Introduction
Bulk high-throughput genomic profiling studies have
improved the understanding of cancer biology and facilitated the
development of novel therapeutics. However, there is increasing
awareness that bulk profiling approaches do not adequately produce
information concerning tumor heterogeneity, an improved insight
into which may facilitate the development of more effective
therapeutic strategies (1).
Genomic profiling of individual single cells is
currently technically available and recent reports of the highly
parallel expression profiling of thousands of cells suggest that
single-cell genomic profiling for clinical applications may become
a reality (2,3). Notably, single-cell profiling using flow
cytometry for immunophenotyping is currently a routine
hematological diagnostic assay (4).
Single-cell genomic profiling is therefore, in theory, potentially
of clinical utility in the diagnostic work-up of a hematological
malignancy such as acute myeloid leukemia (AML).
AML is a malignant disease of abnormally
differentiated cells of the hematopoietic system (5). It is a clonally complex disease that is
characterized by the presence of multiple clonal populations in the
primary cancer, any of which may evolve to result in relapse
(6).
Single-cell genomic profiling enables the
distinction of tumor heterogeneity, and may improve clinical
diagnostics through the identification of putative subclonal
populations and their respective drug sensitivity profiles
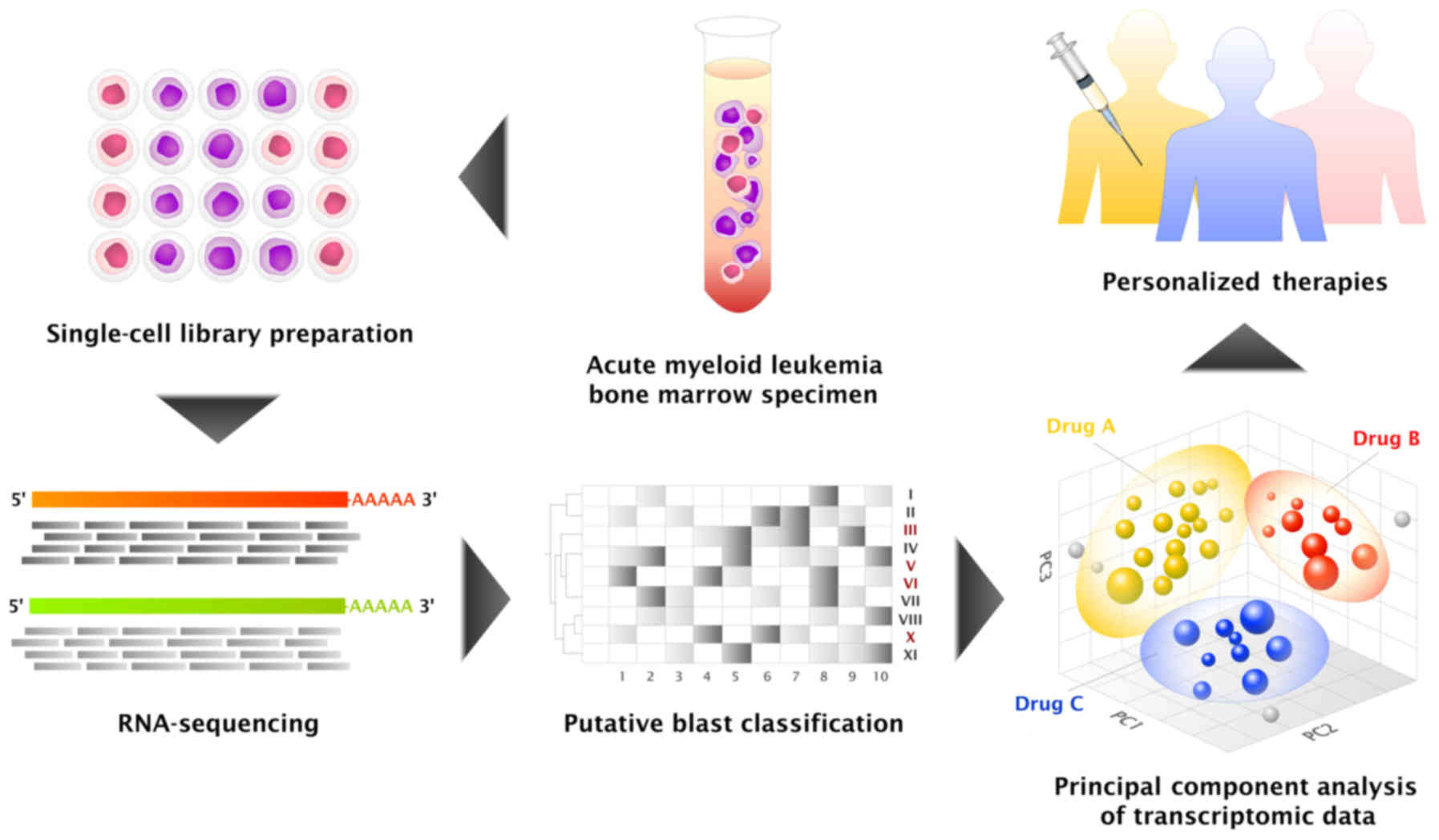
(Fig. 1). In an attempt to develop a
clinically relevant single-cell genomic profiling protocol, a pilot
study of single-cell RNA-sequencing (RNA-seq) of an acute myeloid
leukemia (AML) sample was performed.
Materials and methods
Sample
An AML bone marrow sample, which was harvested in
February 2011 from a 35-year-old female patient, was obtained from
the archives of the Department of Hematology-Oncology (National
University Hospital, Singapore). Ethical approval was obtained for
the present study (Domain Specific Review Boards; National
Healthcare Group, Singapore; ref. 2016/00547). Informed consent was
obtained from the subject participating in the present study.
Flow cytometry
A total of 250,000 events were acquired for
multiparametric analysis using a lyse-wash method on bone marrow
cells (7). Blasts were identified
using a cluster of differentiation (CD) 45/CD34/CD117/human
leukocyte antigen-antigen D related (HLA-DR) combination (7).
Single-cell isolation for RNA-seq
A total of 5×106 cells were incubated
with anti-CD45 antibody (Miltenyi Biotec, Inc., Cambridge, MA, USA;
cat. no. 130-080-201; clone, 5B1) for 1 h at 4°C to stain white
blood cells, while Hoechst 33342 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA; cat. no. H3570) was used to stain nuclei by
adding the staining solution to the cells for 1 h at 4°C. Cells
were loaded at the optimal concentration (250,000 cells/ml, as
recommended by the manufacturer) into the microfluidics chip.
Single cells were isolated into individual chambers
using an integrated fluidic circuit (IFC) on the Automated
Microfluidic C1 system (Fluidigm Corporation, San Francisco, CA,
USA). Cells positive for CD45 and Hoechst were lysed, and RNA
isolation and complementary DNA (cDNA) synthesis was performed
using the SMART-Seq® v4 Ultra® Low Input RNA
kit for Sequencing (Clontech Laboratories, Inc., Mountainview, CA,
USA; cat. no. 634888) which was preamplified using a unique SMARTer
II A oligonucleotide and template switch primer (both reagents
being present in the SMARTer Ultra Low RNA kit; Clontech
Laboratories, Inc.; cat. no. 634833), according to the
manufacturer's protocol. The cDNA was harvested manually by
retrieving 3.5 µl of cDNA from the wells of the IFC for library
preparation. Notably, only RNA strands with polyadenylated
[poly(A)] tails were converted to cDNA and used for downstream
processing.
Library preparation and
next-generation sequencing
Using the coordinates from the imaging, 20 cells
that stained positive for the leukocyte marker CD45 and had intact
nuclei, as observed using the Hoechst stain, were selected. Library
preparation was performed using the Nextera XT DNA Sample
Preparation kit (Illumina, Inc., San Diego, CA, USA; cat. no.
FC-131-1096), according to the manufacturer's protocol. The 20
libraries were processed individually, with each library being
assigned a unique barcode for pooled multiplex sequencing using the
Illumina HiSeq 2000 platform (Illumina, Inc.), according to the
manufacturer's protocol. Paired-end 100 bp reads were generated for
analysis.
Mapping and quantification of
single-cell RNA-seq data
Paired-end FASTQ files were initially mapped to the
reference human Hg19 transcriptome (ftp.ensembl.org/pub/release-75/gtf/homo_sapiens/Homo_sapiens.GRCh37.75.gtf.gz)
(8) using Tophat2 (version 2.1.0;
Johns Hopkins University, Baltimore, MD, USA) (9). Aligned reads (BAM files) were
subsequently sorted and indexed using SAMtools (version 1.2;
Wellcome Trust Sanger Institute, Cambridge, UK) (10). Cufflinks (version 2.2.1; University of
Washington, Seattle, WA, USA) (11)
was utilized for final transcriptome assembly (cufflink and
cuffmerge function), and abundance estimation and
normalization in Fragments Per Kilobase of transcript per Million
mapped reads (FPKM) units (cuffnorm function).
Cell-type classification
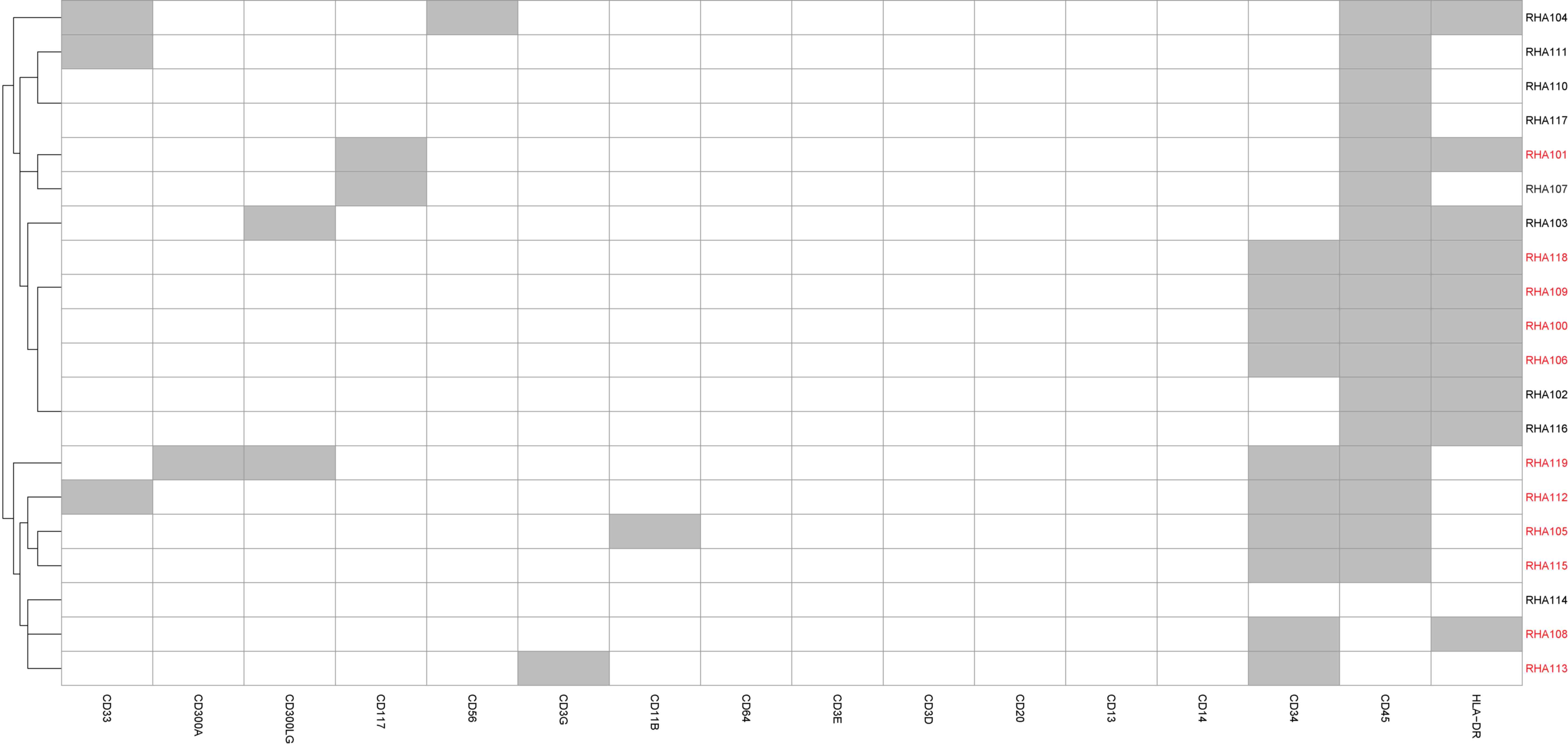
A heatmap was generated for the key cell-type
specific markers (typically used inimmunophenotyping using flow
cytometry, including CD34 and CD45) based on their expression
levels. For any gene, the presence of a transcript with an FPKM
normalized expression value >0 is indicative of gene expression,
while a FPKM normalized expression value of 0 indicates absence of
expression. The presence and absence of the cell-type specific
markers were plotted in a heatmap generated using the ‘pheatmap’
package (version 1.0.8) produced by the R Programming Environment
(www.r-project.org).
Based on the gene expression profiles, cells that
were CD34-positive, or HLA-DRA- and CD117-positive, were classified
as ‘putative blasts’ (12).
Principal component analysis
Principal component analysis was carried out on the
log2-transformed FPKM normalized expression values of
all transcripts using the prcomp function of the R
Programming Environment.
Targeted DNA-sequencing (DNA-seq)
Targeted DNA-seq was performed as previously
described (13,14). A total of 50 ng of genomic DNA was
extracted from the AML bone marrow sample and processed using the
TruSight Myeloid Sequencing Panel (Illumina, Inc.). A total of 54
genes known to be mutated inmyeloid neoplasms, including fms
related tyrosine kinase 3 (FLT3), nucleophosmin
(NPM1) and DNA methyltransferase 3 alpha (DNMT3A),
were assessed. The TruSeq Amplicon (BaseSpace Workflow; version
1.1.0.0; Illumina, Inc.) was used to generate the BAM and VCF
files. Visualization of reads was performed using the Integrative
Genomics Viewer (version 2.3.69; Broad Institute, Cambridge, MA,
USA) (15). Pindel (version 0.2.5a8;
McDonnell Genome Institute, Washington University School of
Medicine, St. Louis, MO, USA) (16)
was used to identify the presence of FLT3 internal tandem
duplications.
Variant calling of RNA-seq data
Variant calling analysis was carried out on the
aligned paired end reads using the Genome Analysis Toolkit (Broad
Institute) (version3.4.46; Haplotype Caller function) with
reference to the aforementioned human Hg19 genome (17,18).
Variants identified from the analysis were annotated using the
SeattleSeq Annotation webserver (snp.gs.washington.edu/SeattleSeqAnnotation138)
(19). Visualization of reads was
performed using the SAMtools tview function (10).
Results
Number of RNA-seq reads per cell. The number of
reads per cell was between 4.5 million and 11.4 million (Table I), which is consistent with previous
single-cell RNA-seq studies (20–23).
 | Table I.Number of RNA-sequencing reads per
cell. |
Table I.
Number of RNA-sequencing reads per
cell.
| Cell number | Number of reads,
millions |
|---|
| RHA100 |
6.3 |
| RHA101 |
9.9 |
| RHA102 |
9.4 |
| RHA103 |
5.1 |
| RHA104 |
4.7 |
| RHA105 |
8.2 |
| RHA106 | 11.4 |
| RHA107 |
7.4 |
| RHA108 |
7.8 |
| RHA109 |
9.2 |
| RHA110 |
5.5 |
| RHA111 |
5.2 |
| RHA112 |
5.2 |
| RHA113 |
5.9 |
| RHA114 |
4.5 |
| RHA115 | 11.4 |
| RHA116 |
9.3 |
| RHA117 |
6.8 |
| RHA118 |
9.3 |
| RHA119 | 10.1 |
Cell-type classification
Immunophenotyping using flow cytometry demonstrated
the blast population to comprise of ~65% the total number of cells.
Based on the single-cell gene expression profile, 11/20 cells were
identified to be putative blasts (Fig.
2).
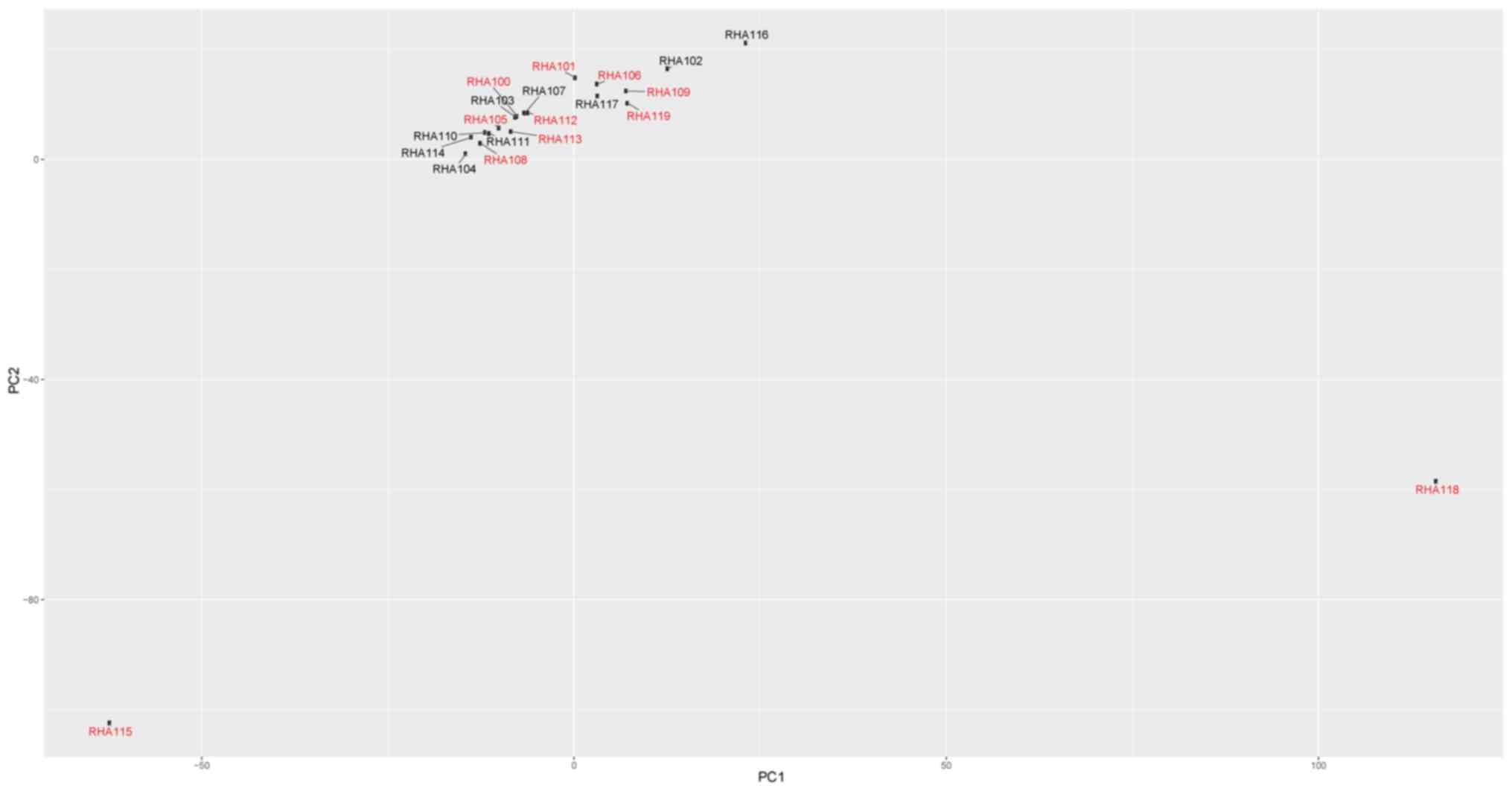
Principal component analysis
Principal component analysis was performed in an
attempt to identify potential subclonal populations (Fig. 3). Two outlier cells were identified,
RHA115 and RHA118. Based on their gene expression profile (Fig. 2), these cells were classified as
putative blasts.
Variant calling of RNA-seq data
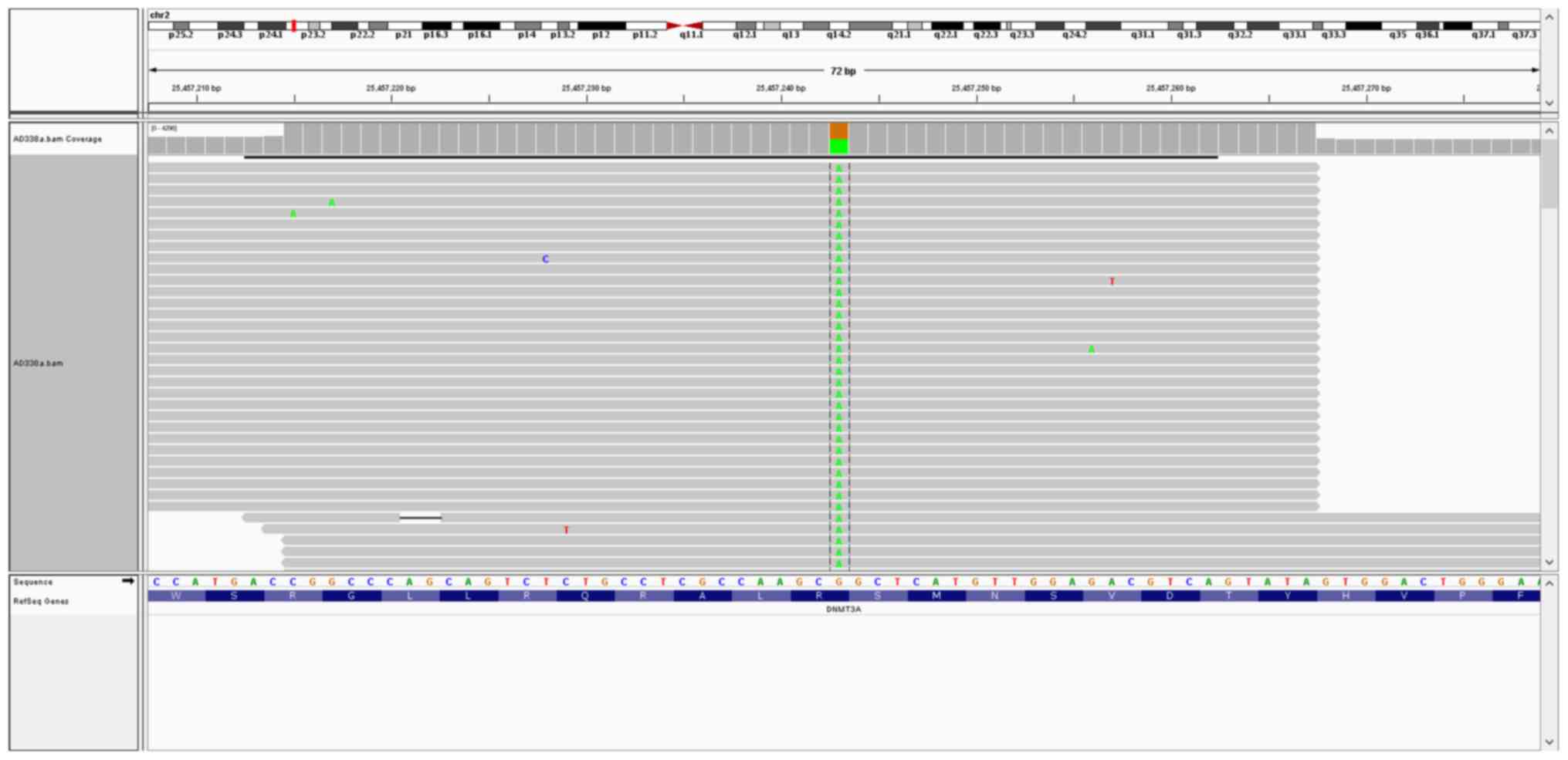
Targeted DNA-seq revealed the presence of
aDNMT3A mutation (c.2644C>T; p.Arg882Cys; Fig. 4); an NPM1 mutation
(c.859_860insTCTG; p.Trp288CysfsTer12); and a 108 bp FLT3
internal tandem duplication (data not shown).
Variant calling of the RNA-seq data did not identify
cells with any of the aforementioned NPM1 and FLT3
mutations. The DNMT3A mutation (c.2644C>T; p.Arg882Cys)
was identified in one cell (RNA human AML119) (Fig. 5). Coverage analysis was performed in
an attempt to understand the apparent absence of NPM1 and
FLT3 transcript mutations, and low abundance of
DNMT3A transcript mutations across the 20 cells. This
revealed the reason to be the absence of transcripts mapping to the
relevant mutation site, potentially secondary to stochastic
transcript dropout (24).
Discussion
Single-cell genomic analysis of AML has been
previously reported (25,26). However, these studies involved only
DNA analysis. To the best of our knowledge, the present study is
the first single-cell transcriptomic analysis of AML.
In the present study, a clinical workflow for
single-cell transcriptomic profiling has been piloted. Using
single-cell RNA-seq, putative blasts were identified based on the
gene expression profile of conventional immunophenotypic markers
used in routine flow cytometry. For flow cytometric analysis, ~20
markers are typically used for profiling. There is a large contrast
with transcriptomic analysis, as there are in principle ≥20,000
markers (genes) (27) that can be
utilized, and individual cellular characterization is able
theoretically to be highly detailed.
In addition to information derived from expression
profiling, mutational (variant) data provides further information
that maybe useful for individual cell categorization (26). Variant identification is most commonly
performed on DNA-seq data (28).
However, variant identification has also been performed on bulk
(29) and single-cell RNA-seq data
(23,30). In the present study, the
DNMT3Ap.Arg882Cys mutation was identified in the transcript,
providing evidence that the mutant transcript is expressed.
High-dimensional data presents an opportunity for
increased cellular characterization and the potential
identification of subclonal populations. Principal component
analysis of the dataset in the present study revealed two putative
blasts that did not cluster with the other blasts. In future
studies, the authors of the present study aim to investigate the
possibility of predicting the drug sensitivity of putative
subclonal populations based on high-dimensional characterization,
as has been performed in previous studies (31).
One of the primary limitations of the protocol
proposed in the present study is the stochastic RNA loss, in which
between 60 and 90% of poly (A) RNA may be lost during sample
preparation (24). In the
presentstudy, FLT3 and NPM1 transcript mutations were
not identified in the 20 cells, while the DNMT3A
(c.2644C>T; p.Arg882Cys) transcript mutation was identified in
one cell. Following further analysis, this observation maybe
explained by the absence of transcripts mapping to the relevant
mutation site. Significant methodological improvements and protocol
optimization are required to overcome this limitation.
Another current limitation is the relatively low
throughput of the protocol proposed in the present study. Due to
reasons of cost and logistics, routine clinical genomic profiling
of a hundred cells is currently challenging (32). By contrast, flow cytometric
immunophenotyping typically involves profiling tens of thousands of
cells (33). With the development of
higher-throughput platforms (2,3), there is
the potential that the cost of single-cell genomic profiling will
decrease significantly to a point where it becomes viable for
clinical implementation.
Despite the aforementioned limitations, single-cell
genomic profiling may lead to the improved diagnosis and
theragnosis of various types of cancer, including AML. In the
present study, a possible single-cell genomic profiling protocol
was piloted for clinical diagnostics. In future studies, a larger
number of cells may need to be profiled to identify distinct
subclonal populations and predict respective drug sensitivity
profiles based on subclonal genomic signatures.
Acknowledgements
The present study was supported by the Clinician
Scientist Award (grant no. NMRC/CSA/046/2012) and the Clinician
Scientist Individual Research Grant (grant no. CIRG/1379/2013) from
the National Medical Research Council (Singapore) awarded to
R.F.
References
|
1
|
Alizadeh AA, Aranda V, Bardelli A,
Blanpain C, Bock C, Borowski C, Caldas C, Califano A, Doherty M,
Elsner M, et al: Toward understanding and exploiting tumor
heterogeneity. Nat Med. 21:846–853. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Klein AM, Mazutis L, Akartuna I,
Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA and Kirschner
MW: Droplet barcoding for single-cell transcriptomics applied to
embryonic stem cells. Cell. 161:1187–1201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Macosko EZ, Basu A, Satija R, Nemesh J,
Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck
EM, et al: Highly parallel genome-wide expression profiling of
individual cells using nanoliter droplets. Cell. 161:1202–1214.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Herzenberg LA, Parks D, Sahaf B, Perez O,
Roederer M and Herzenberg LA: The history and future of the
fluorescence activated cell sorter and flow cytometry: A view from
Stanford. Clin Chem. 48:1819–1827. 2002.PubMed/NCBI
|
|
5
|
Döhner H, Weisdorf DJ and Bloomfield CD:
Acute myeloid leukemia. N Engl J Med. 373:1136–1152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ding L, Tey TJ, Larson DE, Miller CA,
Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan
MD, et al: Clonal evolution in relapsed acute myeloid leukaemia
revealed by whole-genome sequencing. Nature. 481:506–510. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Dongen JJ, Lhermitte L, Böttcher S,
Almeida J, van der Velden VH, Flores-Montero J, Rawstron A, Asnafi
V, Lécrevisse Q, Lucio P, et al: EuroFlow antibody panels for
standardized n-dimensional flow cytometric immunophenotyping of
normal, reactive and malignant leukocytes. Leukemia. 26:1908–1975.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cunningham F, Amode MR, Barrell D, Beal K,
Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G,
Fitzgerald S, et al: Ensembl 2015. Nucleic acids Res. 43:D662–D669.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim D, Pertea G, Trapnell C, Pimentel H,
Kelley R and Salzberg SL: TopHat2: Accurate alignment of
transcriptomes in the presence of insertions, deletions and gene
fusions. Genome Bio. 14:R362013. View Article : Google Scholar
|
|
10
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R: 1000 Genome
Project Data Processing Subgroup: The Sequence Alignment/Map format
and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Trapnell C, Williams BA, Pertea G,
Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ and Pachter
L: Transcript assembly and quantification by RNA-Seq reveals
unannotated transcripts and isoform switching during cell
differentiation. Nat Biotechnol. 28:511–515. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Craig FE and Foon KA: Flow cytometric
immunophenotyping for hematologic neoplasms. Blood. 111:3941–3967.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan B, Hu Y, Ng C, Ban KH, Tan TW, Huan
PT, Lee PL, Chiu L, Seah E, Ng CH, et al: Coverage analysis in a
targeted amplicon-based next-generation sequencing panel for
myeloid neoplasms. J Clin Pathol. 69:801–814. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan B, Ng C, Moshi G, Ban K, Lee PL, Seah
E, Chiu L, Koay ES, Liu TC, Ng CH, et al: Myelodysplastic features
in a patient with germline CEBPA-mutant acute myeloid leukaemia. J
Clin Pathol. 69:652–654. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thorvaldsdóttir H, Robinson JT and Mesirov
JP: Integrative Genomics Viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ye K, Schulz MH, Long Q, Apweiler R and
Ning Z: Pindel: A pattern growth approach to detect break points of
large deletions and medium sized insertions from paired-end short
reads. Bioinformatics. 25:2865–2871. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The Genome Analysis Toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: The Genome Analysis Toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.1–33. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ng SB, Turner EH, Robertson PD, Flygare
SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler
EE, et al: Targeted capture and massively parallel sequencing of 12
human exomes. Nature. 461:272–276. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pollen AA, Nowakowski TJ, Shuga J, Wang X,
Leyrat AA, Lui JH, Li N, Szpankowski L, Fowler B, Chen P, et al:
Low-coverage single-cell mRNA sequencing reveals cellular
heterogeneity and activated signaling pathways in developing
cerebral cortex. Nat Biotechnol. 32:1053–1058. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shalek AK, Satija R, Shuga J, Trombetta
JJ, Gennert D, Lu D, Chen P, Gertner RS, Gaublomme JT, Yosef N, et
al: Single-cell RNA-seq reveals dynamic paracrine control of
cellular variation. Nature. 510:363–369. 2014.PubMed/NCBI
|
|
22
|
Treutlein B, Brownfield DG, Wu AR, Neff
NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA and Quake SR:
Reconstructing lineage hierarchies of the distal lung epithelium
using single-cell RNA-seq. Nature. 509:371–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim KT, Lee HW, Lee HO, Kim SC, Seo YJ,
Chung W, Eum HH, Nam DH, Kim J, Joo KM and Park WY: Single-cell
mRNA sequencing identifies subclonal heterogeneity in anti-cancer
drug responses of lung adenocarcinoma cells. Genome Biol.
16:1272015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim JK, Kolodziejczyk AA, Illicic T,
Teichmann SA and Marioni JC: Characterizing noise structure in
single-cell RNA-seq distinguishes genuine from technical stochastic
allelic expression. Na Commun. 6:86872015. View Article : Google Scholar
|
|
25
|
Hughes AE, Magrini V, Demeter R, Miller
CA, Fulton R, Fulton LL, Eades WC, Elliott K, Heath S, Westervelt
P, et al: Clonal architecture of secondary acute myeloid leukemia
defined by single-cell sequencing. PLoS Genet. 10:e10044622014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paguirigan AL, Smith J, Meshinchi S,
Carroll M, Maley C and Radich JP: Single-cell genotyping
demonstrates complex clonal diversity in acute myeloid leukemia.
Sci Transl Med. 7:281re22015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
International Human Genome Sequencing
Consortium, . Finishing the euchromatic sequence of the human
genome. Nature. 431:931–945. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Papaemmanuil E, Gerstung M, Bullinger L,
Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F,
Bolli N, et al: Genomic classification and prognosis in acute
myeloid leukemia. N Engl J Med. 374:2209–2221. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li M, Wang IX, Li Y, Bruzel A, Richards
AL, Toung JM and Cheung VG: Widespread RNA and DNA sequence
differences in the human transcriptome. Science. 333:53–58. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Patel AP, Tirosh I, Trombetta JJ, Shalek
AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT,
Martuza RL, et al: Single-cell RNA-seq highlights intratumoral
heterogeneity in primary glioblastoma. Science. 344:1396–1401.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mitra AK, Mukherjee UK, Harding T, Jang
JS, Stessman H, Li Y, Abyzov A, Jen J, Kumar S, Rajkumar V and Van
Ness B: Single-cell analysis of targeted transcriptome predicts
drug sensitivity of single cells within human myeloma tumors.
Leukemia. 30:1094–1102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shapiro E, Biezuner T and Linnarsson S:
Single-cell sequencing-based technologies will revolutionize
whole-organism science. Nat Rev Genet. 14:618–630. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee D, Grigoriadis G and Westerman D: The
role of multiparametric flow cytometry in the detection of minimal
residual disease in acute leukaemia. Pathology. 47:609–621. 2015.
View Article : Google Scholar : PubMed/NCBI
|