Introduction
A total of two major isoforms of human cyclin D1 are
generated via alternative splicing: cyclin D1a and cyclin D1b
(1). Cyclin D1a mRNA has a coding
region of 882 bp within five exons (1). The cyclin D1b mRNA lacks exon 5 but has
an extended exon 4 due to nonsplicing; within intron 4, there is a
translation stop codon (1). The
cyclin D1b mRNA encodes a 275-amino acid protein. It differs at the
C-terminus from the 294-amino acid protein encoded by cyclin D1a
mRNA (1). Cyclin D1a is expressed
ubiquitously and has been shown to control the G1-S transition of
the cell cycle (2,3). It binds to cyclin-dependent kinase
(CDK)4/6 to phosphorylate the retinoblastoma (Rb) protein (2,3). Although
expression of cyclin D1b is barely detectable in normal tissue, it
has been detected in various types of cancer, including bladder
cancer, esophageal cancer, colon cancer, B-lymphoid malignancies,
breast cancer, and prostate cancer, Ewing's sarcoma and mantle cell
lymphoma (4). Gene transfer
experiments have indicated that cyclin D1b is more oncogenic than
cyclin D1a (5–8).
A total of two distinct mRNA transcripts, isoform a
and b, were produced by a polymorphism, A870G, located at the
splice donor region in the exon-intron 4 boundary (9,10). The
G870 allele generates the well-described cyclin D1a transcript
(9,10). An alternative spliced product, cyclin
D1b, results from the A870 allele. This allele hinders splicing and
allows for read through into intron 4 and a premature termination
of transcription (9,10). As individuals who are homozygous for
G/G continue to produce a cyclin D1b transcript (9,11), it is
difficult to explain the production of the cyclin D1b transcript by
the mechanism of polymorphism. Recently, Paronetto et al
(12) showed that the RNA-binding
protein Sam68 regulates alternative splicing of cyclin D1.
Previously, it has been demonstrated that ectopic
expression of cyclin D1b promotes cell invasiveness and
anchorage-independent growth in human bladder cancer cells
(13). However, the introduced cyclin
D1b was not able to associate with CDK4 and enhance Rb
phosphorylation (13), showing that
the function of cyclin D1b is independent of Rb phosphorylation in
the enhancement of cell invasiveness and anchorage-independent
growth.
The present authors previously constructed cyclin
D1b transgenic (Tg) mice to clarify the in vivo oncogenic
potential of cyclin D1b and observed that rectal tumors developed
in 62.5% of the female Tg mice (14).
All rectal tumors in cyclin D1b Tg mice revealed histological
characteristics similar to human sessile serrated adenoma/polyps
(SSA/Ps) (15–20). Adenocarcinomas were also detected in
53% of these rectal tumors (14).
This suggested that these adenocarcinomas arose from the SSA/P-like
lesions. No rectal tumors developed in the ovariectomized female
cyclin D1b Tg mice, showing that ovarian hormones are critical for
rectal carcinogenesis in these Tg mice (14). Phosphorylation of extracellular
signal-regulated kinase (Erk), without activation of
mitogen-activated protein kinase (MAPK)/Erk kinase (MEK), and
expression of estrogen receptor (ER)-β were increased in the rectal
tumors of female cyclin D1b Tg mice in comparison with normal
rectums of female wild-type (WT) mice. Activation of Erk was also
observed in mouse embryo fibroblast (MEF) cells ectopically
expressing cyclin D1b. Furthermore, a tumor cell line, D1bTgRT, was
established from a rectal cancer of a female cyclin D1b Tg mouse
(14). Knockdown of cyclin D1b by
small interfering (si)RNA in this cell line suppressed
phosphorylation of Erk, anchorage-independent growth, cell
invasiveness and tumorigenicity in nude mice. These results
demonstrate that the expression of cyclin D1b has a significant
role in female-specific rectal carcinogenesis via Erk activation
and expression of ER-β in this mouse model.
In the present study, the effects of cyclin D1b on
cell transformation and the mechanism of Erk activation independent
of MEK activation in MEF, 293T and D1bTgRT cells were examined and
it was observed that cyclin D1b activates Erk through Akt. In
addition, the present study investigated the role of Akt activation
in the rectal tumorigenesis of cyclin D1b Tg mice and showed that
enhanced phosphorylation of Akt by cyclin D1b contributes to rectal
tumorigenicity.
Materials and methods
Expression plasmids
The cloning of cyclin D1b complementary DNA and
construction of the expression plasmids, pCR3.1-cyclin D1b-Flag and
pCR3.1-cyclin D1a-Flag, and pCR3.1-cyclin D1-T286A-Flag were
performed as previously described (13).
Cell culture
A tumor cell line, D1bTgRT, was established from a
rectal tumor of a cyclin D1b Tg mouse. D1bTgRT cells were cultured
in RPMI-1640 medium (Nacalai Tesque, Inc., Kyoto, Japan)
supplemented with 10% fetal calf serum (FCS) (Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany), penicillin (100 U/ml) and
streptomycin (100 µg/ml) in humidified air containing 5%
CO2 at 37°C. MEF cells prepared from the embryos of WT
and cyclin D1b Tg mice were immortalized by the large T antigen of
Simian vacuolating virus 40 (SV40) (14). Furthermore, MEF cells were infected
with retrovirus expressing activated K-ras, known as
pBABEpuro-K-Ras (21). 293T cells
derived from human embryonic kidney were used for plasmid
transfection. These cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) (Nacalai Tesque, Inc.) supplemented with 10%
FCS, penicillin (100 U/ml) and streptomycin (100 µg/ml) in
humidified air containing 5% CO2 at 37°C.
Cell proliferation assay
Cell proliferation assay was performed by culturing
the cells in 35-mm culture dishes. A total of 2×104 of
the cells were cultured in triplicate into each dish and incubated
at 37°C. Viable cells were trypsinized and counted using a
hemocytometer.
In vitro invasion assay
The in vitro cell invasiveness was evaluated
by a Matrigel™ Basement Membrane Matrix Invasion Chamber (chamber
size, 6.4 mm; membrane surface area, 0.3 cm2; pore size,
8 µm; BD Biosciences, Franklin Lakes, NJ, USA), according to the
manufacturer's protocol (13). A
total of 500 µl of cell suspension (5×104 cells/ml) was
added to each chamber. The chambers containing the cells were
incubated for 4 days in a CO2 (5%) incubator.
Noninvasive cells were removed from the upper surface of the
membrane. The invasive cells on the underside were stained with
Diff-Quik™ stain (Kokusai-Shiyaku, Kobe, Japan) and counted under a
microscope (TMS; Nikon Corporation, Tokyo, Japan). Each cell sample
was evaluated in triplicate.
Soft agar assay
Anchorage-independent growth of the cells was
evaluated by colony-forming ability in soft agar (13). A total of 10,000 cells were inoculated
into a 60-mm dish containing 0.4% Noble agar containing DMEM
supplemented with 10% FCS and incubated at 37°C. The number of
colonies (>0.15 mm in diameter) on each plate was scored after 3
weeks of incubation. Each assay was performed in triplicate.
Tumorigenicity in nude mice
Tumorigenicity of the cells was examined by
subcutaneous injection of 3×106 cells per 0.2 ml PBS (−)
into 6-week-old BALB/c-nu/nu female nude mice (20 g). The animals
were housed in a specific pathogen-free room with controlled
temperature (20–22°C), humidity (50–60%) and a pre-set light-dark
cycle (12:12 h). They were allowed ad libitum access to food (CE-2;
CLEA Japan, Inc., Tokyo, Japan) and water. A total of four weeks
subsequent to injection, the tumorigenic potential of the cells was
assessed by measuring the weight and volume of tumors. The use of
the animals in the experimental protocols was reviewed and approved
by the Committee of Research Center for Animal Life Science at
Shiga University of Medical Science (Otsu, Japan; approval no.
2014-12-5).
Transfection
Transfection of plasmid DNA was performed using
Lipofectamine™ PLUS reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA), according to the manufacturer's protocol.
Briefly, 5×105 cells were inoculated into a 60-mm dish
and, upon overnight incubation at 37°C, 2 µg of plasmid DNA was
transfected.
Immunoblotting
Lysates of cells and tissues were prepared by
Laemmli-sodium dodecyl sulfate (SDS) buffer containing 62.5 mM
Tris-HCl (pH 6.8), 10% glycerol, 5% 2-mercaptoethanol, 2% SDS,
0.01% bromophenol blue and 5 mM ethylenediaminetetraacetic acid.
Each cell lysate (20 µg of protein) underwent 10 or 12% SDS-PAGE,
and the proteins were electrotransferred to Immobilon-P membranes
(EMD Millipore). Following blocking with TBS-T [10 mM Tris-HCl (pH
7.6), 150 mM sodium chloride and 0.1% Tween-20] containing 5%
bovine serum albumin (BSA) (Nacalai Tesque, Inc.), the membranes
were incubated with the primary antibodies diluted 1:1,000 in TBS-T
containing 2% BSA. Following washes with TBS-T, the membranes were
incubated for 1 h at room temperature in horseradish
peroxidase-conjugated anti-mouse or or anti-rabbit immunoglobulin G
(NA931 and NA934, respectively; GE Healthcare Bio-Sciences,
Pittsburgh, PA, USA) diluted 1:20,000 in TBS-T containing 2% BSA.
Following washing with TBS-T, immunoreactivity was detected by an
enhanced chemiluminescence system (GE Healthcare Bio-Sciences)
using X-ray films.
Antibodies
The primary antibodies used for immunoblotting were
as follows: Anti-α-tubulin (DM1A) (T9026) and anti-FLAG (M2)
(F3165) monoclonal antibodies were obtained from Sigma-Aldrich
(Merck Millipore). Anti-phospho Akt (T308 and S473, and D25E6 and
D9E) (#13038 and #4060, respectively) rabbit monoclonal, anti-Akt
rabbit polyclonal (#9272), anti-phospho Erk1/2 (p44/42 MAPK) rabbit
monoclonal (T202/Y204, D13.14.4E) (#9101), anti-Erk1/2 (p44/42
MAPK) rabbit monoclonal (137F5) (#4695), anti-phospho-MEK1/2 rabbit
monoclonal (S221, 166F8) (#16211), anti-MEK1/2 rabbit monoclonal
(47F6) (#9126) and anti-cyclin D1 rabbit monoclonal (92G2) (#2978)
primary antibodies, which were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Anti-E-cadherin (#610181) and
anti-N-cadherin (#13116) monoclonal primary antibodies were
purchased from BD Biosciences, and the anti-vimentin (C9) (sc-6260)
mouse monoclonal antibody was obtained from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). All antibodies were used at
a dilution of 1:1,000.
In vivo evaluation of tumor growth in
nude mice treated by Akt inhibitor injection into tumors
The use of the animals in the experimental protocols
was reviewed and approved by the Ethics Committee of the Research
Center for Animal Life Science at Shiga University of Medical
Science (Otsu, Japan). The right dorsal flank of each 6-week-old
BALB/c-nu/nu female nude mouse was injected subcutaneously with
3×106 D1bTgRT cells. Following, establishment of
palpable tumors (>100 mm3), external tumor volume was
determined on days 0, 7 and 11. A total of six mice carrying
palpable tumors were randomized into two groups of three mice each.
Each Akt inhibitor at a dose of 25 pmol/mg (calculated tumor
weight) or a similar volume of dimethyl sulfoxide (DMSO) was
injected into the tumor of each mouse on days 0 and 7. Tumor volume
was calculated using the formula: AxBxC/2, where A, B and C
represent the diameters of length, height and width, respectively.
The volume of injection for a tumor was prepared as 100 µl.
Statistical analyses
Using the paired Student's t-test, the present study
compared the invasiveness of D1bTgRT cells treated with Akt
inhibitor with those treated with DMSO as a control, and the in
vivo tumorigenicity of D1bTgRT cells treated with Akt inhibitor
with those treated with DMSO as a control, in the same nude mouse.
P≤0.05 was considered to indicate a statistically significant
difference. All statistical analyses were performed using the R
statistical software package, version 2.6.2 (The R Project for
Statistical Computing, Vienna, Austria).
Results
Activation of Akt in MEF cells
expressing cyclin D1b
To investigate the role of cyclin D1b in cell
transformation and the signaling pathway resulting in Erk
activation, the present study established WT and cyclin
D1b-expressing MEF cell lines, named WT-LT and D1b-LT,
respectively, by introducing the large T antigen of SV40 into
embryonic fibroblasts derived from WT and cyclin D1b-Tg mouse.
Significant differences were not observed between WT-LT and D1b-LT
cells in terms of cell proliferation (Fig. 1A and B). However, D1b-LT cells had a
slightly more spindle-shaped appearance compared with WT-LT cells.
Subsequently, to investigate whether the expression of cyclin D1b
elevates the sensitivity to in vitro cell transformation, an
activated K-ras oncogene was introduced into the cell lines
using the retrovirus vector pBABE-puro and established vector and
K-ras-expressing WT-LT and D1b-LT cell lines, named
WT-LT/BP, WT-LT/K-ras, D1b-LT/BP and D1b-LT/K-ras.
The morphology of D1b-LT/K-ras cells was significantly
altered, although the morphology of the WT-LT/K-ras cells
was also transformed into a more spindle-like appearance, as
compared with D1b-LT cells (Fig. 1A).
The proliferation of D1b-LT/K-ras cells was increased
compared with any of the other cells (Fig. 1B). The present study subsequently
investigated the anchorage-independent growth of these cells; as
shown in Fig. 1C, D1b-LT/K-ras
cells produced a large number of colonies in soft agar, whereas
WT-LT/BP cells scarcely produced any colonies and
WT-LT/K-ras and D1b-LT/BP cells slightly produced colonies.
The present study also investigated the tumorigenicity of these
cells in nude mice. As shown in Fig.
1D, although WT-LT/K-ras and D1b-LT/K-ras cells
demonstrated tumorigenicity in nude mice, the tumor volumes of the
D1b-LT/K-ras cells were significantly increased compared
with that of the WT-LT/K-ras cells (P=0.039). Neither
WT-LT/BP nor D1b-LT/BP cells produced tumors in nude mice.
WT-LT/K-ras and D1b-LT/BP cells showed a more invasive
phenotype compared with WT-LT cells in the Matrigel assay, and
expression of cyclin D1b and K-ras synergistically enhanced
cell invasiveness (Fig. 1E). These
results indicate that cyclin D1b by itself has weak transformation
activity, and cyclin D1b-expressing MEF cells are more sensitive to
transformation by the activated K-ras oncogene than WT MEF
cells, suggesting that cyclin D1b cooperates with activated
K-ras to transform MEF cells.
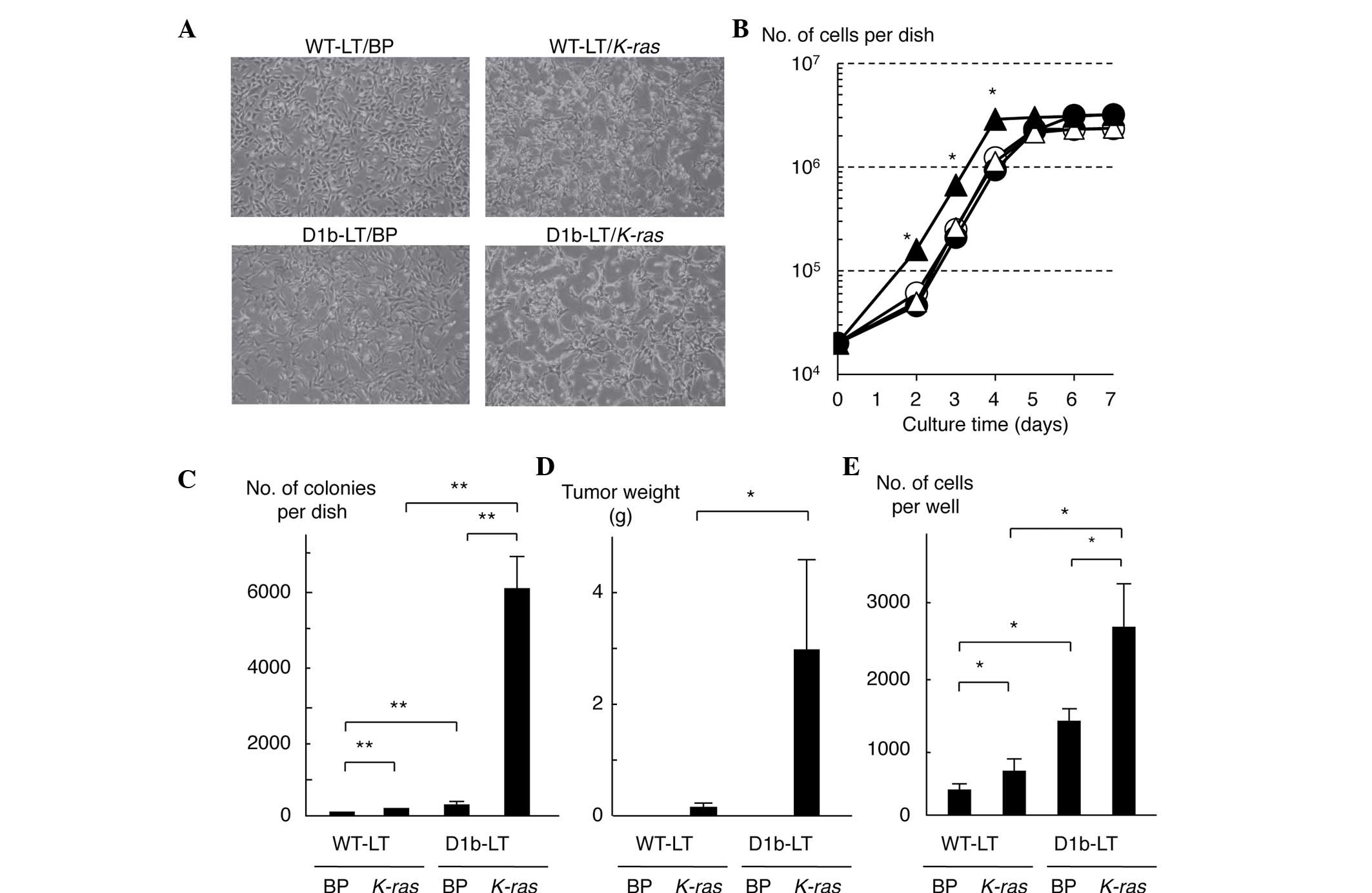 | Figure 1.Cooperation of cyclin D1b with
activated K-ras in the transformation and tumorigenicity of
mouse embryo fibroblast cells. (A) Morphology of MEF cells
expressing cyclin D1b and/or activated K-ras. Magnification,
×100. (B) Growth curves of MEF cells expressing cyclin D1b and/or
activated K-ras. White circle, black circle, white triangle
and black triangle represent WT-LT/BP, WT-LT/K-ras,
D1b-LT/BP and D1b-LT/K-ras cells, respectively. Each sample
was assayed in triplicate, and the bar represents the mean ± SD.
*P≤0.05, for D1b-LT/K-ras cells compared with the remaining
cell lines at days 2, 3 and 4. (C) Anchorage-independent growth of
MEF cells expressing cyclin D1b and/or activated K-ras. Each
sample was assayed in triplicate, and the bar represents the mean ±
SD. **P<0.01. (D) Tumorigenicity in nude mice of MEF cells
expressing cyclin D1b and/or activated K-ras. Each sample
was assayed in triplicate, and the bar represents the mean ± SD.
*P=0.039. (E) Invasiveness of MEF cells expressing cyclin D1b
and/or activated K-ras. Each sample was assayed in
triplicate, and the bar represents the mean ± SD. *P<0.05.
K-ras, v-K-ras2 Kirsten rat sarcoma viral oncogene homolog;
MEF, mouse embryo fibroblast; WT, wild-type; SD, standard
deviation; BP, pBABE puro. |
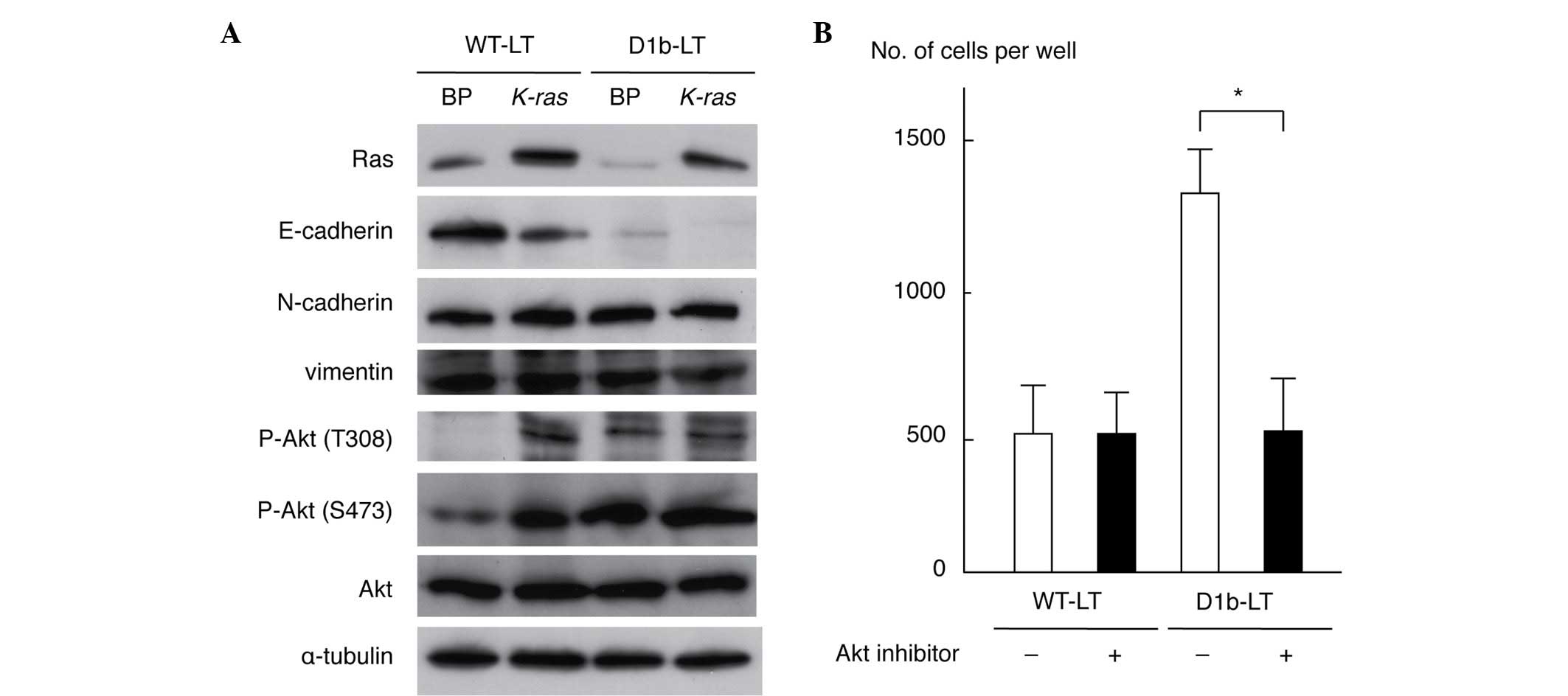
As cyclin D1b enhanced cell invasiveness, the
present study investigated alterations to the expression of
proteins involved in the epithelial-to-mesenchymal transition and
Akt phosphorylation by immunoblot analyses with specific
antibodies. As shown in Fig. 2A,
expression of E-cadherin was downregulated in WT-LT/K-ras
and D1b-LT/BP cells compared with that in WT-LT/BP cells; its
expression was completely lost in D1b-LT/K-ras cells. This
result is compatible with the morphological alterations observed in
transformed MEF cells (Fig. 1A). The
levels of N-cadherin and vimentin were similar in all cell lines.
Phosphorylation of Akt (T308 and S473 residues) was enhanced in
WT-LT/K-ras, D1b-LT/BP and D1b/K-ras cells when
compared with WT-LT/BP cells. The levels of total Akt and α-tubulin
protein were similar in all cell lines. These results indicate that
cyclin D1b is able to suppress E-cadherin expression and to
activate Akt in MEF cells. To clarify whether Akt activation is
necessary for an increase of cell invasiveness by cyclin D1b, a
Matrigel invasion assay of WT-LT and D1b-LT MEF cells treated with
an Akt inhibitor (25 µM) was performed. As shown in Fig. 2B, the Akt inhibitor significantly
suppressed the invasiveness of D1b-LT MEF cells (P=0.0116), but not
of WT-LT MEF cells (P=0.9947), indicating that Akt activation has a
significant role for the enhancement of cell invasiveness in cyclin
D1b-expressing MEF cells.
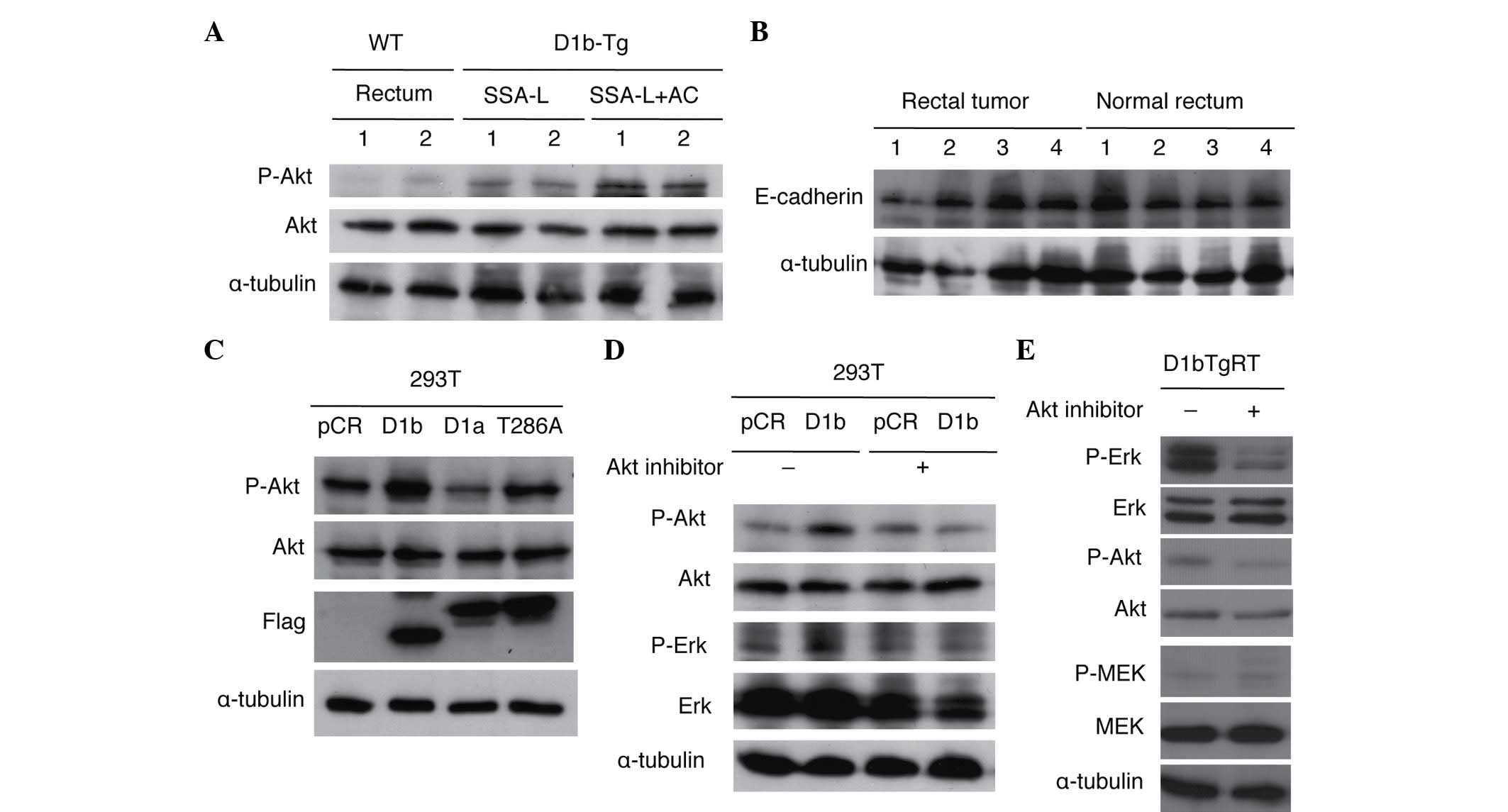
Akt activation in rectal tumors of
cyclin D1b Tg mice and cyclin D1b-expressing 293T cells
To clarify the role of Akt in the rectal
tumorigenicity of cyclin D1b Tg mice, the present study examined
the phosphorylation of Akt in normal rectum and rectal tumors
(SSA/Ps and adenocarcinomas) of cyclin D1b Tg mice. As shown in
Fig. 3A, phosphorylation of Akt was
increased in rectal tumors as compared with normal rectal tissue.
As previously reported, Erk was also activated in rectal tumors
without activation of MEK (14).
Although the present study also examined the expression of
E-cadherin in normal rectum and rectal tumors, alteration of its
expression was not observed in the rectal tumors of these Tg mice
(Fig. 3B). The present study also
investigated Akt activation in 293T cells transfected with cyclin
D1b, cyclin D1a and cyclin D1-T286A. Cyclin D1-T286A is a mutant of
cyclin D1a, in which threonine-286 (T286), the site of glycogen
synthase kinase-3β phosphorylation that is required to enhance both
the nuclear export of cyclin D1 and its turnover, is replaced with
alanine (22). The mutation of T286
to a nonphosphorylatable residue promotes a constitutively nuclear
localization of the cyclin D1a protein, with enhanced oncogenic
potential (23). As shown in Fig. 3C, increased expression of cyclin D1b
and cyclin D1-T286A by transfection accelerated the phosphorylation
of Akt, suggesting that nuclear localization of these cyclin D1
proteins is critical for Akt activation. The present authors have
previously reported that expression of cyclin D1b increases Erk
phosphorylation without activating MEK in 293T cells (14). To clarify whether activation of Erk by
cyclin D1b is mediated through Akt activation, the present study
investigated the effects of an Akt inhibitor on Erk activation by
cyclin D1b in 293T cells. As shown in Fig. 3D, treatment with an Akt inhibitor
suppressed the enhanced phosphorylation of Erk and Akt in 293T
cells expressing cyclin D1b. The present study also investigated
the effects of the Akt inhibitor on Erk activity in D1bTgRT cells
from a rectal tumor of the cyclin D1b Tg mouse. As shown in
Fig. 3E, treatment of the Akt
inhibitor suppressed phosphorylation of Erk and Akt in D1bTgRT
cells. These results show that cyclin D1b activates Erk via Akt to
contribute to tumor formation.
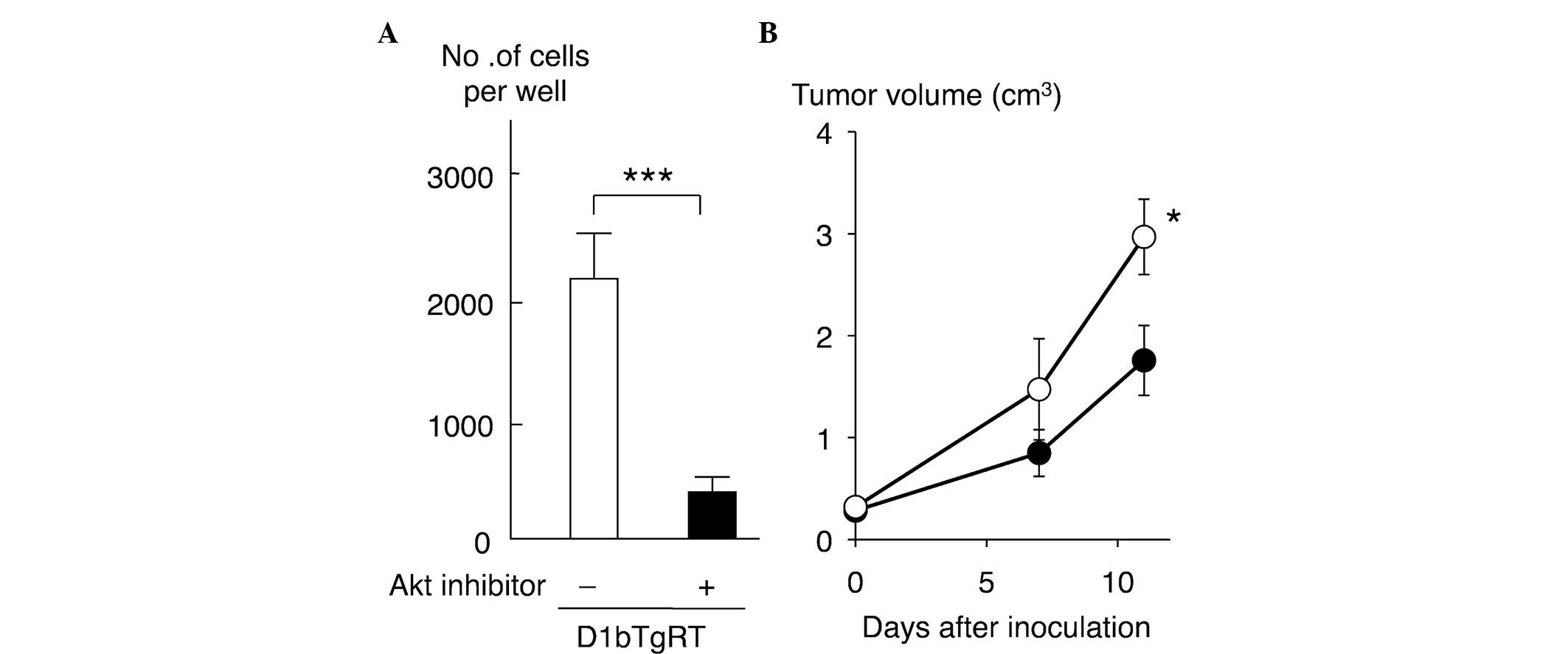
Effect of Akt inhibitor on cell
invasiveness and tumorigenicity of D1bTgRT cells
To clarify the role of Akt in the cell invasiveness
of rectal tumor cells expressing cyclin D1b, the present study
examined the effect of an Akt inhibitor on the cell invasiveness of
D1bTgRT cells. As shown in Fig. 4A,
treatment with the Akt inhibitor (25 µM) significantly reduced the
cell invasiveness of D1bTgRT cells (P=0.0007). Furthermore, the
present study examined the effect of Akt inhibition on tumor growth
in nude mice. As shown in Fig. 4B,
the tumor growth of D1bTgRT cells was significantly decreased by
Akt inhibitor treatments (P=0.0425). These results indicate that
enhanced phosphorylation of Akt by cyclin D1b contributes to cell
invasiveness and tumor formation of the rectal tumor cell line,
D1bTgRT.
Discussion
In the present study, it was initially demonstrated
that cyclin D1b has the ability to enhance cell invasiveness by
itself; it is also able to increase cell invasiveness,
anchorage-independent growth and tumorigenicity in cooperation with
an activated K-ras oncogene in MEF cells. These results are
consistent with the in vivo activity of cyclin D1b that
leads to rectal tumorigenesis in the cyclin D1b-Tg mouse (14). The present study also observed that
phosphorylation of Akt increased in cyclin D1b-expressing MEF
cells. Phosphorylation of Akt was also enhanced in the rectal tumor
tissues of the cyclin D1b Tg mice. Furthermore, Akt was also
activated by transfection of cyclin D1b, but not cyclin D1a, in
human 293T cells. These results suggest that Akt activation is
correlated with Erk activation, as previously described (14). Treatment with an Akt inhibitor
suppresses activation of Erk in 293T cells expressing cyclin D1b.
Furthermore, the Akt inhibitor also suppresses activation of Erk
and the cell invasiveness and tumorigenicity of D1bTgRT cells
derived from a rectal tumor of the cyclin D1b Tg mouse. These
results show that cyclin D1b activates Erk via Akt and suggest that
activation of Akt contributes to the tumorigenicity induced by
cyclin D1b.
The present authors previously constructed cyclin
D1b Tg mice and observed that rectal tumors, including SSA and
adenocarcinoma, were generated at a high frequency (60%) in the
female Tg mice (14). It was
additionally observed that the phosphorylation of Erk was enhanced
without activation of MEK and hotspot mutations of K-ras and
B-raf in the rectal tumors from the Tg mice and in MEF cells
expressing cyclin D1b (14).
siRNA-induced cyclin D1b knockdown in D1bTgRT cells reduced
phosphorylation of Erk and the malignant phenotypes, including
anchorage-independent growth, cell invasiveness and tumorigenicity,
indicating that Erk activation by cyclin D1b is critical for tumor
formation (14). Furthermore, the
transient expression of cyclin D1b enhanced phosphorylation of Erk
without activation of MEK in 293T cells (14). However, in this previous study, it was
not possibly to clarify how cyclin D1b activates Erk without the
activation of upstream signal pathway elements, including MEK,
B-Raf and K-Ras (14). As activation
of Akt by cyclin D1b was observed in MEF cells, the present study
focused on the role of Akt in Erk activation via a MEK-independent
signaling pathway by cyclin D1b and demonstrated this in rectal
tumors of cyclin D1b Tg mice, in a tumor cell line, D1bTgRT,
derived from a rectal tumor in a Tg mouse, and in cyclin
D1b-expressing 293T cells. Previously, Aksamitiene et al
(24) have demonstrated that
phosphoinositide 3-kinase (PI3K)/Akt activates Erk via
T-lymphokine-activated killer cell-originated protein kinase or an
unidentified kinase downstream of the EGF receptor in breast
cancer. Akt is known to associate with a variety of substrate and
nonsubstrate proteins that regulate its kinase activity (25). It is possible that cyclin D1b binds to
Akt or one of its regulating molecules to regulate Akt activity.
Although the present authors attempted to detect the binding of
Akt, PI3K and 3-phosphoinositide dependent protein kinase-1 with
cyclin D1b in 293T cells expressing cyclin D1b, it was not possible
to demonstrate the binding of these proteins under the present
experimental conditions. As cyclin D1b protein is predominantly
localized in the nucleus, this protein may activate the PI3K/Akt
signaling pathway in an indirect manner through transcriptional
regulation of Akt-activating proteins. A constitutively nuclear
cyclin D1a mutant protein, cyclin D1-T286A, also accelerates
phosphorylation of Akt, supporting this idea. Through whichever
mechanism, Akt is considered to have a significant role in Erk
activation and the tumorigenicity of cyclin D1b, although further
investigation is required for the identification of the direct
target protein(s) of cyclin D1b and the clarification of the
molecular mechanism(s) of Akt activation by cyclin D1b.
In addition to the Erk signaling pathway, which is
predominantly involved in regulation of cell proliferation,
PI3K/Akt signaling has diverse functions, including suppression of
apoptosis, cell survival, cell growth and regulation of cell
metabolism, and has been shown to have a role in the progression of
cancer (26–28). In numerous types of human cancer,
including bladder cancer, esophageal cancer, colorectal cancer,
B-lymphoid malignancies, breast cancer and prostate cancer
(4), all of which have been shown to
express the cyclin D1b variant, inhibitors targeting this signaling
pathway are expected to have therapeutic potential.
Acknowledgements
The authors would like to thank Mrs. Akiyo Ushio
(Division of Microbiology and Infectious Diseases, Shiga University
of Medical Science, Otsu, Japan) for her technical assistance with
experiments. The study was supported by the Japan Society for the
Promotion of Science KAKENHI (Grants-in-aid for Scientific
Research) (grant nos. 19591843 and 24590480).
References
|
1
|
Knudsen KE, Diehl JA, Haiman CA and
Knudsen ES: Cyclin D1: Polymorphism, aberrant splicing and cancer
risk. Oncogene. 25:1620–1628. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sherr CJ: D-type cyclins. Trends Biochem
Sci. 20:187–190. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lukas J, Parry D, Aagaard L, Mann DJ,
Bartkova J, Strauss M, Peters G and Bartek J:
Retinoblastoma-protein-dependent cell-cycle inhibition by tumour
suppressor p16. Nature. 375:503–506. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hosokawa Y, Gadd M, Smith AP, Koerner FC,
Schmidt EV and Arnold A: Cyclin D1 (PRAD1) alternative transcript
b: Full-length cDNA cloning and expression in breast cancers.
Cancer Lett. 113:123–130. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu F, Gladden AB and Diehl JA: An
alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear
oncogene. Cancer Res. 63:7056–7061. 2003.PubMed/NCBI
|
|
7
|
Solomon DA, Wang Y, Fox SR, Lambeck TC,
Giesting S, Lan Z, Senderowicz AM, Conti CJ and Knudsen ES: Cyclin
D1 splice variants. Differential effects on localization, RB
phosphorylation, and cellular transformation. J Biol Chem.
278:30339–30347. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burd CJ, Petre CE, Morey LM, Wang Y,
Revelo MP, Haiman CA, Lu S, Fenoglio-Preiser CM, Li J, Knudsen ES,
et al: Cyclin D1b variant influences prostate cancer growth through
aberrant androgen receptor regulation. Proc Natl Acad Sci USA.
103:2190–2195. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Holley SL, Parkes G, Matthias C, Bockmühl
U, Jahnke V, Leder K, Strange RC, Fryer AA and Hoban PR: Cyclin D1
polymorphism and expression in patients with squamous cell
carcinoma of the head and neck. Am J Pathol. 159:1917–1924. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Howe D and Lynas C: The cyclin D1
alternative transcripts[a] and [b] are expressed in normal and
malignant lymphocytes and their relative levels are influenced by
the polymorphism at codon 241. Haematologica. 86:563–569.
2001.PubMed/NCBI
|
|
11
|
Bala S and Peltomäki P: Cyclin D1 as a
genetic modifier in hereditary nonpolyposis colorectal cancer.
Cancer Res. 61:6042–6045. 2001.PubMed/NCBI
|
|
12
|
Paronetto MP, Cappellari M, Busà R,
Pedrotti S, Vitali R, Comstock C, Hyslop T, Knudsen KE and Sette C:
Alternative splicing of the cyclin D1 proto-oncogene is regulated
by the RNA-binding protein Sam68. Cancer Res. 70:229–239. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim CJ, Nishi K, Isono T, Okuyama Y, Tambe
Y, Okada Y and Inoue H: Cyclin D1b variant promotes cell
invasiveness independent of binding to CDK4 in human bladder cancer
cells. Mol Carcinog. 48:953–964. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim CJ, Tambe Y, Munkaisho K, Sugihara H,
Isono T, Sonoda H, Shimizu T, Kondoh G and Inoue H: Female-specific
rectal carcinogenesis in cyclin D1b transgenic mice.
Carcinogenesis. 35:227–236. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Snover DC, Ahnen D, Burt R and Odze RD:
Serrated polyps of the colon and rectum and serrated polyposisWHO
classification of tumors of the digestive system. Bozman FT,
Carneiro F, Hruban RH and Theise ND: 4th. Springer-Verlag; Berlin,
Germany: pp. 160–165. 2010
|
|
16
|
Snover DC, Jass JR, Fenoglio-Preiser C and
Batts KP: Serrated polyps of the large intestine: A morphologic and
molecular review of an evolving concept. Am J Clin Pathol.
124:380–391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
East JE, Saunders BP and Jass JR: Sporadic
and syndromic hyperplastic polyps and serrated adenomas of the
colon: Classification, molecular genetics, natural history, and
clinical management. Gastroenterol Clin North Am. 37:25–46. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Leggett B and Whitehall V: Role of the
serrated pathway in colorectal cancer pathogenesis.
Gastroenterology. 138:2088–2100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Snover DC: Update on the serrated pathway
to colorectal carcinoma. Hum Pathol. 42:1–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rex DK, Ahnen DJ, Baron JA, Batts KP,
Burke CA, Burt RW, Goldblum JR, Guillem JG, Kahi CJ, Kalady MF, et
al: Serrated lesions of the colorectum: Review and recommendations
from an expert panel. Am J Gastroenterol. 107:1315–1329; quiz 1314,
1330. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tambe Y, Hasebe M, Kim CJ, Yamamoto A and
Inoue H: The drs tumor suppressor regulates glucose metabolism via
lactate dehydrogenase-B. Mol Carcinog. 55:52–63. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alt JR, Cleveland JL, Hannink M and Diehl
JA: Phosphorylation-dependent regulation of cyclin D1 nuclear
export and cyclin D1-dependent cellular transformation. Genes Dev.
14:3102–3114. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Diehl JA, Cheng M, Roussel MF and Sherr
CJ: Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis
and subcellular localization. Genes Dev. 12:3499–3511. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Aksamitiene E, Kholodenko BN, Kolch W,
Hoek JB and Kiyatkin A: PI3K/Akt-sensitive MEK-independent
compensatory circuit of ERK activation in ER-positive PI3K-mutant
T47D breast cancer cells. Cell Signal. 22:1369–1378. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brazil DP, Park J and Hemming BA: PKB
binding proteins. Getting in on the Akt. Cell. 111:293–303. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase-Akt pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Downward J: Targeting Ras signaling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Engelman JA: Targeting PI3K signaling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|