Introduction
Spindle cell tumors are a rare form of neoplasm that
are often difficult to diagnose, with benign lesions occasionally
exhibiting malignant and infiltrative features (1). Infantile rhabdomyofibrosarcoma is
positioned intermediately between truculent spindle cell
rhabdomyosarcoma and indolent, infantile fibrosarcoma in terms of
clinical presentation, immunohistochemistry, behavior, morphology
and ultrastructural features (2). It
was first reported in 1993 (3).
Infantile rhabdomyofibrosarcoma exhibits a few round
rhabdomyoblastic cells with abundant cytoplasmic eosinophilia that
are scattered within a uniform proliferation of spindle-shaped
cells in a fasciculated pattern. By contrast, infantile
fibrosarcoma has uniform, solidly-packed spindle cells arranged in
a fascicular or herringbone pattern similar to the vascular pattern
observed with hemangiopericytoma (4).
Infantile rhabdomyofibrosarcoma may be identified as childhood
spindle-cell sarcoma with a low degree of rhabdoid differentiation
and aggressive clinical behavior (5).
Overlooking the diagnosis of infantile rhabdomyofibrosarcoma may
increase the chances of local recurrence or metastatic disease;
therefore, aggressive multimodality treatment is required for these
patients (6). Reports of
rhabdomyofibrosarcoma cases are limited in the literature. The
present case report describes a 26-month-old patient who presented
with a slowly progressive, soft-tissue mass in the right chest
wall, which was successfully resected.
Case report
A 26-month-old female was admitted to the Department
of Plastic and Reconstructive Surgery at The Children's Hospital of
Zhejiang University School of Medicine (Hangzhou, China) in October
2012 for the management of a large neoplasm in the right chest
wall. The patient had been born at 38 weeks, weighing 3 kg,
following an uncomplicated pregnancy and delivery. The lesion had
been present since birth and was ~35×80 mm in size. Over the
previous 2 years, the mass had been painless and had slowly
increased in size until 1 month prior to admission. No audible
bruit, fluctuation or thrill was present, and the skin was normal
on examination. Further physical examination was unremarkable, with
the exception of a 6-cm soft-tissue mass present in the right chest
wall. Laboratory analysis indicated the following results: White
blood cell count, 6.96×109 cells/l (normal range,
4.0–12×109 cells/l); hemoglobin count, 113 g/l (normal
range, 110–140 g/l); platelet count, 256×109 cells/l
(normal range, 100–400×109 cells/l); and fibrinogen
level, 2.15 g/l (normal range, 1–5 g/l). Doppler ultrasound (iU
Elite; Philips Healthcare, Amsterdam, The Netherlands) identified
fluid collection in the subcutaneous tissue and muscle planes of
the right chest wall. The tumor was initially suspected to be a
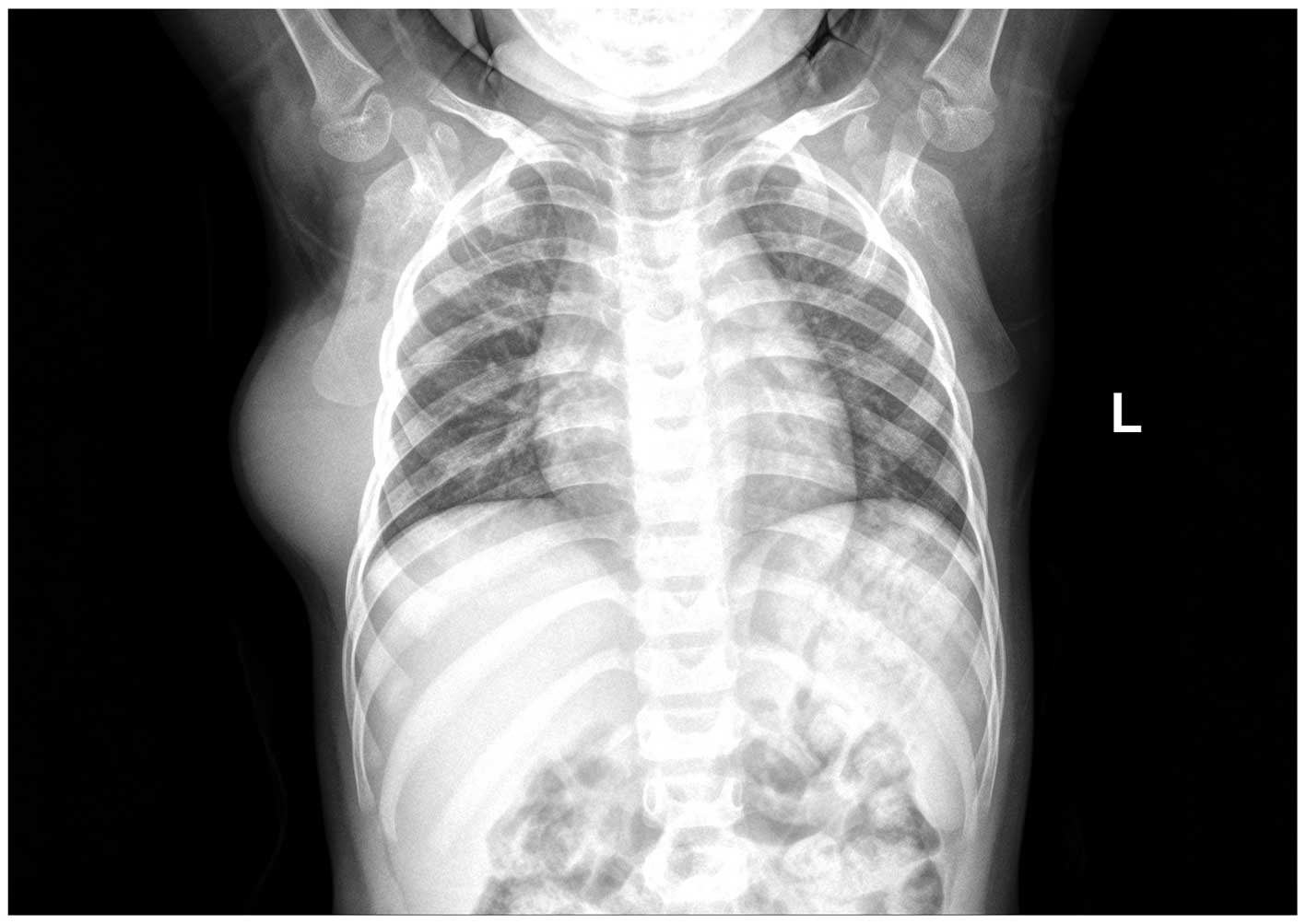
lymphatic hemangioma. A posterior-anterior view of the chest was
obtained using X-ray, which demonstrated that each lung field was
expanded. There was no evidence of a lung mass or infiltrate, and
the bilateral apices, bilateral pleural spaces and cardiac contour
were all normal. The mediastinum and bilateral hila were also
normal. A semicircle shadow with uniform density was observed to be
protruding outwards from the soft tissue of the right chest wall.
The lesion was ~33×77 mm in size, and no marked sorption or
destruction of the ribs were detected (Fig. 1). The patient underwent a wide, local
excision of the tumor, but did not undergo any chemotherapy
following the surgical procedure. The surgical specimen was fixed
with 10% neutral formaldehyde for >24 hour and paraffin embeded.
The tissue slices (thickness, 4 µm) were stained with hematoxylin
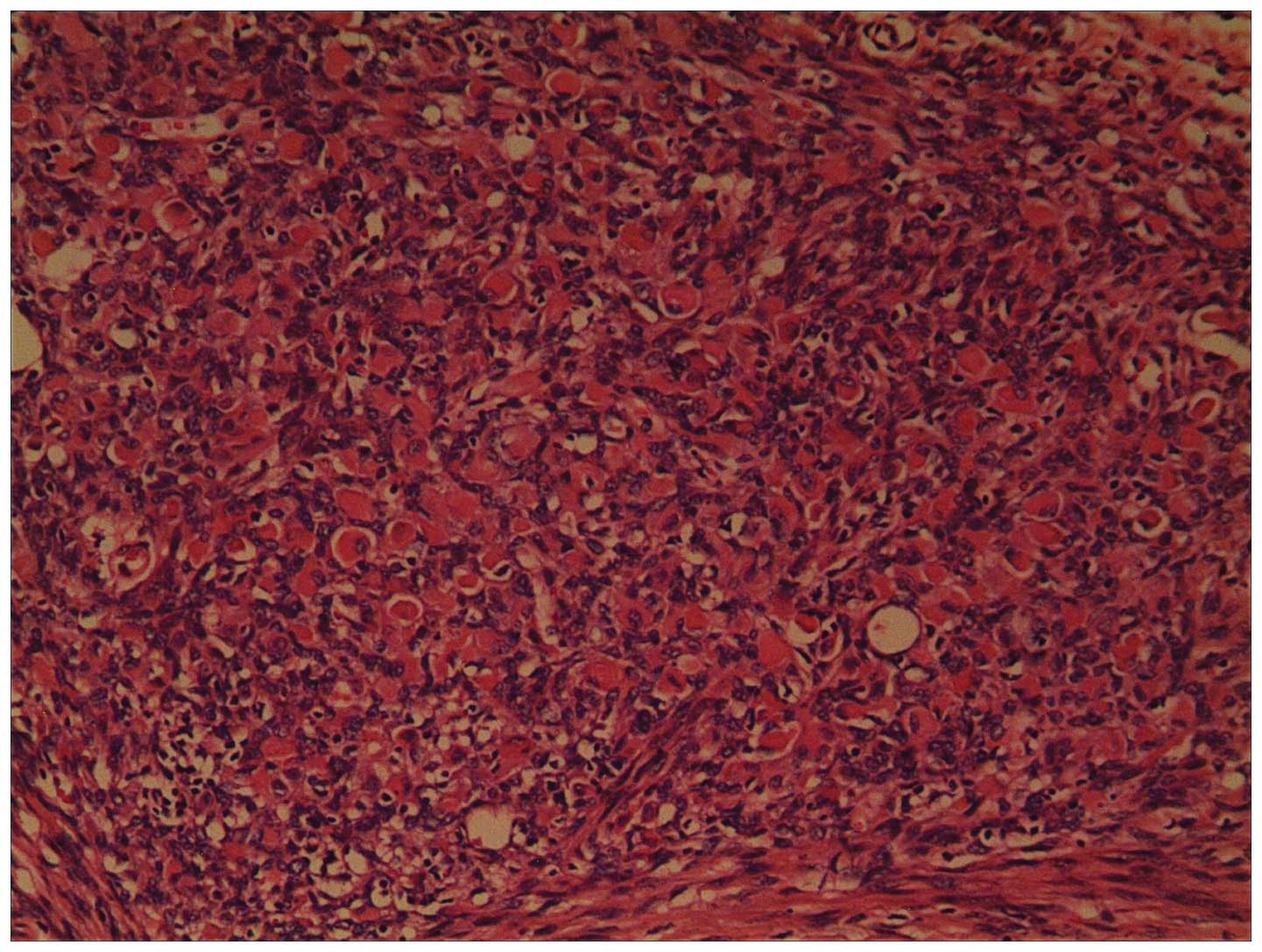
and eosin. Histopathology of the resected tumor identified that the
spindle cells were arranged in fascicles with a herringbone
pattern, and exhibited pale, eosinophilic cytoplasm and mitoses. A
few round rhabdomyoblastic cells with abundant cytoplasmic
eosinophilia that were scattered amoung uniform spindle-shaped
cells, which were in a fasciculated pattern. (Fig. 2). However, no marked cross striation
was observed in the rhabdomyoblastic cells. Immunohistochemical
staining demonstrated strong positivity for vimentin, smooth muscle
actin and desmin, and negativity for myogenin D1, S-100 and cluster
of differentiation 34. According to the histopathological and
immunohistochemical evidence the tumor was diagnosed as infantile
rhabdomyofibrosarcoma. A computed tomography scan (SOMATOM Emotion
16; Siemens Healthcare, Erlangen, Germany) performed post-surgery
did not reveal any signs of recurrence. Despite the aggressive
nature of the lesion, there has been no evidence of recurrence or
metastasis after 2 years of follow-up. Currently, the patient is
alive and well. Written informed consent was obtained from the
family of the patient for the publication of the present study.
Discussion
Infantile rhabdomyofibrosarcoma occupies an
intermediate position between infantile fibrosarcoma and
rhabdomyosarcoma in its clinical presentation, behavior morphology,
immunohistochemical characteristics and ultrastructural features.
Lundgren et al (3) described
the first three cases of infantile rhabdomyofibrosarcoma, which
were initially diagnosed as infantile fibrosarcoma. Two of the
three patients with metastatic spread succumbed to the disease
within 2 years of the primary operation, whereas the third patient
is currently alive with a local recurrence. The three tumors
deviated from infantile fibrosarcoma due to their clinical,
ultrastructural, immunocytochemical and cytogenetic
characteristics. An extensive comparative study was performed on
these cases along with other cases of infantile fibrosarcoma
(3).
Chaudhary et al (6) reviewed the limited number of infantile
rhabdomyofibrosarcoma cases that were available in the literature.
The median age of patients was 24 months (range, 13–48 months),
with a male to female ratio of 5:2 with no specific predilection
(6). In the study by Chaudhary et
al, the cells were immunohistochemically positive for vimentin,
smooth muscle actin and desmin, and negative for myoblobin. The
present case demonstrated similarities with these previously
reported studies, with the exception of the type of treatment
undertaken. Myogenin has been reported to be highly specific and
considered the most reliable marker for myogenesis, and Kumar et
al (7) has hypothesized that
myogenin is 100% specific for rhabdomyosarcoma. In addition,
myogenin D1 is positive in the majority of rhabdomyoblastic cells
and certain spindle cells (5). By
contrast, myogenin and myogenin D1 are not expressed in infantile
fibrosarcoma (8). Therefore, if a
spindle-cell lesion has a high degree of muscle differentiation
cross-striations and nuclear staining for myogenin, the tumor would
best diagnosed and classified as infantile
rhabdomyofibrosarcoma.
Generally, aggressive multimodality treatment,
including surgery and adjuvant chemotherapy, is required for the
treatment of rhabdomyofibrosarcoma. However, in the present case,
the patient was successfully treated with wide local excision
alone, and presented no signs of recurrence after 2 years of
follow-up. Although the patient was recommended to undergo adjuvant
chemotherapy, her parents initially refused, but eventually agreed
to chemotherapy with follow-up. Another case of infantile
rhabdomyofibrosarcoma was treated at The Children's Hospital of
Zhejiang University School of Medicine, in which an 8-month-old
male patient presented with a tumor in the back. The patient
underwent surgery with no follow-up chemotherapy. However, the
neoplasm recurred at the same site 3 months later. In consequence,
the patient underwent a second surgery, with immunohistochemical
examination of the mass displaying similar features to the first
tumor. The patient underwent one cycle of chemotherapy, which
consisted of an intravenous drip of vindesine (3 mg/m2)
and ifosfamide (1300 mg/m2) on the 1st day, then
etoposide (100 mg/m2) on days 1–3. The patient was
followed-up for 3 years and had a good response to treatment
(9).
In the present case, early and aggressive local
surgery to remove the tumor, in addition to multidisciplinary
cooperation, were successful in treating the disease. Close
follow-up is highly recommended for a good prognosis in cases of
infantile rhabdomyofibrosarcoma. Since the number of cases of
infantile rhabdomyofibrosarcoma is extremely limited, additional
accumulation of case data and extensive comparative analysis are
required.
Acknowledgements
The authors would like to thank Mr. John Evans for
reviewing the original manuscript (Zhejiang Ivy International High
School, Hangzhou, China).
References
|
1
|
Gupta A, Maddalozzo J, Win Htin T, Shah A
and Chou PM: Spindle cell rhabdomyosarcoma of the tongue in an
infant: A case report with emphasis on differential diagnosis of
childhood spindle cell lesions. Pathol Res Pract. 200:537–543.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rao SI, Uppin SG, Ratnakar KS, Sundaram C
and Senthil RP: Infantile rhabdomyofibrosarcoma: A distinct variant
or a missinglink between fibrosarcoma and rhabdomyosarcoma? Indian
J Cancer. 43:39–42. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lundgren L, Angervall L, Stenman G and
Kindblom LG: Infantile rhabdomyofibrosarcoma: A high-grade sarcoma
distinguishable from infantile fibrosarcoma and rhabdomyosarcoma.
Hum Pathol. 24:785–795. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Weiss SW and Goldglum JR:
RhabdomyosarcomaSoft Tissue Tumors. Enzinger FM and Weiss SW: 4th.
1st. Mosby Inc.; Maryland Heights, MO: pp. 785–835. 2001
|
|
5
|
Miki H, Kobayashi S, Kushida Y, Sasaki M,
Haba R, Hirakawa E, Ogura K and Ohmori M: A case of infantile
rhabdomyofibrosarcoma with immunohistochemical,
electronmicroscopical, and genetic analyses. Hum Pathol.
30:1519–1522. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chaudhary N, Shet T and Borker A:
Infantile rhabdomyofibrosarcoma: A potentially underdiagnosed
aggressive tumor. Int J Appl Basic Med Res. 3:66–68. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kumar S, Perlman S, Harris CA, Raffeld M
and Tsokos M: Myogenin is a specific marker for rhabdomyosarcomas:
An imunohistochemical study in paraffin-embedded tissues. Mod
Pathol. 13:988–993. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang HF, Wang TL, Gu WZ, Lin L and Li MJ:
Infantile rhabdomyofibrosarcoma. Zhonghua Bing Li Xue Za Zhi.
34:607–608. 2005.(In Chinese). PubMed/NCBI
|
|
9
|
Cessna MH, Zhou H, Perkins SL, Tripp SR,
Layfield L, Daines C and Coffin CM: Are myogenin and myo-D1
expression specific for rhabdomyosarcoma? A study of 150 cases with
emphasis on spindle cell mimics. Am J Surg Pathol. 25:1150–1157.
2001. View Article : Google Scholar : PubMed/NCBI
|