Introduction
RNA interference (RNAi) based on small interfering
RNA (siRNA) has been widely exploited for the suppression of gene
expression in model organisms such as Caenorhabditis elegans
and Drosophila melanogaster (1), as well as in mammalian cells (2). Furthermore, RNAi technology based on
short hairpin RNAs (shRNAs) has been widely used in recent years.
As compared with shRNAs, siRNAs are easy to handle and require less
time to synthesize (3). Although Shan
et al (4) used small molecules
containing different concentrations of siRNA to enhance RNAi,
mammalian cells are often insensitive to the transfection methods
used to introduce synthesized siRNAs into cells (5). Furthermore, the effects of synthesized
siRNAs are transient and do not allow for stable inhibition of a
specific gene (6).
Therefore, an alternative approach for introducing
plasmids or viruses carrying shRNAs into mammalian cells for
large-scale, loss-of-function screens is required. Furthermore,
this approach should achieve stable and highly effective gene
suppression in a variety of mammalian cell types. However, the gene
inhibitory effects are limited by the variable efficiencies and
specificities of the empirically designed siRNA or shRNA constructs
(7). Previous studies have screened
for the optimum shRNA construct and have demonstrated that some
siRNAs have ‘off-target’ effects, such as interfering with the
expression or function of other genes or proteins (8–10). To
circumvent these limitations, several different sequence fragments
of a target gene were introduced into a cell concurrently, which
resulted in an enhanced silencing efficiency when attempting to
inhibit the function of a single gene (11).
Lentiviral shRNA vectors are currently the most
appealing tool for the efficient delivery and stable suppression of
genes in nearly all cell types, and many researchers have used this
approach to achieve highly effective gene suppression in mammalian
cells (5,12–14).
However, this technique has inherent limitations associated with
the use of lentiviruses; in particular, it is unsafe for
researchers to be recurrently exposed to lentiviruses, especially
if the laboratory is not equipped with a biological safety cabinet.
Therefore, the present study selected the enhanced green
fluorescent protein (EGFP)-C1 plasmid (pEGFP-C1) for the
construction of multi-shRNA RNAi vectors to provide an effective
and safe method for introducing shRNA into cells.
The authors of the present study have performed
several studies using shRNA plasmid vectors. In our previous study,
two shRNA interference vectors were used to silence one gene, and
it was demonstrated that this method had better silencing effects
than when a single shRNA vector was used (15). In another study, a multi-shRNA vector
was constructed to circumvent the challenges associated with
transfecting some human cancer cells with more than one vector
(16). This vector contained three U6
promoters and at least three shRNA fragments that were able to
simultaneously inhibit one gene at multiple sites. Based on this
idea, a multi-shRNA vector platform able to direct the synthesis of
shRNAs in human cells was developed in the present study.
The current study also addressed the silencing
efficiency of endogenous and exogenous genes. First, the
construction strategy of multi-shRNA vectors was described. Second,
the silencing effects of the multi-shRNA vector on an exogenously
expressed gene (DsRed) in HEK293 cells were compared to those of
vectors containing one and two shRNAs. Third, the potential
off-target effects of these RNAi processes, which may occur when
dsRNAs that are longer than 30 bp are introduced into mammalian
cells (17,18), were tested. Finally, the silencing and
off-target effects of the multi-shRNA vector targeting the Akt2
gene in SKOV3 human ovarian cancer cells were verified and compared
with vectors containing one or two shRNAs targeting the same gene.
In addition, the viability and apoptosis rate of SKOV3 cells
treated with paclitaxel (PTX) were evaluated 48 h after silencing
of the Akt2 gene using the various shRNA vectors.
The results of the present study suggested that the
multi-shRNA vector was more effective at gene suppression than the
single- or double-assembled shRNA vectors. Therefore, the technique
of multiple shRNA vector construction may be applied to RNAi
library generation and loss-of-function studies in mammalian
cells.
Materials and methods
Plasmid construction
The pEGFP-C1-U6 vector, which encodes three human U6
shRNA promoters, was constructed at our laboratory and was based on
the pEGFP-C1 vector from Promega Corporation (Madison, WI, USA).
The pSIREN-DNR-DSRed-Express vector was purchased from BD
Biosciences (Franklin Lakes, NJ, USA).
Three target sites for silencing the DsRed gene were
designed by referring to the technical information provided by
Ambion (Thermo Fisher Scientific, Inc., Waltham, MA, USA). These
sequences were as follows: 5′-AAGGAGTTCATGCGCTTCAAG-3′ (25–46 bp),
5′-AAGTTCATCGGCGTGAACTTC-3′ (367–388 bp) and
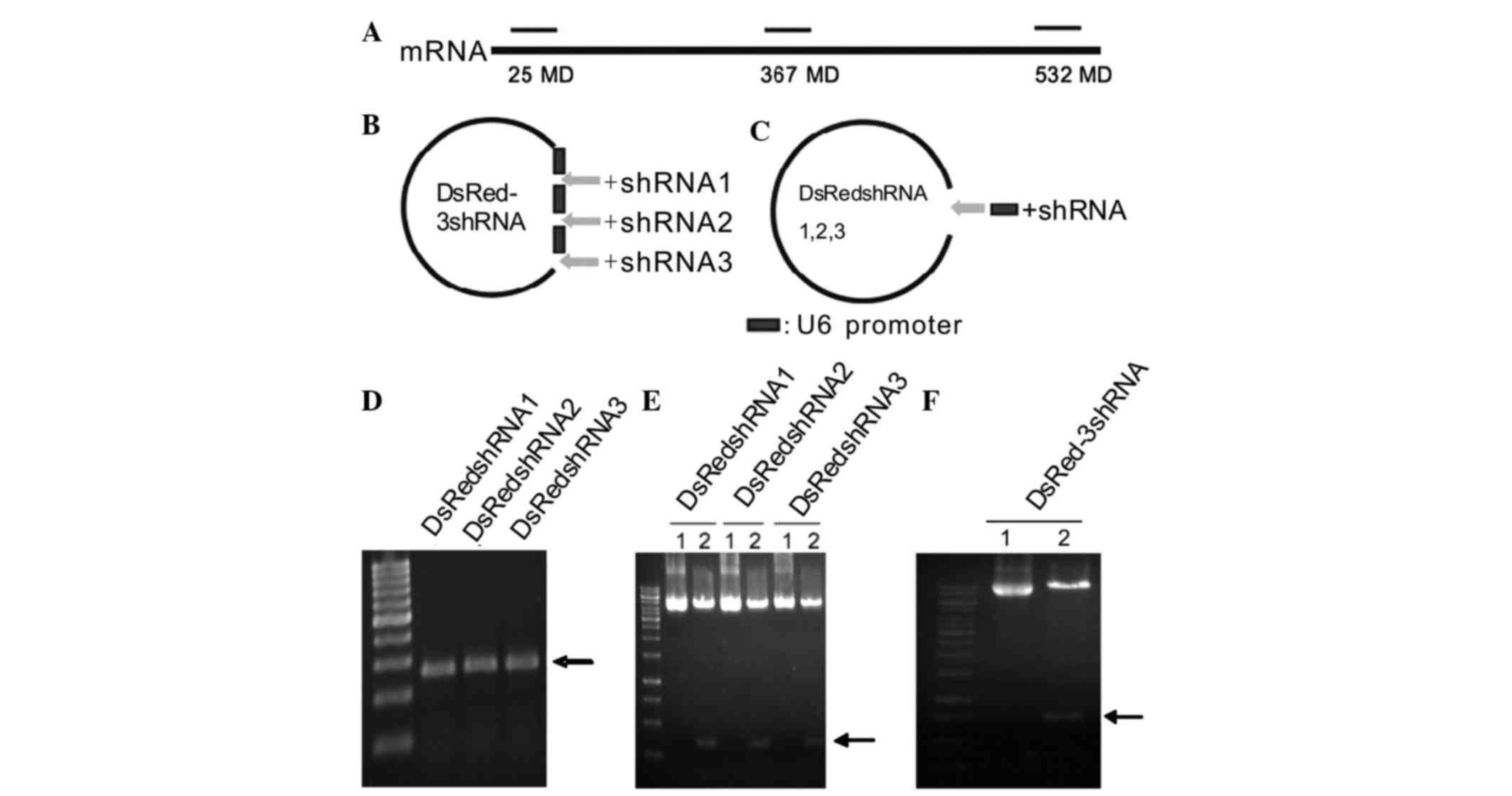
5′-AAGTCCATCTACATGGCCAAG-3′ (532–553 bp) (Fig. 1A). To prevent interference with the
expression of off-target genes, a Basic Local Alignment Search Tool
(http://blast.ncbi.nlm.nih.gov/Blast.cgi) analysis was
performed to confirm that the selected oligonucleotide sets showed
no homology with any other genes.
By ligating three fragments of annealed, forward and
reverse oligonucleotides encoding shRNA sequences into the
pEGFPC1-U6 vector (one after each U6 promoter), the multi-shRNA
vector was constructed (Fig. 1B). The
sequences of the shRNAs targeting DsRed are shown in Table I. The annealed shRNAs were ligated
into the Bam HI and Pst I (shRNA1), Sac I and
Kpn I (shRNA2) or Hind III and Mlu I (shRNA3)
sites of the pEGFPC1-U6 vector using the corresponding restriction
enzymes. The restriction maps of these shRNAs were provided in our
previous study (16). This multiple
shRNA-targeting DsRed plasmid was termed DsRed-3shRNA.
 | Table I.Sequences of oligonucleotides
encoding the human DsRed-3shRNAs, with different flanking ends. |
Table I.
Sequences of oligonucleotides
encoding the human DsRed-3shRNAs, with different flanking ends.
| shRNA name | Oligonucleotide
sequence |
|---|
| shRNA1 sense |
5′-GATCCGGAGTTCATGCGCTTCAAGTTCAAGAGACTTGAAGCGCATGAACTCCTTTTTTCTGCA-3′ |
| shRNA1
antisense |
5′-GAAAAAAGGAGTTCATGCGCTTCAAGTCTCTTGAACTTGAAGCGCATGAACTCCG-3′ |
| shRNA2 sense |
5′-CGTTCATCGGCGTGAACTTCTTCAAGAGAGAAGTTCACGCCGATGAACTTTTTTGGTAC-3′ |
| shRNA2
antisense |
5′-CAAAAAAGTTCATCGGCGTGAACTTCTCTCTTGAAGAAGTTCACGCCGATGAACGAGCT-3′ |
| shRNA3 sense |
5′-AGCTTGTCCATCTACATGGCCAAGTTCAAGAGACTTGGCCATGTAGATGGACTTTTTTA-3′ |
| shRNA3
antisense |
5′-CGCGTAAAAAAGTCCATCTACATGGCCAAGTCTCTTGAACTTGGCCATGTAGATGGACA-3′ |
The production strategy for the vectors containing
single shRNAs is shown in Fig. 1C.
These vectors were designated DsRedshRNA1, DsRedshRNA2 and
DsRedshRNA3. The shRNA fragments containing one human U6 promoter
and one target site were obtained in vitro using polymerase
chain reaction (PCR) and the QIAquick PCR Purification kit (Qiagen
GmbH, Hilden, Germany); each PCR fragment was 267 bp in length
(Fig. 1D). The three fragments were
obtained using the same forward primer
(5′-GGAATTCAAGCGGCCGCATAACTTCG-3′), but different reverse primers
(shown in Table II). The full-length
DsRed plasmid (maintained by our lab) was used as the template for
PCR, and the PCR products were double-digested with Eco RI
and Mlu I and ligated into the Eco RI and Mlu
I cloning sites of pEGFP-C1 to form the single-site shRNA vectors.
pEGFP-C1 containing a non-specific sequence was used as a
control.
| Table II.Primers for DsRed single-shRNA
vectors. |
Table II.
Primers for DsRed single-shRNA
vectors.
| Vectors | Forward
primers | Reverse
primers |
|---|
| DsRedshRNA1 |
5′-GGAATTCAAGCGGCCGCATAACTTCG-3′ |
5′-CGACGCGTAAAAAAGGAGTTCATGCGCTTCAAGTCTCTTGAACTTGAAGCGCATGAACTCCTGTCCTTTCCACAAGA-3′ |
| DsRedshRNA2 |
5′-GGAATTCAAGCGGCCGCATAACTTCG-3′ |
5′-CGACGCGTAAAAAAGTTCATCGGCGTGAACTTCTCTCTTGAAGAAGTTCACGCCGATGAACTCGTCCTTTCCACAAGA-3′ |
| DsRedshRNA3 |
5′-GGAATTCAAGCGGCCGCATAACTTCG-3′ |
5′-CGACGCGTAAAAAAGTCCATCTACATGGCCAAGTCTCTTGAACTTGGCCATGTAGATGGACTCGTCCTTTCCACAAGA-3′ |
Vectors containing the inserted fragments were
selected following digestion with Eco RI and Mlu I
enzymes and analysis of the plasmids by 1.5% agarose gel
electrophoresis (Fig. 1E and F).
Sequencing was performed to confirm the lack of mutations.
Production of the multi-shRNA RNAi vector targeting
Akt2, designated Akt2-3shRNA, the two-site shRNA vector, designated
Akt2-2shRNA, and the single-site shRNA vector, designated
Akt2-shRNA, was described in our previous studies (15,16).
Cell lines and cultures
The HEK293 human embryonic kidney cell line and the
SKOV3 human ovarian cancer cell line were purchased from American
Type Culture Collection (Manassas, VA, USA). HEK293 cells were
maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco;
Thermo Fisher Scientific, Inc.) containing 10% fetal bovine serum
(FBS) (Gibco; Thermo Fisher Scientific, Inc.), and SKOV3 cells were
maintained in McCoy's 5A medium (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 20% FBS, at 37°C in a humidified atmosphere
containing 5% CO2. The cells were routinely subcultured
twice per week.
Establishment of stable HEK293 cell
lines expressing DsRed
HEK293 cells (1×106/ml) were seeded into
6-well plates in 2 ml DMEM containing 10% FBS without antibiotics 1
day prior to transfection. Cells at 80% confluency were transfected
with 4 µg full-length DsRed plasmid using 5 µl Lipofectamine 2000
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) per well,
according to the manufacturer's protocol. After 24 h, the cells
were split to 30–40% confluency, and 300 µg/ml hygromycin B
(Calbiochem® Small Molecules; Merck Millipore,
Darmstadt, Germany) was added to the medium for screening over the
following 2 weeks until red-fluorescent monoclones were formed. The
red monoclones were isolated, seeded into a 24-well plate and grown
until they reached 30% confluency. These cells were then treated
with DMEM containing 10% FBS and 300 µg/ml hygromycin B for another
week. When the red-fluorescent cells were >90% confluent, the
concentration of hygromycin B in the medium was decreased to 100
µg/ml and the red cells were allowed to grow for another 2 weeks to
form the stable HEK293/DsRed cell line.
Transfection of cells
For RNAi, HEK293/DsRed and SKOV3 cells that had been
cultured in 6-well plates were transfected with the indicated
plasmids using Lipofectamine 2000. After 6 h, the transfection
solution was removed and replaced with fresh complete growth
medium. The cells were then assayed for the expression of the
shRNAs at various time points following transfection. In the
present study, 2 µl exogenous IFN-β (Sigma-Aldrich; Merck
Millipore) into the medium of 293/DsRed/DsRedshRNA1,
293/DsRed/DsRedshRNA2, 293/DsRed/DsRedshRNA3,
293/DsRed/DsRed-3shRNA and 293/DsRed cells for more than 16 h at
37°C in a humidified atmosphere containing 5% CO2 to
induce the IFN signaling pathways. The cells were seeded onto
6-well plates at a cell density of 1×106 cells/ml per
well in 2 ml DMEM with 10% FCS.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and semiquantitative
RT-PCR
Total RNA was extracted from the cells at various
time points following transfection using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). RT-qPCR was performed
using the Stratagene Mx3000P System (Agilent Technologies, Inc.,
Santa Clara, CA, USA) and the SYBR Green Realtime PCR Master Mix
(Toyobo Co., Ltd., Osaka, Japan). All primer combinations that were
used are shown in Table III. The
primers were produced by Yingjun Biotechnology Corporation
(Shanghai, China). The PCR cycling conditions were 95°C for 1 min,
followed by 40 cycles of 95°C for 15 sec, 60°C for 15 sec and 72°C
for 30 sec. GAPDH served as an endogenous control. All reactions
were run in triplicate, and the fold-amplification of the genes was
determined using the 2−ΔΔCq method (19).
| Table III.Primer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table III.
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Name | Forward
primers | Reverse
primers |
|---|
| DsRed |
5′-CGGTCTGGGTGCCCTCGTAG-3′ |
5′-CAAGGAGTTCATGCGCTTCA-3′ |
| GFP |
5′-GGATCCCGCCACCATGGTGAGCAAG-3′ |
5′-GAATTCCTTGTACAGCTCGTCCATG-3′ |
| GAPDH |
5′-AGAAGGCTGGGGCTCATTTA-3′ |
5′-AGGGGCCATCCACAGTCTTC-3′ |
| OAS1 |
5′-TCAGAAGAGAAGCCAACGTGA-3′ |
5′-CGGAGACAGCGAGGGTAAAT-3′ |
| AKT1 |
5′-GTGGACCAACGTGAGGCTC-3′ |
5′-GAAGGTGCGTTCGATGACAG-3′ |
| AKT2 |
5′-CAAGCGTGGTGAATACATCAAGA-3′ |
5′-GCCTCTCCTTGTACCCAATGA-3′ |
| AKT3 |
5′-TCTTACACATAGCAGGGGCACCTTC-3′ |
5′-CAGTAGCAGCAACAGCATGAGACC-3′ |
For the semiquantitative RT-PCR, the cycling
conditions were as follows: 94°C for 5 min; 30 cycles at 94°C for
30 sec, 60°C for 30 sec and 72°C for 30 sec; and 72°C for 10 min.
PCR products (10 µl) were analyzed by 1.5% agarose gel
electrophoresis in the presence of ethidium bromide for UV light
transilluminator visualization.
Western blotting
The harvested cells were lysed using lysis buffer
(Beyotime Institute of Biotechnology, Haimen, China), and protein
lysates (50 µg) were denatured in SDS sample buffer at 100°C for 10
min. The proteins were then separated by 10% SDS-PAGE and
transferred onto nitrocellulose membranes. The membranes were
blocked with 5% non-fat milk in TBST (25 mmol/l Tris-HCl, pH 7.5,
137 mmol/l NaCl, 2.7 mmol/l KCl and 0.05% Tween-20) for 1 h at
37°C, followed by incubation with mouse anti-signal transducer and
activator of transcription (STAT1; 1:1,000; cat. no. MA1-037X;
NeoMarkers; Thermo Fisher Scientific, Inc.), rabbit anti-Akt2
(1:1,000; cat. no. 3063; Cell Technology, Inc., Danvers, MA, USA)
and mouse anti-β-actin (1:1,000; cat. no. sc-69879; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) primary antibodies overnight
at 4°C. The membrane was washed three times with TBST, followed by
incubation with horseradish peroxidase-conjugated goat IgG
secondary antibodies (1:1,000; cat. nos. sc-2004 and sc-2005; Santa
Cruz Biotechnology, Inc.) for 2 h at room temperature. The
antibodies were visualized using NBT/BCIP/buffer (1:1:50; Roche
Diagnostics, Basel, Switzerland). The band intensities were
quantified using ImageJ 1.48 software (National Institutes of
Health, Bethesda, MD, USA).
Flow cytometric analysis
The DsRed fluorescence intensity and apoptosis of
HEK293/DsRed cells was evaluated using flow cytometry on a FACSort™
flow cytometer (BD Biosciences). The cells were harvested using
0.2% trypsin, washed twice with PBS and resuspended in PBS at a
density of 1×106 cells. For analysis of apoptosis, the
cells were fixed in 80% ice-cold ethanol overnight at −20°C and
then incubated in 500 ml PBS containing 50 g/ml propidium iodide
and 20 µg/ml RNase for 30 min. The data were analyzed using
CellQuest version 3.3 software (BD Biosciences). The experiments
were repeated three times.
MTT assays for cell viability
The cytotoxic effect of PTX was determined using MTT
assays. Briefly, SKOV3 cells were cultured in 96-well plates at at
a density of 5×103 cells/well and then treated with
various concentrations of PTX (1, 30, 90, 300, 1,000 or 3,000 nM;
Sigma-Aldrich; Merck Millipore) for 48 h. Subsequently, 20 ml MTT
(5 mg/ml; Sigma-Aldrich; Merck Millipore) was added to the wells,
followed by incubation at 37°C for 4 h. The supernatants were then
aspirated, 100 ml DMSO was added to the wells and the cells were
incubated at 37°C for an additional 20 min. The absorbance of the
wells was then read using a microplate reader at a test wavelength
of 570 nm and a reference wavelength of 630 nm. Appropriate
controls that lacked the cells were included to determine the
background absorbance. The response to PTX treatment was assessed
by standardizing the responses of the treatment groups to that of
an untreated control.
Statistical analysis
All experiments were repeated at least three times,
and the data are expressed as the mean ± standard error.
Statistical analyses were performed using SPSS 13.0 software for
Windows and included Student's t-tests or one-way analysis of
variance followed by Least Significant Difference and or
Student-Newman-Keuls tests. P<0.05 was considered statistically
significant.
Results
Strategies for constructing
single-site and multi-site shRNA vectors
In the present study, a platform to construct
multi-shRNA vectors targeting a single gene was designed. The
modified p-EGFP-C1 vector containing three human U6 promoters was
used as the blank vector. One target site (annealed forward and
reverse oligonucleotides) was inserted after each U6 promoter. The
selected target oligonucleotides annealed to the 5′-end, 3′-end or
to the middle of the full-length DsRed gene (Fig. 1A). One shRNA was inserted after each
of the U6 promoters (Fig. 1B). This
process is much safer and easier than constructing lentiviral shRNA
vectors. PCR was used to obtain the target fragment and an empty
plasmid was selected as the blank vector. This method avoided the
requirement for lentivirus packaging, reduced the mutation rate
during PCR and permitted the same efficiency of silencing as when
using lentiviral shRNA vectors.
The construction process for the three single-site
shRNA vectors is shown in Fig. 1C.
PCR was used to obtain a 267-bp shRNA fragment containing a
promoter sequence, a target sequence, a loop sequence, a
complementary sequence, a termination signal and a cloning site
(Fig. 1D). The PCR products (Fig. 1E) that had been digested with
Eco RI and Mlu I were ligated into the non-modified
p-EGFP-C1 vectors.
For each shRNA, stem sequences matching a 21-base
region of the target transcript, with an intervening 6-base ‘loop’,
and which contained the corresponding cloning site, were
designed.
Multi-shRNA vector has a higher gene
silencing efficiency than single-shRNA vectors for exogenous DsRed
expression in HEK293 cells
The abilities of the multi-shRNA vector and three
single-shRNA vectors to silence the DsRed exogenous gene in HEK293
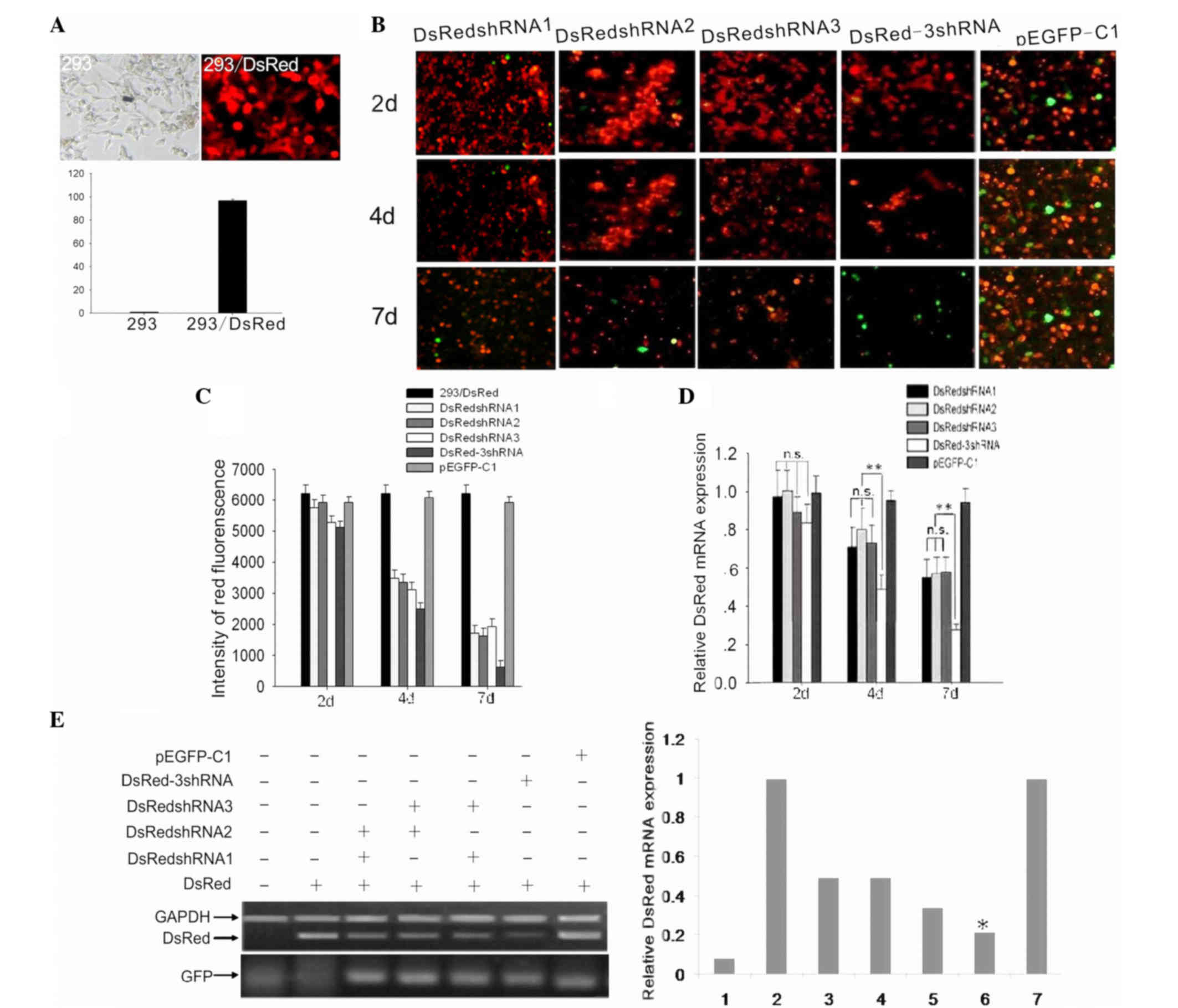
cells was examined. Following establishment of the stable
HEK293/DsRed cell line (Fig. 2A),
four shRNA vectors, DsRed-3shRNA, DsRedshRNA1, DsRedshRNA2 and
DsRedshRNA3, were transfected into the HEK293/DsRed cells. The RNAi
plasmid p-EGFP-C1, which contained no target sequence, was used as
a control.
A conventional optical fluoroscope was used to
ensure that the DsRed plasmid was transfected into HEK293 cells,
and it was observed that >90% of the cells were red. Flow
cytometry was then used to determine the percentage of red
HEK293/DsRed cells (Fig. 2A). Several
methods were used to examine the RNAi effects, and the RNAi effects
were observed at multiple time points in order to avoid the effect
of time on the plasmid transfection efficiency. As shown in
Fig. 2B-D, three points of time were
analyzed and, at these points, fluorescent images were obtained
using a camera and a conventional optical fluoroscope (Fig. 2B). Simultaneously, flow cytometric
analysis was performed to calculate the intensity of red
fluorescence and evaluate its attenuation, which indirectly
corresponded to the extent of DsRed gene silencing at the protein
level (Fig. 2C). Using RT-qPCR, the
ability of the different shRNA vectors to silence the mRNA
expression of the DsRed gene was assessed at different time points
following transfection (Fig. 2D).
Finally, using semiquantitative RT-PCR, the relative expression
levels of DsRed in cells transfected with two shRNAs (i.e.,
co-transfected with two single-shRNA vectors), the DsRed-3shRNA
vector or a single-shRNA vector were determined (Fig. 2E). GFP was the tag protein used to
confirm the successful transfection of the shRNA vectors.
After 2 days of transfection, it was evident that
the multi-shRNA vector produced stronger RNAi effects compared with
the single-shRNA vector (P<0.05) and the two-shRNA vector
(P<0.01) at the mRNA and protein expression levels. However,
within the first 2 days, there was no obvious difference in the
fluorescence intensity of the cells transfected with different
shRNA vectors. Additionally, within the first 2 days, the
expression of DsRed did not change significantly following
transfection with the different shRNA vectors at the mRNA and
protein levels (P>0.05).
IFN response assay to verify the
specificity of RNA interference
In most mammalian cells, the use of long (>30
nucleotides) double-stranded (ds)RNA provokes an interferon (IFN)
response; in particular, a non-specific type 1 IFN response that
can lead to nonspecific gene suppression and a general shutdown of
protein synthesis (20). Using our
RNAi library, we designed multi-shRNA vectors that formed 19-bp
dsRNAs in cells. However, the specificity of the RNAi had to be
verified. According to Jaitin and Schreiber (21), if IFN signaling pathways are induced,
the expression of 2′-5′-oligoadenylate synthase 1 (OAS1) at the
mRNA level and STAT1 at the protein level are enhanced. In the
present study, 2 µl exogenous IFN-β was added to HEK293/DsRed cells
transfected with DsRedshRNA1, DsRedshRNA2, DsRedshRNA3 or
DsRed-3shRNA for >16 h to induce the IFN signaling pathways. If
an endogenous IFN response occurred, the expression of OAS1 mRNA
and STAT1 protein should not alter considerably following the
addition of exogenous IFN-β. However, a marked increase in the
expression of OAS1 mRNA (Fig. 3A) and
STAT1 protein (Fig. 3B) were observed
following the addition of exogenous IFN-β (P<0.01), which
suggested that an exogenous, but not an endogenous, IFN response
was generated in the shRNA-transfected HEK293/DsRed cells.
Therefore, the gene silencing effects that were induced by the
different shRNA vectors were specific.
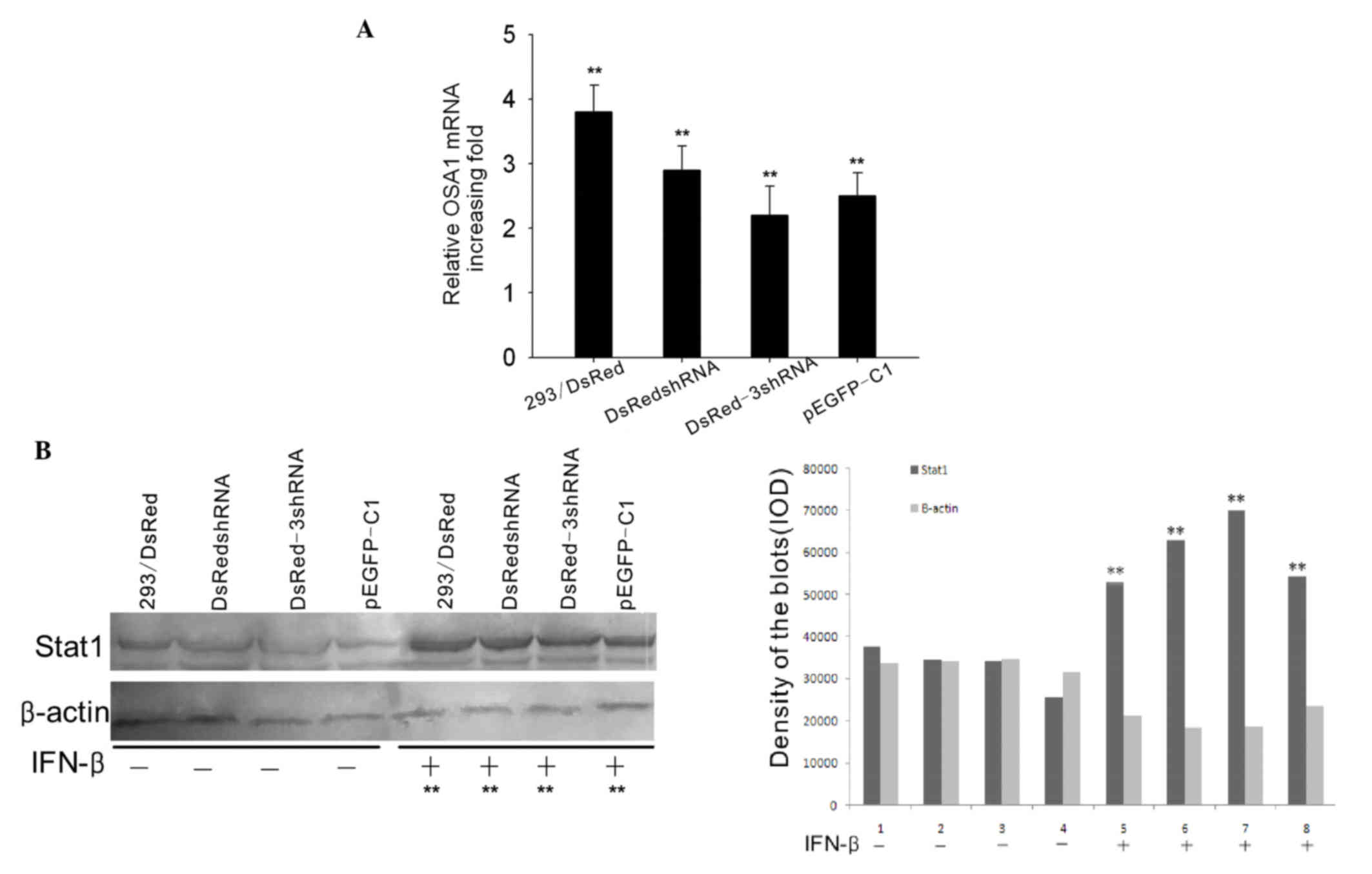 | Figure 3.IFN response assay. The HEK293/DsRed
cell line was transfected with various shRNA vectors and treated
with IFN-β for 16 h. (A) Semiquantitative reverse
transcription-polymerase chain reaction was performed to measure
the mRNA expression level of OAS1. The mRNA expression of OAS1
increased 3.8±0.51-, 2.8±0.34-, 2.3±0.23- and 2.7±0.31-fold in
293/DsRed, 293/DsRed/DsRedshRNA, 293/DsRed/DsRed-3shRNA and
293/DsRed/pEGFP-C1 cells, respectively, when IFN-β was added for 16
h. The bar graph shows the results of three independent
experiments. **P<0.01. (B) The protein expression of STAT1 was
increased 2.26-, 3.43-, 3.85- and 2.83-fold in the 293/DsRed,
293/DsRed/DsRedshRNA, 293/DsRed/DsRed-3shRNA and 293/DsRed/pEGFP-C1
cells, respectively, when IFN-β was added for 16 h, as determined
by western blotting **P<0.01. IFN, interferon; shRNA, short
hairpin RNA; OSA1, 2′-5′-oligoadenylate synthase 1; STAT1, signal
transducer and activator of transcription. |
Multi-shRNA vector silences the
expression of the endogenous Akt2 gene in SKOV3 cells more
effectively than single- and two-site shRNA vectors
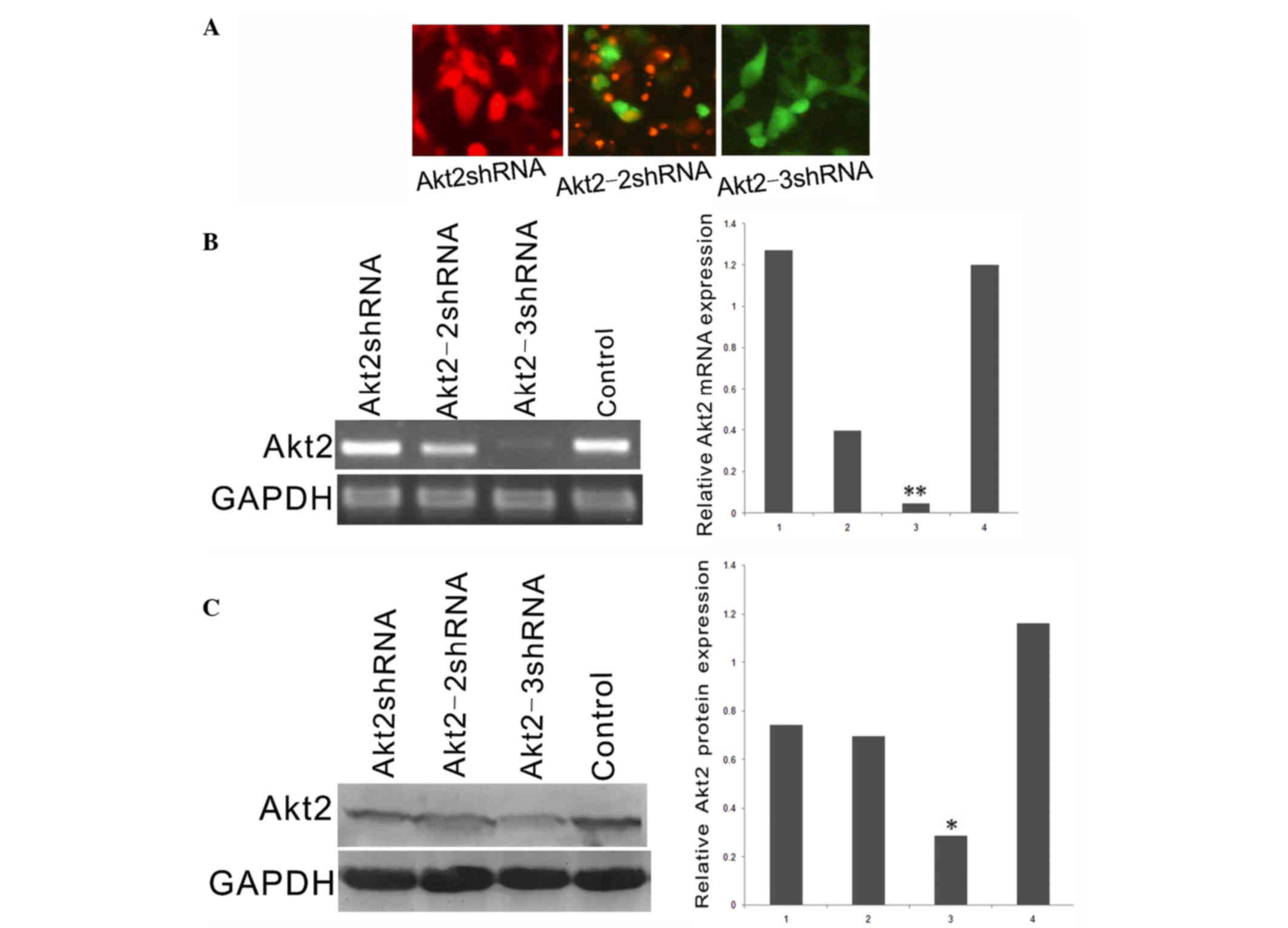
The endogenous gene, Akt2, was selected as the
target because, in our previous studies, it was shown that Akt2 was
highly expressed in the SKOV3 human ovarian cancer cell line
(15,16). First, images of SKOV3 cells
transfected with the Akt2shRNA, Akt2-2shRNA, Akt2-3shRNA or
negative control vectors were obtained using a conventional optical
fluoroscope to determine their transfection efficiencies (Fig. 4A). Subsequently, the mRNA and protein
expression of Akt2 in the shRNA-transfected cells was detected
(Fig. 4B and C). Notably, the
Akt2-3shRNA vector had a stronger silencing effect than the
Akt2shRNA or Akt2-2shRNA vectors, as compared with the control
(P<0.05); however, the Akt2shRNA and Akt2-2shRNA vectors had
stronger silencing effects than the control (P<0.05).
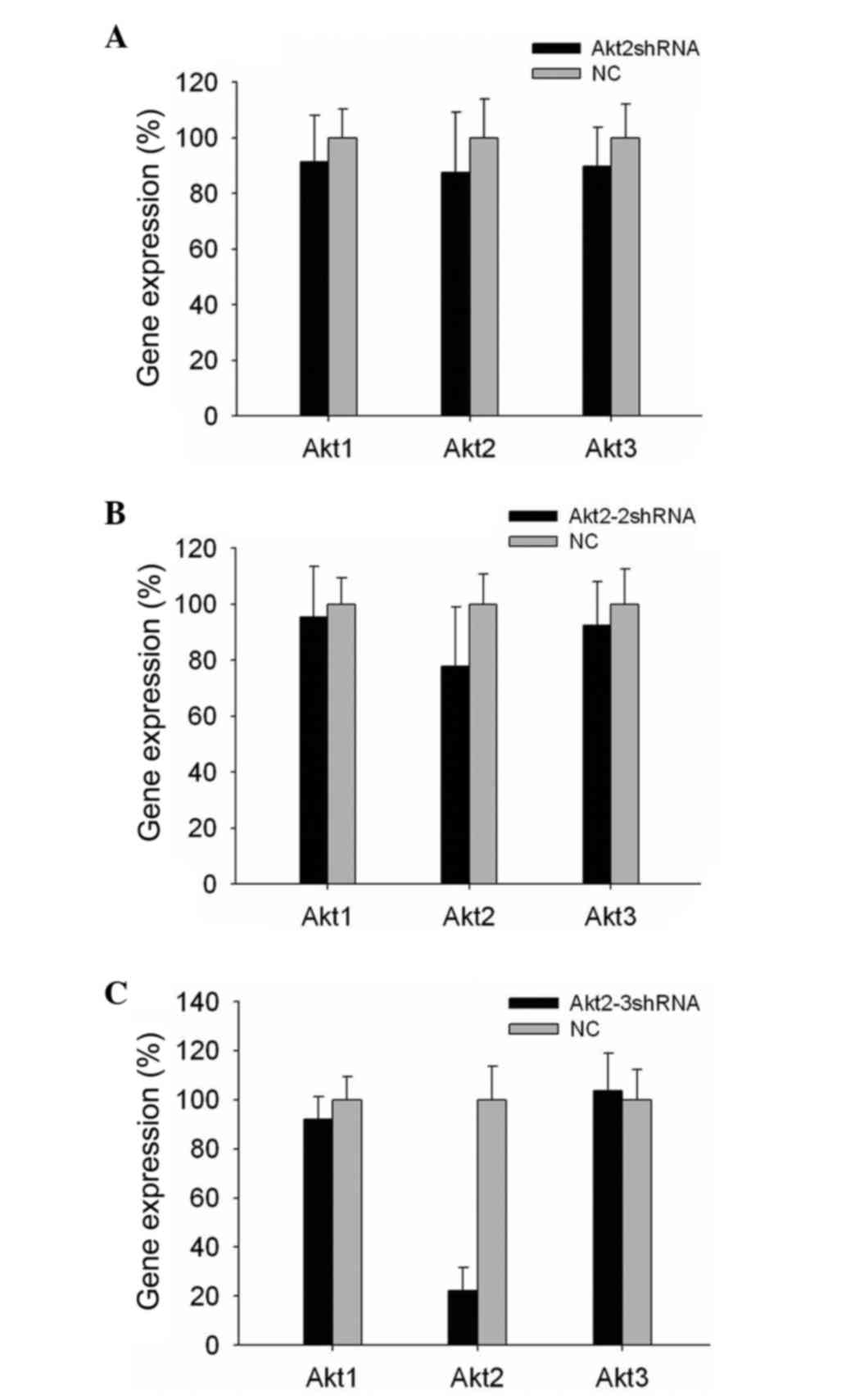
RT-qPCR was used to determine the gene silencing
specificity (Fig. 5). It was found
that, in all shRNA vector-transfected cells, the mRNA expression
levels of Akt1 and Akt3 did not change significantly (P>0.05),
while Akt2 was silenced. In addition, it was observed that the
single-site shRNA vectors produced more frequent off-target effects
than the two-site and multi-site shRNA vectors (P<0.05; Fig. 5). Notably, the same results were
obtained when other genes were silenced (data not shown).
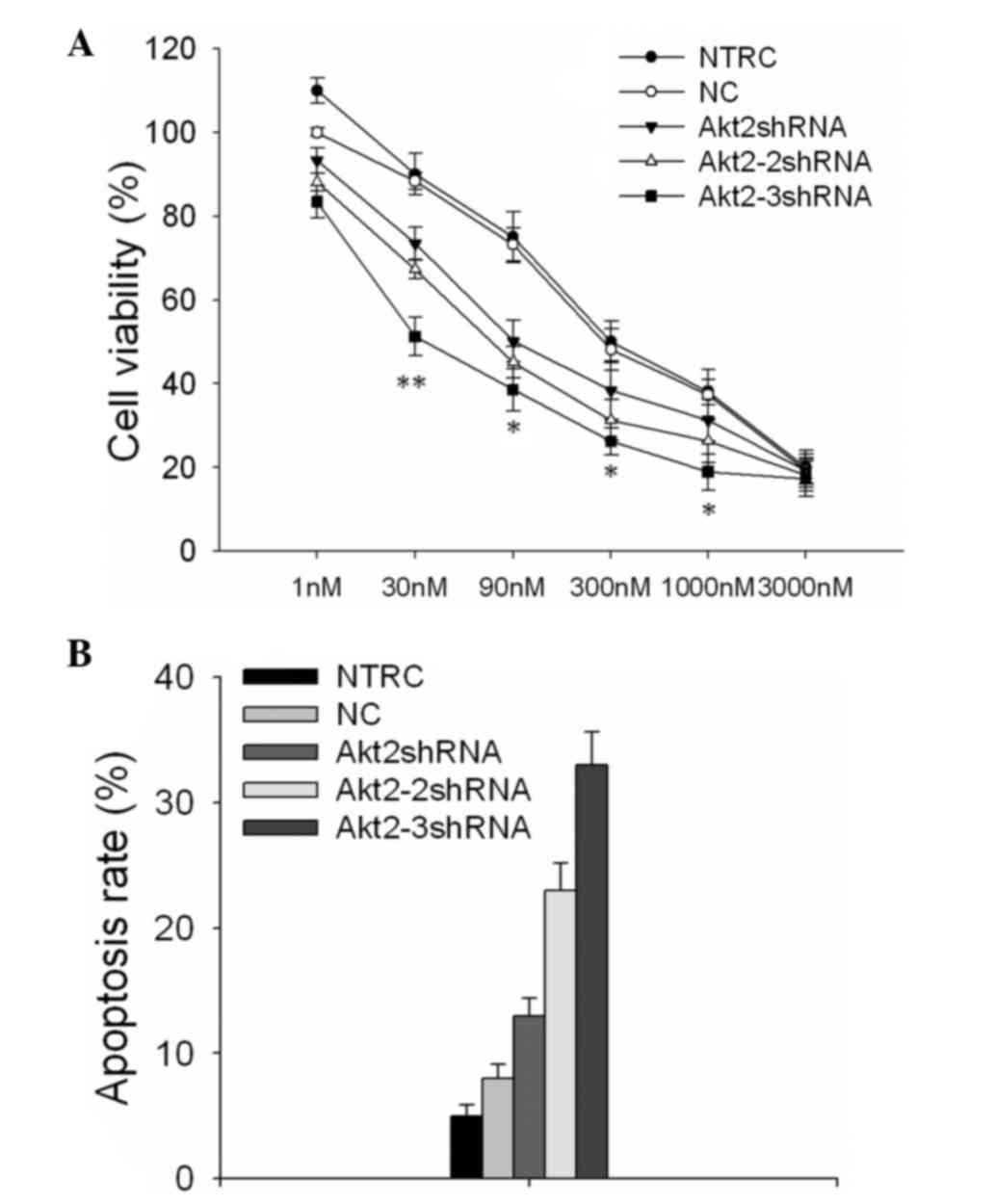
Multi-site shRNA vector shows stronger
loss-of-function of the endogenous Akt2 gene in SKOV3 cells than
the single-shRNA and two-site shRNA vectors
As shown in Fig. 6,
the drug tolerance of SKOV3 cells to PTX following silencing of the
endogenous Akt2 gene with the different vectors was detected.
Following treatment of the shRNA vector-transfected SKOV3 cells
with various concentrations of PTX for 48 h, MTT assays were
performed to detect the cell viability. According to the data,
cells transfected with the Akt2-3shRNA vector showed a markedly
lower viability than the other cells (Fig. 6A). The half maximal inhibitory
concentration values were increased 5.16- and 3.57-fold in cells
transfected with the Akt2-shRNA and Akt2-2shRNA vectors,
respectively (P<0.01), and 17.24- and 21.07-fold in the negative
control and blank control cells, respectively (P<0.01), as
compared with the cells transfected with the Akt2-3shRNA
vector.
The apoptosis rate was evaluated following treatment
with 90 nM PTX for 48 h (Fig. 6B).
Cells transfected with the Akt2-3shRNA vector displayed an
apoptosis rate of 26±2.32%, which was significantly higher than
that of other cells, including an apoptotic rate of 18±3.14% in
SKOV3 cells transfected with Akt2-2shRNA (P=0.02) and that of
15±2.06% in SKOV3 cells transfected with Akt2-shRNA (P<0.001).
The apoptosis rates of the cells transfected with the three shRNA
vectors were all higher than those of the negative (6±1.92%) or
blank controls (5±1.41%), and these differences were statistically
significant (P<0.01).
Discussion
The application of RNAi has revolutionized the study
of gene function in vitro and promises to facilitate
large-scale loss-of-function studies in human cells (22). Recently, siRNA and shRNA vectors have
been used successfully (23), but
many practical and theoretical problems remain before RNAi becomes
routine. To develop a resource that will enable broad and efficient
silencing in human cells, the present study designed a platform for
the construction of multi-site shRNA vectors that contain three
shRNA fragments targeting different sites of a single gene. To
verify the effectiveness of this method, the exogenous reporter
gene DsRed and the endogenous gene Akt2 were selected as the two
target genes and their corresponding multiple and single RNAi
vectors were constructed. The present study demonstrated that the
multi-site shRNA vector had better silencing effects on DsRed at
the mRNA and protein expression levels than the single-site and
two-site vectors after 2 days, although no significant differences
were observed before that time. In addition, the multi-site shRNA
vector exhibited the best loss-of-function effects on the
endogenous Akt2 gene, as it had the most powerful ability to
reverse PTX-induced resistance in SKOV3 cells. Furthermore, the IFN
response to the shRNA vectors was assessed, according to a previous
study (21). Notably, the mRNA
expression of OAS1 and the protein expression of STAT1 were not
readily detected following addition of exogenous IFN-β to the
cells. In addition, when Akt2 was silenced using the various shRNA
vectors, the expression levels of its homologous genes, Akt1 and
Akt3, did not change significantly (P>0.05), and were changed to
the least extent in the cells transfected with the multi-site shRNA
vector, suggesting that this vector decreased the occurrence of
off-target effects.
Currently, the most popular shRNA technique involves
a single shRNA ligated into a plasmid or lentiviral vector
containing polymerase III promoters (24,25).
Transfection of cells with the plasmid or lentiviral vector can
then eliminate the expression of a target gene (26,27).
However, off-target effects, which are the result of partial
homology to other transcripts, complicate the application of shRNAs
(28,29). To avoid this inherent property of
shRNAs, numerous study groups have screened for the optimum shRNA
(5,30). In our previous study, two independent,
19-nucleotide sequences that targeted the Akt2 gene were designed
and were cloned into two different plasmids (15). Subsequently, SKOV3 cells were
co-transfected with the two shRNA vectors and it was demonstrated
that the two-site shRNA reduced the expression of the Akt2 gene
more effectively than the single-site shRNA (15). However, co-transfection is not always
convenient because many mammalian cells are not easily transfected,
and many cells exhibit plasmid incompatibility (31). To mitigate these problems, Berns et
al (11) proposed the concept of
multi-site shRNAs for large-scale RNAi screens in human cells.
Other groups have constructed multiple shRNA vectors to knockdown
multiple genes (32–34), and many groups have constructed
lentiviral shRNA vectors when establishing an RNAi library
(5,35). These improvements resulted in
on-target effects by the shRNA vectors on the corresponding genes.
Therefore, multi-site shRNA plasmids are popular and have been
widely applied in human cells (36,37);
however, few studies have investigated the properties of multi-site
shRNA vectors targeting a single gene.
The present study compared the suppression abilities
of different-site shRNA vectors. Previously, endogenous genes have
always been assessed (36); thus, the
present study included the exogenous reporter gene DsRed and an
endogenous gene as targets. First, the construction methods of
multi-site shRNA vectors were demonstrated, which involved
reconstructing the pEGFP-C1 into the pEGFP-C1-U6-U6-U6 vector,
which carries three human U6 promoters. The shRNA double
oligonucleotides were synthesized in vitro, annealed and
ligated into the reconstructed vector to form the multi-site RNAi
vector. This strategy bypassed the mutation rate problem of PCR
and, therefore, further simplified the construction procedure.
After the construct was successfully made, whether
the multi-site shRNA vector was more advantageous than the
single-site and two-site shRNA vectors for silencing an exogenous
gene in vitro was investigated. Since few studies have
performed silencing of exogenous genes in mammalian cells, the
post-inhibition time points were broadened to ensure that the
silencing effects of the various shRNA vectors were observed. The
results demonstrated that the multi-site shRNA vector could
maintain a longer and higher RNAi effect than the single-site and
two-site shRNA vectors.
When targeting the endogenous Akt2 gene, it was
determined that the 2-day time point was the optimum time point for
observing the effects. Within 2 days, the multi-site shRNA vector
had suppressed the expression of Akt2 at the mRNA and protein
levels and exhibited a significantly greater loss-of-function than
the single-site and two-site shRNA vectors. The off-target effects
of the vectors were assessed by detecting the expression of genes
that were homologous to Akt2. While the expression levels of Akt1
and Akt3 decreased marginally in the cells transfected with the
single-site shRNA, the expression levels of these genes were
unchanged in the two-site and multi-site groups. These results
suggested that multi-site shRNA interference could simultaneously
enhance suppression efficiency and reduce off-target effects.
Various limitations associated with RNAi still
exist, even though steps toward improvement were made in this
study. The present study did not performe research on cells from
organisms other than humans and, therefore, it may not be assumed
that the RNAi effects using this strategy are the same in other
organisms as in human cells. Further studies of a braod array of
cell types are required. Furthermore, although plasmids are safer
tools than lentiviruses, when using suspended cells, such as blood
and stem cells, lentiviruses may be the better choice for silencing
genes.
In summary, the present study produced a highly
efficient, multi-site shRNA vector that targeted exogenous and
endogenous genes in human cells. This method was much safer, easier
to perform and more suitable for constructing an RNAi library, as
compared with methods involving co-transfection of cells with two
or three shRNAs to silence a single gene. In addition, the time
requirement was reduced by not having to screen for the best shRNA
and position problems were avoided during the process of
construction. These accomplishments will likely broaden the
application of these novel vectors for studying gene function in
vivo. Furthermore, a specific advantage was the possibility of
targeting three different genes simultaneously, which overcomes the
difficulty of co-transfection. Using this system, the function of
more than one gene can be monitored and controlled without the risk
of off-target effects. The multi-site shRNA vector that is
described here is particularly beneficial when searching for a wide
range of novel genes and is valuable for evaluating signaling
pathways involved in tumorigenesis and the mechanisms of
chemoresistance, as well as the exploitation of adenovirus
restructuring drugs.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (nos. 30973184 and 81101971),
Guangdong Natural Science Foundation (nos. B2011295 and
S2011040006012) and the ‘973’ Program of China (no.
2009CB521800).
References
|
1
|
Fire A, Xu S, Montgomery MK, Kostas SA,
Driver SE and Mello CC: Potent and specific genetic interference by
double-stranded RNA in Caenorhabditis elegans. Nature. 391:806–811.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elbashir SM, Harborth J, Lendeckel W,
Yalcin A, Weber K and Tuschl T: Duplexes of 21-nucleotide RNAs
mediate RNA interference in cultured mammalian cells. Nature.
411:494–498. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Crombez L and Divita G: A non-covalent
peptide-based strategy for siRNA delivery. Methods Mol Biol.
683:349–360. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shan G, Li Y, Zhang J, Li W, Szulwach KE,
Duan R, Faghihi MA, Khalil AM, Lu L, Paroo Z, et al: A small
molecule enhances RNA interference and promotes microRNA
processing. Nat Biotechnol. 26:933–940. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moffat J, Grueneberg DA, Yang X, Kim SY,
Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK,
et al: A lentiviral RNAi library for human and mouse genes applied
to an arrayed viral high-content screen. Cell. 124:1283–1298. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ku SH, Jo SD, Lee YK, Kim K and Kim SH:
Chemical and structural modifications of RNAi therapeutics. Adv
Drug Deliv Rev. 104:16–28. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Khandelwal N, Breinig M, Speck T, Michels
T, Kreutzer C, Sorrentino A, Sharma AK, Umansky L, Conrad H,
Poschke I, et al: A high-throughput RNAi screen for detection of
immune-checkpoint molecules that mediate tumor resistance to
cytotoxic T lymphocytes. EMBO Mol Med. 7:450–463. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cho JS, Kim YC and Morrison SL: Inhibitors
of MyD88-dependent proinflammatory cytokine production identified
utilizing a novel RNA interference screening approach. PLoS One.
4:e70292009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Du C, Ge B, Liu Z, Fu K, Chan WC and
McKeithan TW: PCR-based generation of shRNA libraries from cDNAs.
BMC Biotechnol. 6:282006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scherer LJ, Yildiz Y, Kim J, Cagnon L,
Heale B and Rossi JJ: Rapid assessment of anti-HIV siRNA efficacy
using PCR-derived pol III shRNA cassettes. Mol Ther. 10:597–603.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Berns K, Hijmans EM, Mullenders J,
Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M,
Nijkamp W, Weigelt B, et al: A large-scale RNAi screen in human
cells identifies new components of the p53 pathway. Nature.
428:431–437. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stewart SA, Dykxhoorn DM, Palliser D,
Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et
al: Lentivirus-delivered stable gene silencing by RNAi in primary
cells. RNA. 9:493–501. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Klinghoffer RA, Roberts B, Annis J,
Frazier J, Lewis P, Linsley PS and Cleary MA: An optimized
lentivirus-mediated RNAi screen reveals kinase modulators of
kinesin-5 inhibitor sensitivity. Assay Drug Dev Technol. 6:105–119.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang YH, Wang ZX, Qiu Y, Xiong J, Chen YX,
Miao DS and De W: Lentivirus-mediated RNAi knockdown of
insulin-like growth factor-1 receptor inhibits growth, reduces
invasion, and enhances radiosensitivity in human osteosarcoma
cells. Mol Cell Biochem. 327:257–266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xing H, Weng D, Chen G, Tao W, Zhu T, Yang
X, Meng L, Wang S, Lu Y and Ma D: Activation of
fibronectin/PI-3K/Akt2 leads to chemoresistance to docetaxel by
regulating survivin protein expression in ovarian and breast cancer
cells. Cancer Lett. 261:108–119. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weng D, Song X, Xing H, Ma X, Xia X, Weng
Y, Zhou J, Xu G, Meng L, Zhu T, et al: Implication of the
Akt2/survivin pathway as a critical target in paclitaxel treatment
in human ovarian cancer cells. Cancer Lett. 273:257–265. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Manche L, Green SR, Schmedt C and Mathews
MB: Interactions between double-stranded-RNA regulators and the
protein-kinase DAI. Mol Cell Biol. 12:5238–5248. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Williams BR: Role of the double-stranded
RNA-activated protein kinase (PKR) in cell regulation. Biochem Soc
Trans. 25:509–513. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Uprichard SL: The therapeutic potential of
RNA interference. FEBS Lett. 579:5996–6007. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jaitin DA and Schreiber G: Upregulation of
a small subset of genes drives type I interferon-induced antiviral
memory. J Interferon Cytokine Res. 27:653–664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leung RK and Whittaker PA: RNA
interference: From gene silencing to gene-specific therapeutics.
Pharmacol Ther. 107:222–239. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Elbashir SM, Harborth J, Weber K and
Tuschl T: Analysis of gene function in somatic mammalian cells
using small interfering RNAs. Methods. 26:199–213. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rubinson DA, Dillon CP, Kwiatkowski AV,
Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus
MT, et al: A lentivirus-based system to functionally silence genes
in primary mammalian cells, stem cells and transgenic mice by RNA
interference. Nat Genet. 33:401–406. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tiscornia G, Singer O and Verma IM: Design
and cloning of lentiviral vectors expressing small interfering
RNAs. Nat Protoc. 1:234–240. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Roelz R, Pilz IH, Mutschler M and Pahl HL:
Of mice and men: Human RNA polymerase III promoter U6 is more
efficient than its murine homologue for shRNA expression from a
lentiviral vector in both human and murine progenitor cells. Exp
Hematol. 38:792–797. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miest T, Saenz D, Meehan A, Llano M and
Poeschla EM: Intensive RNAi with lentiviral vectors in mammalian
cells. Methods. 47:298–303. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jackson AL, Burchard J, Leake D, Reynolds
A, Schelter J, Guo J, Johnson JM, Lim L, Karpilow J, Nichols K, et
al: Position-specific chemical modification of siRNAs reduces
‘off-target’ transcript silencing. RNA. 12:1197–1205. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sledz CA, Holko M, de Veer MJ, Silverman
RH and Williams BR: Activation of the interferon system by
short-interfering RNAs. Nat Cell Biol. 5:834–839. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boudreau RL, Monteys AM and Davidson BL:
Minimizing variables among hairpin-based RNAi vectors reveals the
potency of shRNAs. RNA. 14:1834–1844. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Feinbaum R: Introduction to plasmid
biology. Curr Protoc Mol Biol Chapter. 1:Unit1.5. 2001. View Article : Google Scholar
|
|
32
|
Xia XG, Zhou H and Xu Z: Multiple shRNAs
expressed by an inducible pol II promoter can knock down the
expression of multiple target genes. Biotechniques. 41:64–68. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stove V, Smits K, Naessens E, Plum J and
Verhasselt B: Multiple gene knock-down by a single lentiviral
vector expressing an array of short hairpin RNAs. Electron J
Biotechnol. 9:572–579. 2006. View Article : Google Scholar
|
|
34
|
Xu XM, Yoo MH, Carlson BA, Gladyshev VN
and Hatfield DL: Simultaneous knockdown of the expression of two
genes using multiple shRNAs and subsequent knock-in of their
expression. Nat Protoc. 4:1338–1348. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Root DE, Hacohen N, Hahn WC, Lander ES and
Sabatini DM: Genome-scale loss-of-function screening with a
lentiviral RNAi library. Nat Methods. 3:715–719. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Song J, Giang A, Lu Y, Pang S and Chiu R:
Multiple shRNA expressing vector enhances efficiency of gene
silencing. BMB Rep. 41:358–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim SM, Lee KN, Lee SJ, Ko YJ, Lee HS,
Kweon CH, Kim HS and Park JH: Multiple shRNAs driven by U6 and CMV
promoter enhances efficiency of antiviral effects against
foot-and-mouth disease virus. Antiviral Res. 87:307–317. 2010.
View Article : Google Scholar : PubMed/NCBI
|