Introduction
Ovarian cancer is the fourth most common type of
cancer in the female genital tract, but has the highest mortality
rate of all gynecological tumors. Ovarian carcinoma accounts for
>90% of cases of ovarian cancer and may arise from relatively
pluripotent cells of the coelomic epithelium or ‘modified
mesothelium’ that covers the ovary (1). The coelomic epithelium is the
mesodermal-derived epithelial lining of the intraembryonic coelom
that invaginates to give rise to the mülllerian ducts, the
primordia for the epithelia of the fallopian tube, the endometrium
and the endocervix (1). There is now
compelling evidence that certain carcinomas can also originate from
the fallopian tubes and the endometriosis (1,2). Ovarian
carcinoma are a heterogeneous group of neoplasms that exhibit a
wide range of tumor morphologies and clinical manifestations
(2). According to the World Health
Organization classification, the most common tumor subtypes are
serous, endometrioid and mucinous, and are characterized by their
morphological resemblance to various mucosal tissues of the female
reproductive tract (3). Cytogenetic
and molecular analysis indicates that multiple genetic and
epigenetic changes are involved in the pathogenesis of ovarian
carcinoma (2,4); however, it remains unclear how these
alterations lead to the development of ovarian cancer. Better
understanding of the molecular mechanisms responsible for the
different types of ovarian cancer may improve diagnosis and
treatment according to the biological characteristics of the
cancer.
T-box genes constitute a family encoding
transcription factors characterized by a highly conserved
DNA-binding region, the T-box. These genes have a role in
regulating the proliferation and differentiation of tissue-specific
stem and progenitor cells during organogenesis (5,6). Previous
studies have indicated that altered T-box gene expression may be
associated with human cancer (7–10). For
example, TBX15, a member of the T-box family, appears to be
involved in mesodermal differentiation (11,12),
putatively through its interaction with co-repressors of the
Groucho family (13) and, therefore,
modulation of the Notch, Wingless/Wnt and decapentaplegic/bone
morphogenetic protein/transforming growth factor-β signaling
pathways (14). Furthermore,
TBX15 hypermethylation was observed in prostate carcinoma
and may be correlated to a negative prognosis (15,16).
As stated, the TBX15 gene is involved in
mesodermal differentiation. Furthermore, the ovary and the female
reproductive system are of mesodermal origin. Therefore, the
present study analyzed the TBX15 gene, in particular the
epigenetic alteration of the TBX15 gene promoter, in ovarian
carcinoma in order to investigate its role in the pathogenesis of
this neoplasia. This alteration could be used to predict tumor
development, progression, recurrence and therapeutic effects.
First, the percentage of DNA methylation at CpG sites was
determined by bisulfite-pyrosequencing, as well as by cloning and
sequencing, to refine the methylation pattern at the level of
individual DNA molecules. Second, immunohistochemistry was
performed to determine the correlation between DNA methylation and
expression of the TBX15 protein. Differences between the
subtypes of ovarian carcinoma, and a possible correlation to
pathological parameters and prognosis were also investigated.
Materials and methods
Patients and tissues
The present study was conducted on tissues obtained
from ovaries removed surgically from women with ovarian carcinoma
who were not subjected to preoperative chemotherapy. For control
cases, 17 normal ovarian and tubal tissues were obtained from women
treated surgically for benign gynecological conditions with no
evidence of cancer (median age, 58 years). The 80 tumor samples
included 29 serous, 23 mucinous and 28 endometrioid carcinomas, the
three most frequent subtypes of carcinoma. All tissues were
obtained via total abdominal hysterectomy and bilateral
salpingo-oophorectomy performed between January 1995 and April 2010
at Pathological Anatomy Unit of the University of Modena and Reggio
Emilia (Modena, Italy). For cases of mucinous carcinoma, a careful
evaluation of the clinical history, in search of a primary mucinous
carcinoma at another site, was performed, and these types of
patients were excluded. Furthermore, cases with immunohistochemical
positivity for CDX2, CEA or CK20, or negativity for CA125 were
excluded from the study. The clinical and pathological features of
patients are summarized in Table I.
The stage was established according to the International Federation
of Gynaecology and Obstetrics criteria (17). The pathological grade was specified
according to the Silverberg grading system (18) for all tumors excluding serous tumors.
For serous tumors, low and high grade were determined according to
the proposed 2-tier grading system (19). Recurrences, metastasis and survival
were established by the investigation of clinical files and
pathological reports of patients. The study was approved by ethics
committee of The University of Modena and Reggio Emilia and written
informed consent was obtained from all patients.
 | Table I.Clinical and pathological features of
ovarian carcinoma. |
Table I.
Clinical and pathological features of
ovarian carcinoma.
|
|
|
| Histological
grade | Stage |
|---|
|
|
|
|
|
|
|---|
| Neoplasia | Patients, n | Median age,
years | Low | High | I | II | III | I | II | III | IV |
|---|
| Serous
carcinoma | 29 | 57 | 5 | 24 | – | – | – | 6 | 5 | 17 | 1 |
| Mucinous
carcinoma | 23 | 59 | – | – | 13 | 4 | 6 | 19 | 1 | 3 | 0 |
| Endometrioid
carcinoma | 28 | 61 | – | – | 14 | 8 | 6 | 22 | 3 | 3 | 0 |
Tissues were formalin-fixed and paraffin-embedded,
sectioned at 3–4 µm, stained with hematoxylin and eosin, and
reviewed by two pathologists (Professor Lorena Losi and Professor
Francesco Rivasi) in order to confirm the histopathological
diagnosis.
Microdissection, DNA extraction and
bisulfite conversion
Deparaffinized and rinsed sections (OTTIX and
alcohol; Diapath S.P.A, Bergamo, Italy) were stained in 0.1%
toluidine blue for 30 sec, washed, air-dried and manually
microdissected using a scalpel blade under a microscopic by a
pathologist (Professor Lorena Losi), in order to enrich the samples
to at least 75% tumor cells. DNA extraction was performed using a
Maxwell 16 FFPE Tissue LEV DNA Purification kit (Promega
Corporation, Madison, WI, USA), according to the manufacturer's
instructions and 100 ng genomic DNA was subjected to bisulfite
conversion using the EpiTect Bisulfite kit (Qiagen, Hilden,
Germany), according to the manufacturer's protocol.
Methylation analysis by
pyrosequencing
Bisulfite-modified genomic DNA was PCR-amplified
using specific primers covering two consecutive regions of the
human TBX15 promoter (2.8 kb upstream of the transcription
start site of NM_152380.2), as previously described (20). The primers were as follows: Forward,
5′-ATGGGATAGTATAATTGATTTGGAATTT-3′ and reverse,
5′-AAAAACCTTTCACCCCCATAA-3′ for the upstream region (Amp1); and
forward, 5′-GGTATTGGGGTAAGAGGAGA-3′ and reverse,
5′-ACCACACAAAACTCCCTTTAT-3′ for the downstream region (Amp2). Both
reverse primers were biotinylated at the 5′-end. PCR was performed
using 3 µl of DNA, MgSO4 at a final concentration of 3
mM and each primer at a final concentration of 0.25 µM using 0.1 U
of Platinum Taq DNA Polymerase (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). PCR was performed for a total
of 41 cycles at an annealing temperature of 60°C. The primers of
the reference gene GAPDH were: Forward, 5′-AAGGTGAAGGTCGGAGTCAAC-3′
and reverse, 5′-GAGTTAAAAGCAGCCCTGGTG-3′.
The DNA methylation status of the two regions was
assessed by pyrosequencing using the automated system PyroMark Q24
(Qiagen). Sequencing primers were designed using the PyroMark Assay
Design SW software (version 2.0; Qiagen), as follows: Upstream,
5′-GAGGGAGTGGATTTT-3′ and downstream,
5′-GGAAGTTTAGATTTTATATTTGTGA-3′. The results were analyzed using
Pyromark Q24 software (version 2.0.6; Qiagen) to estimate the
percentage of methylation at each CpG position for Amp1 (n=1,..9)
and Amp2 (n=1,…10). Samples were categorized as follows: <40%,
low level of methylation; 40–70%, medium level of methylation; and
>70% very high level of methylation.
Methylation analysis by cloning and
sequencing
The PCR products (Amp1 and Amp2) were ligated into a
pGEM-T Vector (Promega Corporation), according to the
manufacturer's protocol. The ligation products were transformed
into chemically competent JM109 bacteria (Promega Corporation),
according to the manufacturer's protocol. In total, 100 µl mixture
was plated on agar plates containing ampicillin, X-gal and
isopropyl thio-β-D-galactoside for blue-white screening. The plates
were incubated at 37°C overnight. Finally, DNA was extracted from
15–20 bacterial colonies, and subjected to PCR amplification of
Amp1 and Amp2 followed by analysis by pyrosequencing.
Cloning-sequencing data was then analyzed using BiQ Analyzer
software (http://biq-analyzer.bioinf.mpi-inf.mpg.de/) to
generate corresponding lollipop methylation diagrams (21).
Immunohistochemical analysis
Immunohistochemical analysis of TBX15 was conducted
on all 80 formalin-fixed, paraffin-embedded neoplastic tissue
samples and all control samples. Immunohistochemistry was performed
on a representative tumor sample in which, in addition to
neoplasia, there was a marginal region of normal ovarian tissue,
which was sectioned at 4 µm. The normal controls are referred to
tissues of different patients without ovarian carrcinomas.
Immunohistochemistry was performed using a TBX15 rabbit anti-human
polyclonal (C-terminus) primary antibody (catalog no.,
LS-C81114-100; dilution, 1:100; LifeSpan Biosciences, Inc.,
Seattle, WA, USA) with a BenchMark XT automated staining system
(Ventana, Strasbourg, France), using 3,3′-diaminobenzidine as the
chromogen. The secondary antibody was included in the Ultraview kit
(catalog no., 05269806001; Roche Diagnostics Spa, Milan, Italy),
which was used according to the manufacturer's protocol. At the end
of reaction, slides were counterstained with hematoxylin. Primary
antibody incubation was 37°C for 1 h.
Statistical analysis
Explorative and inferential data analysis was
performed using R program version 3.2.2 (www.R-project.org/) with the following added packages:
Dplyr, reshape2, data.table, extraGrid and ggplot2. One-way
analysis of variance (ANOVA) followed by Tukey's honest significant
difference (HSD) multiple comparisons of means were applied to
analyze the difference in mean methylation between histotypes in
all CpGn sites, for Amp1 (n=1,..9) and Amp2 (n=1,…10)
regions. Immunohistochemistry results were analyzed by two methods:
i) Results were primarily analyzed by linear regression between
mean methylation of pathological histotypes over all CpG sites and
corresponding immunohistochemistry for Amp1, Amp2, and the mean of
Amp1 and Amp2; ii) results were also analyzed by the χ2
test to verify possible associations between pathological features
(grade and stage), patient status (overall and disease-free
survival) and the methylation values of pathological histotypes
(split in two groups by thresholds of ≤40% and >40%
methylation). Data was analyzed from three independent experiments.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Quantitative analysis of TBX15
promoter methylation at multiple CpG sites by pyrosequencing
Methylation levels of 19 CpG sites within the
5′-region of the TBX15 gene were analyzed by bisulfite
pyrosequencing in 80 ovarian tumor samples and 17 control samples.
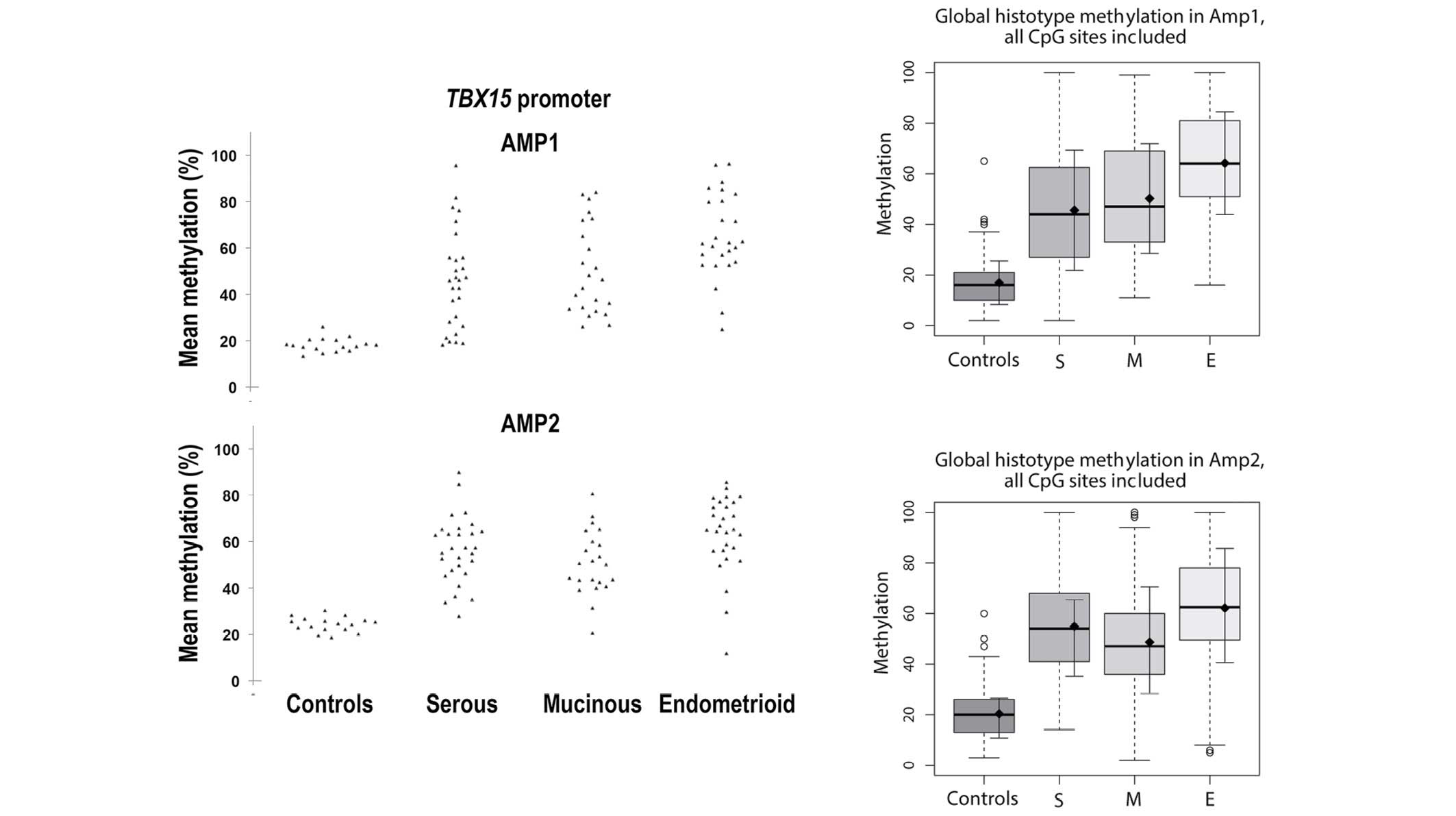
In normal ovarian tissues, a mean methylation level range of 10–30%
was observed in all the 17 samples at all 19 CpG sites analyzed;
this is considered to be a low level of methylation (Fig. 1A and B). For both TBX15
promoter regions investigated (Amp1 and Amp2), a significant
increase in mean methylation (range, 30–80%) was observed in the
different groups of ovarian cancer compared with controls (N) at
all the 19 CpG sites, as shown from Tukey's HSD post-hoc
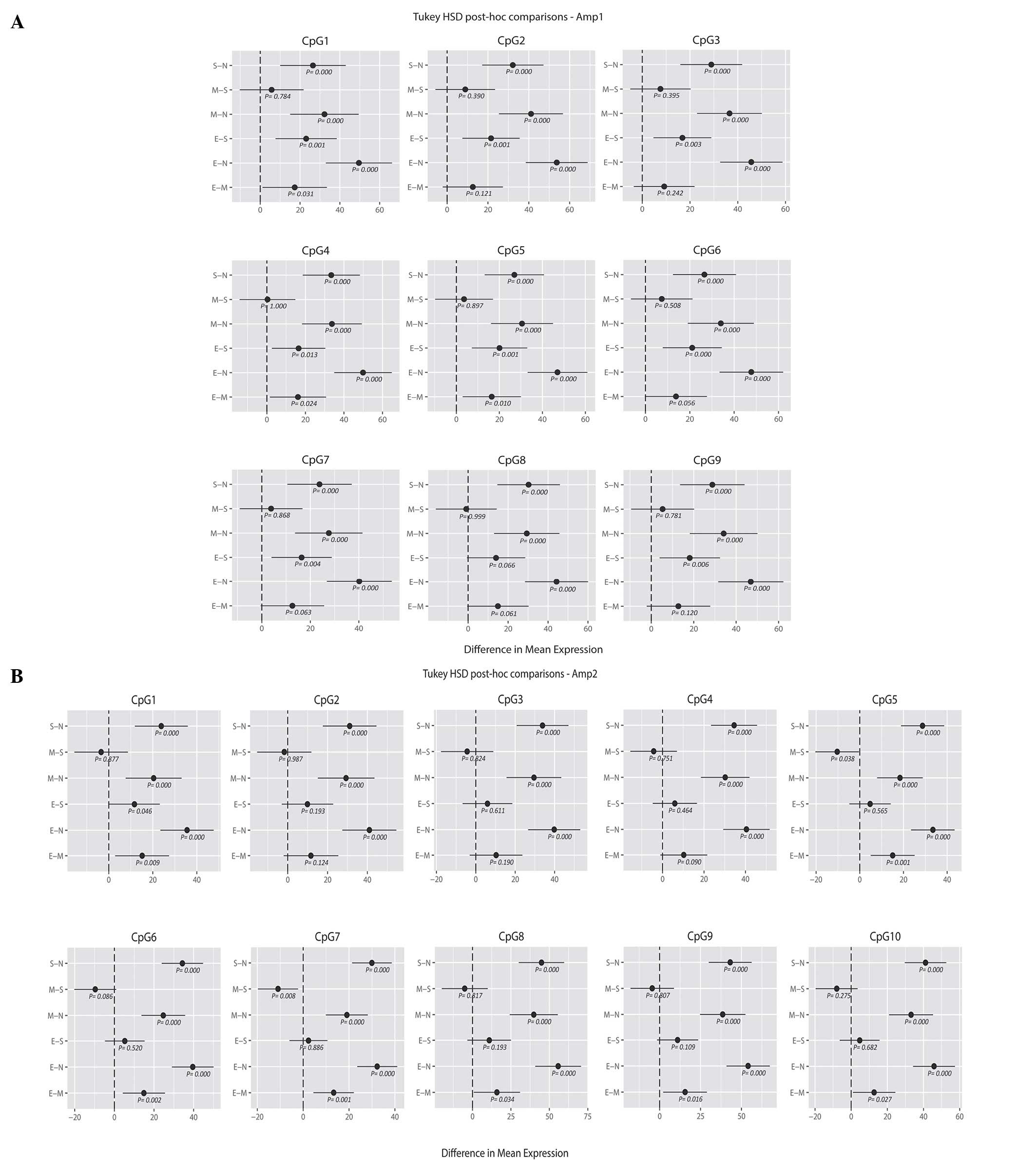
comparisons of means (Fig. 2A and B).
For the upstream region of the promoter (Amp1), the endometrioid
carcinoma samples showed a higher mean percentage of methylation
than the serous and mucinous carcinoma samples for all the analyzed
CpG positions; in particular, with the methylation values for CpGs
1, 2, 3, 4, 5, 6, 7, 9 of the serous histotype (E-S, y-axis;
Fig. 2A) and CpGs 1, 4 and 5 of the
mucinous histotype reached statistical significance compared with
endometrioid carcinoma (E-M, y-axis; P<0.05; Fig. 2A). By contrast, no significant
difference was noted when comparing serous and mucinous cases.
Similarly, the ANOVA and Tukey's HSD analysis revealed differences
in the methylation profile of the downstream region (Amp2) between
the three tumor groups. Again, endometrioid carcinomas showed the
highest mean percentage of methylation. When considering the CpG
positions individually, mucinous tumors presented significantly
lower methylation levels at CpG sites 5 and 7 compared with serous
carcinomas, whereas for endometrioid methylation was significantly
increased at CpG position 1 compared with both mucinous and serous
carcinomas (mucinous, P=0.008; serous, P=0.040; Fig. 2B). Considering all the 19 CpG sites (9
and 10 CpG sites for the Amp1 and Amp2, respectively), the mean ±
standard deviation methylation was 19.21±2.95% for the control
group, while the means were 50.56±16.28, 50.48±15.08 and
63.27±15.56% for serous, mucinous and endometrioid subtypes,
respectively (Fig. 3). Unlike control
samples, tumor samples exhibited a wider distribution of
TBX15 promoter methylation percentages, ranging from cases
showing a methylation percentage similar to control samples to
cases with methylation reaching almost 100%. Notably, when
calculating the median of the methylation percentage for each
group, the values were close to the mean of each group (49.7, 50.9
and 63.5% for serous, mucinous and endometrioid subtypes,
respectively), demonstrating that the distribution of the observed
methylation percentages was considerably homogenous within each
group (Fig. 3).
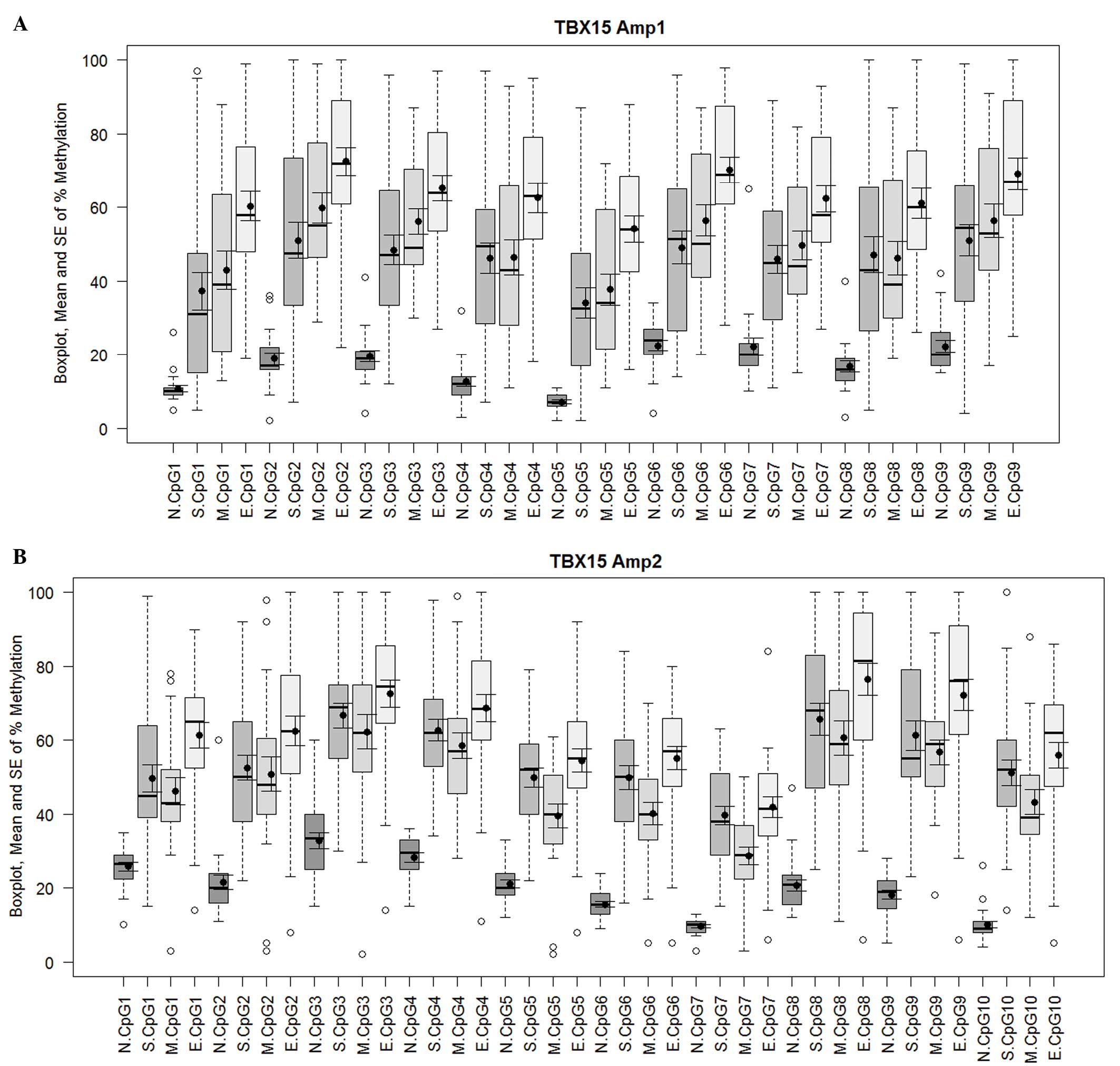 | Figure 1.Methylation percentage of each CpG
site within the (A) Amp1 and (B) Amp2 regions of the TBX15
promoter gene. For both TBX15 promoter regions investigated
(Amp1 and Amp2), a meaningful increase in methylation was observed
in the different groups of ovarian carcinoma (S, M and E) compared
with the controls (N) at all 19 CpG sites, 9 for Amp1 and 10 for
Amp2. Box plots indicate the first and third quartiles, median, and
upper and lower quartiles (whiskers). Outliers are indicated by
empty circles and mean values are indicated by black circles. Data
are also presented as mean ± SE. SE, standard error; N, normal; S,
serous; M, mucinous; E, endometrioid. |
TBX15 promoter methylation patterns of
individual clones
To assess the methylation profile of the
TBX15 promoter at the single molecule level, a
cloning-sequencing procedure was performed on the Amp1 and Amp2 PCR
products. The control cases, with a mean percentage of methylation
of ~20%, exhibited methylation in only a small number of CpG sites
in the Amp1 and Amp2 regions. Indeed, the mean methylation of the
Amp1 and Amp2 regions was 15, 20 and 17% for N5, N6 and N17,
respectively, (Fig. 4).
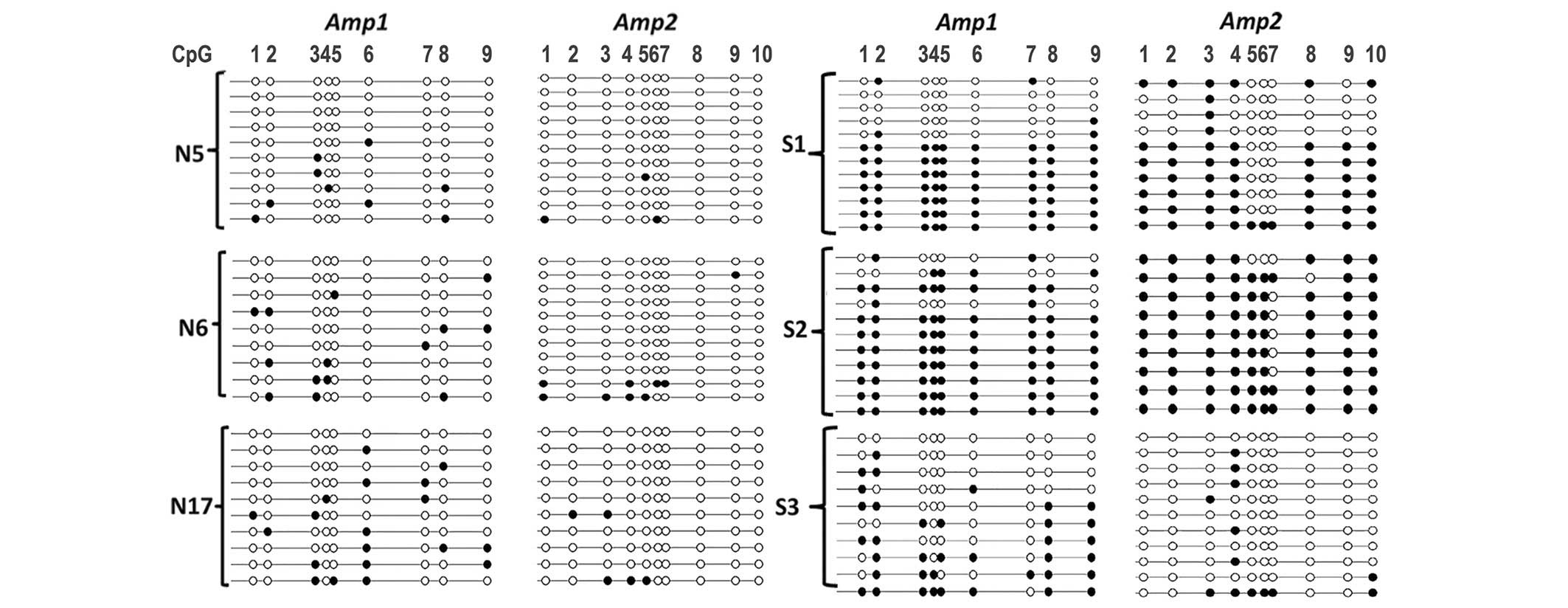 | Figure 4.Lollipop methylation diagram showing
DNA methylation of the Amp1 and Amp2 regions of the TBX15
promoter gene at the molecular level. Amplification,
bisulfite-pyrosequencing and cloning of the Amp1 and Amp2 regions
was performed on genomic DNA extracted from control ovarian (N5,
N6, N17) and carcinoma (S1, S2, S3) samples. A small number of
methylated CpG sites (black circles) were observed in control
tissues, whereas hypermethylation was observed in a large
proportion of S1 and S2 samples. N, normal; S, serous; M, mucinous;
E, endometrioid. |
For tumor samples, both regions (Amp1 and Amp2) were
found to be either fully (or almost fully) methylated or contained
only a small number of methylated CpG sites, as in normal tissues.
Therefore, at the single molecule level, the TBX15 promoter
exhibited either low or high methylation in ovarian carcinoma
samples. When tumor samples were analyzed more in detail, a marked
association was observed between the mean methylation level
measured by pyrosequencing and the percentage of molecules with
either fully (or almost fully) methylated Amp1 and Amp2 regions.
This observation is illustrated in Fig.
4, with three serous tumor samples that exhibited different
mean levels of TBX15 methylation. Similar results were
obtained with mucinous and endometrioid carcinomas (data not
shown). For the S2 tumor sample with strong hypermethylation (80
and 91% for Amp1 and Amp2, respectively), as expected, almost all
the molecules were methylated in almost all CpG sites. The S1
sample, which had an intermediate level of methylation (76 and 53%
for Amp1 and Amp2, respectively), had a mixture of molecules, some
with sporadic methylation, such as in the controls, and some
molecules that were heavily or even fully methylated (Fig. 4). For the S3 serous sample with mild
methylation (37 and 13% for Amp1 and Amp2, respectively), the
situation was similar to that in the controls except that the
number of methylated CpG sites was marginally higher. According to
all the results obtained, hypermethylation of the TBX15
promoter should be considered when mean methylation of at least 40%
is observed in the Amp1 or Amp2 region. In the current study,
pyrosequencing revealed that hypomethylation occurred in all 17
control ovary samples and in 15 ovarian carcinoma samples.
TBX15 hypermethylation was observed in 65/80 (82%) cases of
ovarian cancer, in particular in 83, 74 and 88% of serous, mucinous
and endometrioid carcinomas, respectively.
Immunohistochemical expression of
TBX15
To determine if TBX15 promoter methylation
could be a mechanism for gene silencing, TBX15 protein expression
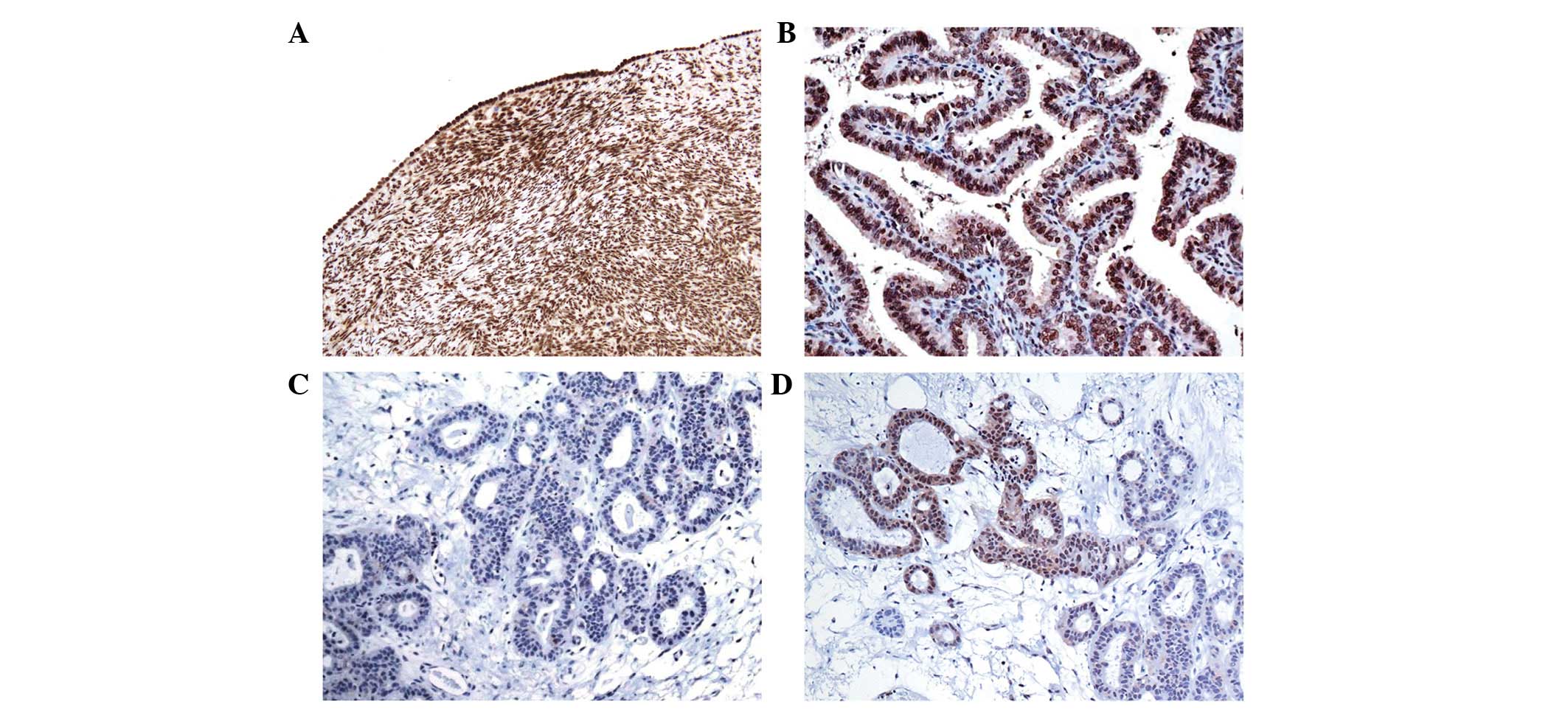
was analyzed by immunohistochemistry. In the control cases, TBX15
protein was present in the nucleus of the ovarian mesothelium,
stromal cells and vascular smooth muscle cells (Fig. 5A). Immunohistochemical positivity was
also present in the epithelium of the fallopian tubes.
For all the neoplastic samples with a low level of
methylation (<40%), the presence of TBX15 expression was
observed in almost all the tumor cells (Fig. 5B). By contrast, an absence of TBX15
protein expression was observed in almost all the tumor cells in
samples with a very high level of methylation (>70%; Fig. 5C). Notably, in samples with a medium
level of methylation (40–70%), only some focal regions exhibited
TBX15 protein expression (Fig. 5D).
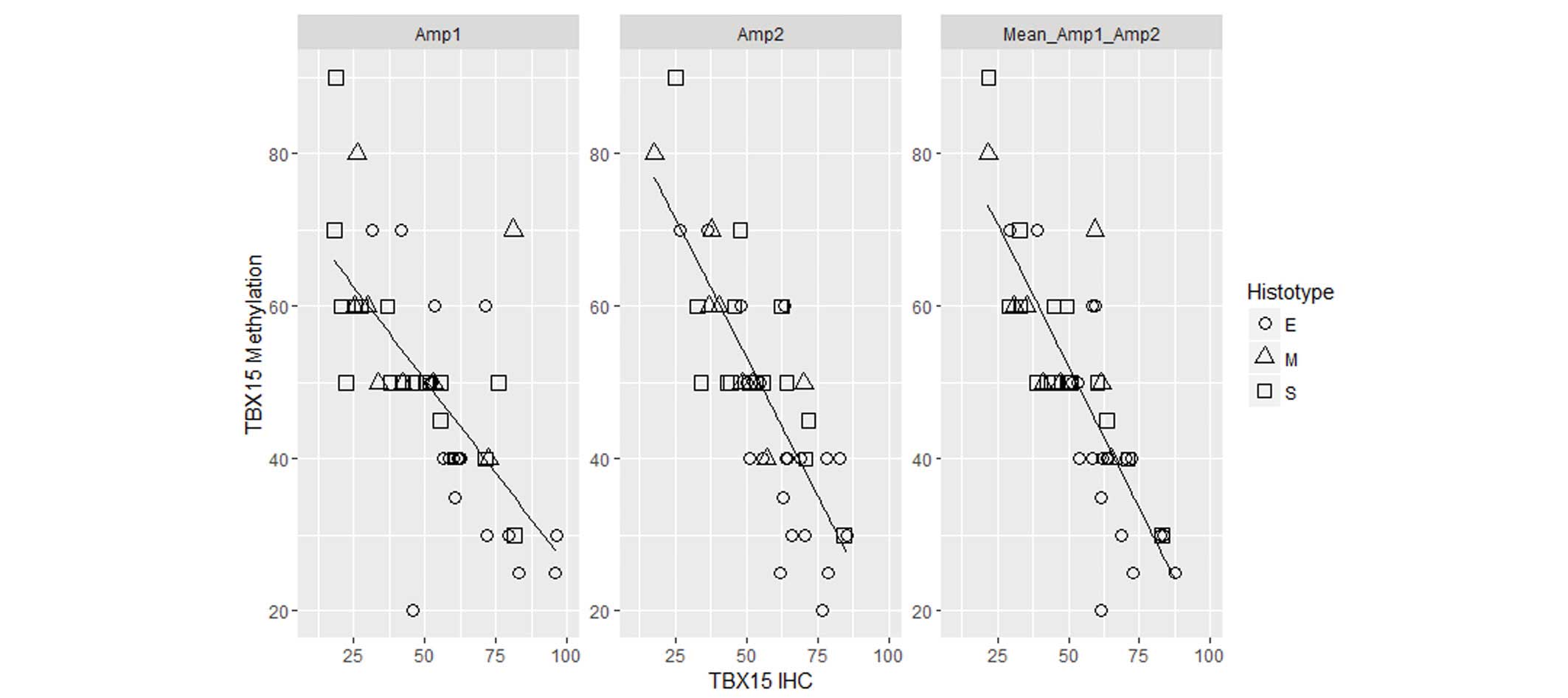
Therefore, in ovarian tumor samples, an inverse correlation was
observed between the level of TBX15 promoter methylation and
the expression of the TBX15 protein (r=−0.7 for Amp1; r=−0.8 for
Amp2; r=−0.83 for the mean between the two regions; P<0.01;
Fig. 6). The two regions, Amp1 and
Amp2, present a comparable correlation coefficient, suggesting that
both regions may be involved in the silencing of the gene.
Correlation of TBX15 methylation with
clinical and pathological parameters
The possible association of methylation results with
pathological and clinical features, such as grade, stage, overall
survival and disease-free survival, was investigated. When cases
with >40% mean methylation were considered to be methylated, the
χ2 test showed that all considered parameters were
independent of TBX15 methylation status (data not shown).
Discussion
In the current study, the methylation status of the
TBX15 promoter gene and its protein expression were
investigated in order to determine its role in the pathogenesis of
ovarian carcinoma according to its implication in mesodermal
tissues, such as the ovary and the other organs of the reproductive
system. In normal ovarian tissues, the TBX15 methylation
level was low and varied between 10–30% at a single CpG site.
Notably, hypermethylation of the TBX15 promoter was observed in 82%
of the total ovarian carcinoma samples, with similar percentages in
various histological subtypes.
The analysis of TBX15 expression by
immunohistochemistry confirmed the correlation between promoter
methylation and the loss of TBX15 expression in ovarian carcinoma.
Therefore, DNA methylation may be the primary mechanism responsible
for TBX15 gene silencing in ovarian carcinoma. Intratumor
heterogeneity was noted by immunohistochemistry and was confirmed
by methylation analysis in approximately half of the ovarian cancer
cases. In the present study, the analyzed tumor samples were
enriched to at least 75% tumor cells by manual microdissection. The
mean methylation level of tumor samples ranged between 30 and 80%,
which indicates that, for some of the carcinoma samples, only
certain tumor cells harbored a hypermethylated TBX15
promoter. Additional information regarding the methylation status
was obtained by bisulfite sequencing after the cloning of PCR
products, which revealed that, in all normal ovarian tissues and in
~15% of cancer tissues, CpG methylation was randomly distributed
over the region and no fully methylated molecules were detected. By
contrast, in neoplastic samples, the mean increase in methylation
compared with the controls resulted in fully or almost fully
methylated molecules. Overall, methylation analysis revealed
heterogeneity at CpG sites and between individual molecules. The
presence of hypomethylated and hypermethylated clones in various
ovarian tumor samples may be associated with the presence of normal
stromal cells, tumor cells at various stages of progression towards
tumorigenesis, or populations of tumor cells subject to clonal
evolution. This observation is in accordance with genetic or
epigenetic intratumoral heterogeneity, as previously observed in
colorectal (22), breast (23) and ovarian (24) cancer.
To date, the role of TBX15 in neoplastic
diseases remains unknown; however, a previous study identified
aberrant methylation of TBX15 in prostate carcinoma,
highlighting its possible role as a prognostic marker. Indeed,
TBX15 methylation was found to be associated with the
pathological stage and Gleason score in prostate carcinoma
(16), suggesting that TBX15
may be useful as a methylation marker in pre- and post-treatment
clinical evaluations. In the present study, no correlation was
identified between TBX15 methylation and the stage or grade
of ovarian carcinoma. Furthermore, the overall and disease-free
survival rates did not appear to be associated with the methylation
and immunohistochemical data. This lack of correlation may be due
to the presence of intratumor heterogeneity in ovarian tumors,
which confounds the validation of single biomarkers, or, more
likely, because this epigenetic alteration occurs in the majority
of ovarian carcinoma cases. It would be useful for future studies
to determine whether hypermethylation of TBX15 could be used
to predict tumor development, progression and therapeutic effects
in ovarian carcinoma.
A previous study observed that specific T-box genes
promote epithelial-mesenchymal transition (EMT) in neoplastic cell
lines and even in certain neoplasia. This process involves events
that convert adherent epithelial cells into individual migratory
cells that can invade the extracellular matrix; such cells may be
critical for cancer progression and the development of metastasis
(25). Furthermore, an association
between cells undergoing EMT and cells with stem cell-like
properties in cancer (cancer stem cells) has been noted (26,27). A
member of the T-box family, Brachyury, which appears to be
regulated by the β-catenin oncogene, can induce the expression of
stemness markers, such as NANOG, CD133, CD166 and CD44, in a
subpopulation of colorectal cancer cells that mimic invasive front
mesenchymal-like cells (28). This
transcription factor may be useful in predicting the outcome of
chemotherapy and radiotherapy, as cancer stem cells are resistant
to these treatments, and could become a target for specific
therapies. A similar course may also occur in ovarian carcinoma and
TBX15 may have a role in this process.
In conclusion, the data obtained in the present
study supports the hypothesis that epigenetic alteration of the
TBX15 gene is implicated in the pathogenesis of the majority
of cases of ovarian carcinoma. The current data also suggests that
TBX15, which is implicated in the development of
mesoderm-derived organs, such as the female reproductive system,
may have a role in the neoplastic ovarian process, independent of
the histological subtype. Therefore, hypermethylation of TBX15 may
represent a potential biomarker for early detection, progression
and response to treatments with a significant impact on reducing
the mortality of this disease.
Acknowledgements
The authors thank Professor Melanie Cripps for the
English revision of the manuscript. Financial support was partially
provided by Banca Popolare dell'Emilia Romagna (Modena, Italy).
References
|
1
|
Auersperg N: Ovarian surface epithelium as
a source of ovarian cancers: Unwarranted speculation or
evidence-based hypothesis? Gynecol Oncol. 130:246–251. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kurman RJ and Shih IeM: The origin and
pathogenesis of epithelial ovarian cancer: A proposed unifying
theory. Am J Surg Pathol. 34:433–443. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kurman RJ, Carcangiu ML, Herrington CS and
Young RH: WHO Classification of Tumours of Female Reproductive
Organs (4th). IARC. Lyon: 2014.
|
|
4
|
Gloss BS and Samimi G: Epigenetic
biomarkers in epithelial ovarian cancer. Cancer Lett. 342:257–263.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Showell C, Binder O and Conlon FL: T-box
genes in early embryogenesis. Dev Dyn. 229:201–218. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takashima Y and Suzuki A: Regulation of
organogenesis and stem cell properties by T-box transcription
factors. Cell Mol Life Sci. 70:3929–3945. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dorfman DM, Hwang ES, Shahsafaei A and
Glimcher LH: T-bet, a T-cell-associated transcription factor, is
expressed in a subset of B-cell lymphoproliferative disorders. Am J
Clin Pathol. 122:292–297. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dorfman DM, Hwang ES, Shahsafaei A and
Glimcher LH: T-bet, a T cell-associated transcription factor, is
expressed in Hodgkin's lymphoma. Hum Pathol. 36:10–15. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ito A, Asamoto M, Hokaiwado N, Takahashi S
and Shirai T: Tbx3 expression is related to apoptosis and cell
proliferation in rat bladder both hyperplastic epithelial cells and
carcinoma cells. Cancer Lett. 219:105–112. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rowley M, Grothey E and Couch FJ: The role
of Tbx2 and Tbx3 in mammary development and tumorigenesis. J
Mammary Gland Biol Neoplasia. 9:109–118. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kispert A, Herrmann BG, Leptin M and
Reuter R: Homologs of the mouse Brachyury gene are involved in the
specification of posterior terminal structures in Drosophila,
Tribolium and Locusta. Genes Dev. 8:2137–2150. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vidricaire G, Jardine K and McBurney MW:
Expression of the Brachyury gene during mesoderm development in
differentiating embryonal carcinoma cell cultures. Development.
120:115–122. 1994.PubMed/NCBI
|
|
13
|
Farin HF, Bussen M, Schmidt MK, Singh MK,
Schuster-Gossler K and Kispert A: Transcriptional repression by the
T-box proteins Tbx18 and Tbx15 depends on Groucho corepressors. J
Biol Chem. 282:25748–25759. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Buscarlet M and Stifani S: The ‘Marx’ of
Groucho on development and disease. Trends Cell Biol. 17:353–361.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kron K, Pethe V, Briollais L, Sadikovic B,
Ozcelik H, Sunderji A, Venkateswaran V, Pinthus J, Fleshner N, van
der Kwast T and Bapat B: Discovery of novel hypermethylated genes
in prostate cancer using genomic CpG island microarrays. PLoS One.
4:e48302009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kron K, Liu L, Trudel D, Pethe V,
Trachtenberg J, Fleshner N, Bapat B and van der Kwast T:
Correlation of ERG expression and DNA methylation biomarkers with
adverse clinicopathologic features of prostate cancer. Clin Cancer
Res. 18:2896–2904. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Prat J: FIGO committee on gynecologic
oncology: Staging classification for cancer of the ovary, fallopian
tube, and peritoneum. Int J Gynaecol Obstet. 124:1–5. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Silverberg SG: Histopathologic grading of
ovarian carcinoma: A review and proposal. Int J Gynecol Pathol.
19:7–15. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Malpica A, Deavers MT, Lu K, Bodurka DC,
Atkinson EN, Gershenson DM and Silva EG: Grading ovarian serous
carcinoma using a two-tier system. Am J Surg Pathol. 28:496–504.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chelbi ST, Doridot L, Mondon F, Dussour C,
Rebourcet R, Busato F, Gascoin-Lachambre G, Barbaux S, Rigourd V,
Mignot TM, et al: Combination of promoter hypomethylation and PDX1
overexpression leads to TBX15 decrease in vascular IUGR placentas.
Epigenetics. 6:247–255. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bock C, Reither S, Mikeska T, Paulsen M,
Walter J and Lengauer T: BiQ analyzer: Visualization and quality
control for DNA methylation data from bisulfite sequencing.
Bioinformatics. 21:4067–4068. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Losi L, Baisse B, Bouzourene H and
Benhattar J: Evolution of intratumoral genetic heterogeneity during
colorectal cancer progression. Carcinogenesis. 26:916–922. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moelans CB, de Groot JS, Pan X, van der
Wall E and van Diest PJ: Clonal intratumor heterogeneity of
promoter hypermethylation in breast cancer by MS-MLPA. Mod Pathol.
27:869–874. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Woloszynska-Read A, Mhawech-Fauceglia P,
Yu J, Odunsi K and Karpf AR: Intertumor and intratumor NY-ESO-1
expression heterogeneity is associated with promoter-specific and
global DNA methylation status in ovarian cancer. Clin Cancer Res.
14:3283–3290. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Imajyo I, Sugiura T, Kobayashi Y, Shimoda
M, Ishii K, Akimoto N, Yoshihama N, Kobayashi I and Mori Y: T-box
transcription factor Brachyury expression is correlated with
epithelial-mesenchymal transition and lymph node metastasis in oral
squamous cell carcinoma. Int J Oncol. 41:1985–1995. 2012.PubMed/NCBI
|
|
26
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Blick T, Hugo H, Widodo E, Waltham M,
Pinto C, Mani SA, Weinberg RA, Neve RM, Lenburg ME and Thompson EW:
Epithelial mesenchymal transition traits in human breast cancer
cell lines parallel the CD44(hi/)CD24 (lo/−) stem cell phenotype in
human breast cancer. J Mammary Gland Biol Neoplasia. 15:235–252.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sarkar D, Shields B, Davies ML, Müller J
and Wakeman JA: BRACHYURY confers cancer stem cell characteristics
on colorectal cancer cells. Int J Cancer. 130:328–337. 2012.
View Article : Google Scholar : PubMed/NCBI
|