
using the LightCycler® FastStart DNA
MasterPLUS SYBR Green I mix (Roche Applied Science,
Roche Diagnostics S.p.A., Monza, Italy). The real-time reactions
were analyzed on a LightCycler® 1.5 (Roche Diagnostics
GmbH, Mannheim, Germany). The concentration of the DNA samples was
adjusted by including two reference genes, EIF3L and KDELR3, that
are located on chromosome 22. Relative quantification, in respect
to a calibration curve used to establish efficiency, was utilized
to detect the number of copies of DNA targets per diploid genome.
All PCR experiments were replicated three times. qPCR demonstrated
the presence of a single copy of the two target genes, POLR3GL
(0.96 ± 0.05) and CD160 (0.97 ± 0.07) per diploid genome of the
fetus, confirming the array-CGH data. The deletion was also
confirmed in the father’s DNA, while the DNA of the mother showed
the presence of two copies of the genes per diploid genome (data
not shown).
Sanger DNA sequencing methods
The sanger method was used to analyze the DNA
sequence of the region spanning the 5′UTR and the first intron of
the RBM8A gene (chr1:145,507,556-145,513,535) in the fetus and
parents. Primers, 5′-ATGGCCACAGAAACACTTCC-3′ (forward) and
5′-GGGCGGAATCTCTA-ATCCAC-3′ (reverse), were selected to include the
two SNPs involved in TAR. The sequencing reactions of the strands
were analyzed on an automatic DNA sequencer Applied Biosystems 3500
Genetic Analyzer for sequence typing and fragment analysis (Life
Technologies Corporation, Foster City, CA, USA). Genotyping of the
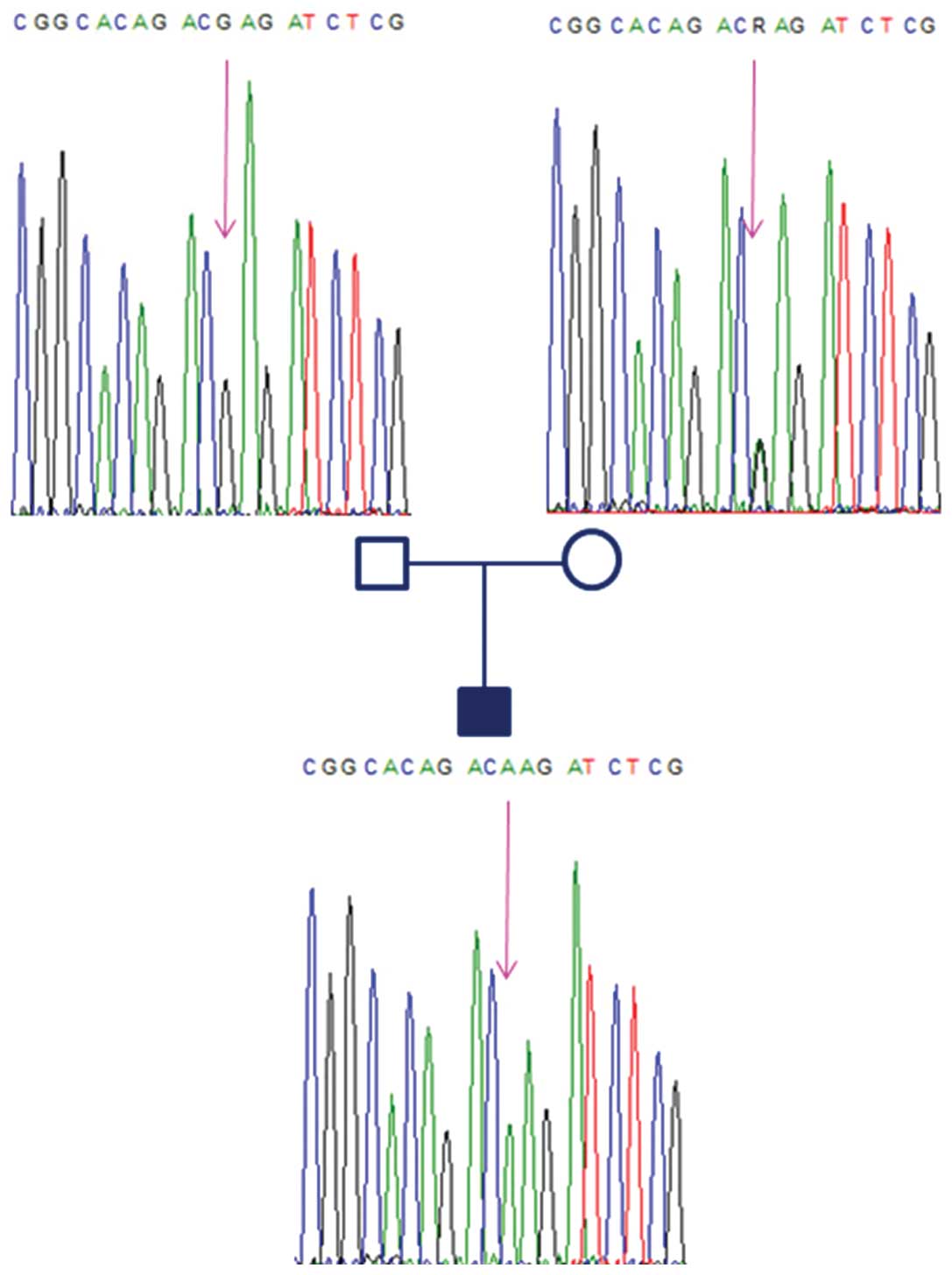
sequence demonstrated the presence of the wild-type G allele in the
father and revealed a heterozygous (G/A) condition at the 5′UTR
polymorphism (rs139428292) in the mother. The father’s genotype was
hemizygous, as he had the deletion of 334 kb, which included the
gene RBM8A. The electropherogram of the fetus only identified the A
allele of maternal origin. Thus, the fetus was affected by TAR
syndrome, as it had inherited the paternal null allele
(microdeletion) and the maternal A allele of the 5′UTR polymorphism
(Fig. 2). All subjects of the
family showed only the wild-type G allele for the intronic SNP
(rs201779890). Following genetic counseling, the parents decided to
terminate the pregnancy at 14 weeks of gestation. The couple did
not consent for fetal pathology assessment.
In the present case the deletion was ~334 kb at
1q21.1 and included nine hypothetical/unknown protein coding genes
according to SWIIS-PROT, TrEMBL and TrEMBL-NEW and their
corresponding mRNAs from Genbank (HFE2, TXNIP, RBM8A, GNRHR2,
PEX11B, ITGA10, PIAS3, CD160 and PDZK1). The RBM8A gene encodes RNA
binding protein Y14, one component of the exon junction complex,
which interacts with mRNA and mediates gene expression (9). The Y14 protein is widely expressed
and highly conserved among species.
Discussion
In the majority of TAR cases there is a compound
inheritance of a microdeletion in 1q.21.1 and of a low-frequency
regulatory SNP. Albers et al(4) reported that of 55 TAR patients, 51
were known carriers of the 200-kb deletion. In two of the patients,
an exonic 4-bp insertion frameshift mutation (605313.0003) and an
exonic null heterozygous mutation (605313.0004) were detected,
respectively, in the RBM8A gene. In all 53 cases, 1 of 2
low-frequency SNPs were detected in regulatory regions of the RBM8A
gene in hetero/hemizygosity. A total of 51 patients carried a
deletion (null allele) and one of the two low frequency SNPs, while
2 patients carried a truncation or frameshift null mutation in
RBM8A and the low frequency 5′ UTR SNP. Consequently, a compound
inheritance mechanism of a rare null allele (deletion or
mutation-frameshift, null) and one of two low-frequency SNPs in the
regulatory regions of RBM8A, causes TAR.
RBM8A has been repeatedly associated with TAR
syndrome and functional studies indicate that SNP rs139428292,
identified in the present study, results in reduced promoter
activity of the gene. A genotype of rs139428292 and a
microdeletion/null allele of the RBM8A leads to lower levels of
Y14, thus disrupting mRNA processing in several tissues and causing
TAR syndrome (4).
The present study describes the first case of
prenatal analysis of TAR syndrome in a fetus with compound
inheritance of a 334-kb deletion in the 1q21.1 region and of a
low-frequency 5′UTR SNP. It also confirms of the causal nature of
the RBM8A gene in the diagnosis of TAR syndrome, as recently
described by Albers et al(4). This study provides additional
information for the understanding of TAR syndrome and a diagnostic
molecular approach that, according to the existing literature, is
able to detect the majority of TAR cases prenatally.
References
|
1
|
Martínez-Frías ML, Bermejo Sánchez E,
García García A, et al: An epidemiological study of the
thrombocytopenia with radial aplasia syndrome (TAR) in Spain. An
Esp Pediatr. 49:619–623. 1998.(In Spanish).
|
|
2
|
Hall JG, Levin J, Kuhn JP, et al:
Thrombocytopenia with absent radius (TAR). Medicine (Baltimore).
48:411–439. 1969.
|
|
3
|
Klopocki E, Schulze H, Strauss G, et al:
Complex inheritance pattern resembling autosomal recessive
inheritance involving a microdeletion in thrombocytopenia-absent
radius syndrome. Am J Hum Genet. 80:232–240. 2007. View Article : Google Scholar
|
|
4
|
Albers CA, Paul DS, Schulze H, Freson K,
et al: Compound inheritance of a low-frequency regulatory SNP and a
rare null mutation in exon-junction complex subunit RBM8A causes
TAR syndrome. Nat Genet. 44:435–439. 2012. View Article : Google Scholar
|
|
5
|
Soranzo N, Spector TD, Mangino M, et al: A
genome-wide meta-analysis identifies 22 loci associated with eight
hematological parameters in the HaemGen consortium. Nat Genet.
41:1182–1190. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rooney DE: Human Cytogenetics:
Constitutional Analysis. Third edition. Oxford University Press;
Oxford: pp. 79–80. 2001
|
|
7
|
Manolakos E, Vetro A, Kefalas K, et al:
Deletion 2q31.2-q31.3 in a 4-year-old girl with microcephaly and
severe mental retardation. Am J Med Genet A. 155A:1476–1482.
2011.
|
|
8
|
D’haene B, Vandesompele J and Hellemans J:
Accurate and objective copy number profiling using real-time
quantitative PCR. Methods. 50:262–270. 2010.PubMed/NCBI
|
|
9
|
Kataoka N, Yong J, Kim VN, et al: Pre-mRNA
splicing imprints mRNA in the nucleus with a novel RNA-binding
protein that persists in the cytoplasm. Mol Cell. 6:673–682. 2000.
View Article : Google Scholar : PubMed/NCBI
|