Introduction
Autophagy is a self-protective cellular mechanism
that occurs during nutrient deficiency, which functions to conserve
vital components in the cell via degradation and recycling of
cytoplasmic constituents (1–4). Furthermore, autophagy may aid cell
survival under conditions of starvation, hypoxia, and metabolic and
chemotherapeutic stress, but superfluous autophagy may trigger cell
death (5). It has been reported that
the inhibition of mTOR and PI3K-mTOR can activate autophagy in
glioma cells, which may promote glioma cell survival (6). It has also been shown that anticancer
drugs that inhibit autophagy are able to promote apoptosis
(7–9),
which suggests that autophagy has a protective role in tumor cells.
This outcome indicates that the role of autophagy acts as a
double-edged sword that has the role of both killing and protecting
cells.
Dichloroacetate (DCA) is a small inhibitor of
pyruvate dehydrogenase kinase (PDK) that activates pyruvate
dehydrogenase (PDH) and increases glucose oxidation by promoting
the influx of pyruvate into the Krebs cycle. DCA facilitates
mitochondrial oxidation of glucose-derived pyruvate to produce ATP.
Moreover, it has recently been demonstrated as a promising nontoxic
antineoplastic agent that promotes apoptosis in breast, colorectal,
and prostate cell carcinoma models (10–13).
However, the correlation between DCA and autophagy is unknown, and
the mechanism by which DCA induces autophagy is the subject of
ongoing investigation. In the present study, we found that DCA
induced autophagy in an esophageal squamous carcinoma cell lines,
TE-1 and TE-8 cells. We observed increased expression of light
chain (LC)-3B-II and minimal apoptosis following DCA treatment.
Additionally, DCA in conjunction with 3-methyladenine (3-MA) to
inhibit autophagy significantly enhanced DCA-induced apoptosis. We
also observed the inhibition of the Akt-mTOR signaling pathway, a
crucial negative regulator of autophagy, which provides insight
into the mechanism of DCA-induced autophagy.
Materials and methods
Cell culture and reagents
Human esophageal squamous carcinoma cells, TE-1,
were cultured in RPMI1640 supplemented with 10% fetal bovine serum
and 100 mg/l streptomycin (Sigma) at 37°C in an atmosphere
containing 5% CO2. The reagents used in the present
study were DCA, 3-MA (both Sigma-Aldrich, St. Louis, MO, USA),
adenovirus (GFP-RFP-LC3; Hanbio Shanghai, China), Lipofectamine
2000 (Invitrogen Life Technologies, Sanghai, China), and an MTT kit
(Sangon Biotech, Sanghai, China). The antibodies used in the
present study were Beclin-1 (Cell Signaling Technology, Inc., MA,
USA), LC3-I/II, P62, Atg5, PARP, Pro-caspase-3 (all Abcam,
Cambridge, UK), β-actin (Santa Cruz Biotechnology Inc., Dallas, TX
USA), Akt, phospho-Akt, mTOR and p-mTOR (all Cell Signaling
Technology,).
Immunofluorescence microscopy
Cells were grown on coverslips in a 24-well plate.
Before the experiment, TE-1 cells were infected with GFP-RFP-LC3
adenovirus. At 24 h post-infection, the media was changed, and the
cells were treated with DCA for 24 h. For immunostaining, the cells
were fixed in 4% paraformaldehyde. Cells were incubated with DAPI
for 5 min and washed three times with PBS. The coverslips were
mounted on slides with Vectashield mounting medium. The images were
captured using an LSM 510 Meta confocal microscope (Carl Zeiss
MicroImaging GmbH, Jena, Germany) and processed using the software
provided by the manufacturer.
Western blot
Proteins were extracted in RIPA buffer (50 mM
Tris-base, 1.0 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1%
sodium deoxycholate, and 1 mM PMSF) and quantified using a BCA
protein assay kit. Samples were separated using 12% SDS-PAGE and
transferred to PVDF membranes. The membranes were blocked for 1 h
with PBS containing 0.1% Tween-20 in 5% skim milk at 37°C and
subsequently probed by the primary antibodies rabbit-anti-Beclin-1
(diluted 1:1,000), rabbit-anti-Atg5 (diluted 1:1,000),
mouse-anti-P62 (diluted 1:1,000), rabbit-anti-LC3 (diluted
1:1,000), rabbit-anti-Procaspase-3 (diluted 1:1,000),
mouse-anti-PARP (diluted 1:1,000), rabbit-anti-Akt,
mouse-anti-Phospho-Akt, rabbit-anti-mTOR and
rabbit-anti-Phospho-mTOR, all used at dilutions of 1:1,000. Blots
were incubated with the respective primary antibodies overnight at
4°C. After washing three times in Tris-buffered saline with
Tween-20 (20 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20), the blots
were incubated with an HRP-conjugated anti-rabbit secondary
antibody (diluted 1:5,000) or an HRP-conjugated anti-mouse
secondary antibody (diluted 1:5,000) for 1 h at room temperature.
Blots were visualized by western chemiluminescence reagents
(Millipore, WBKLS0500). Densitometric quantitation of blotting
strips was performed using the Quality-One software. β-actin was
used as an internal control.
Cell viability, proliferation and
apoptosis assays
For viability assay, cells were seeded at 5,000
cells per well in 96-well plates overnight at 37°C. Subsequently,
DCA and medium control were added to each well, and the cells were
cultured at 37°C for the indicated times. Cell viability was
estimated using an MTT assay. The absorbance was measured at 570 nm
with a microplate reader (Bio-Rad). Cell proliferation was measured
by cell counting. Briefly, cells were re-plated in 24-well plates
at a density of 1×105 cells/well. Cells were incubated for 24 h,
and DCA and medium control were added to each well. Cells were
counted at 24 or 48 h post-treatment with an automated cell
counter. Cell apoptosis was analyzed by flow cytometry using
propidium iodide staining. All experiments were performed in
triplicate.
siRNA preparation and
transfection
At g5 knockdown was accomplished by transfecting
TE-1 cells with targeting siRNA. Atg5 and control siRNA were
synthesized by Sangon Biotech. Cells (3×105) were transfected with
1 µg siRNA using 2.5 µl Lipofectamine 2000 reagent. Scrambled
siRNA, which consists of a scrambled sequence that will not lead to
specific degradation of any known cellular mRNA, was employed as a
negative control.
Statistical analysis
Data are presented as the mean ± SD. Student's
t-test was used for the statistical analysis of the data. All
statistical analyses were conducted using SPSS 19.0 (IBM, SPSS,
Armonk, NY, USA) statistical software. A P-value of <0.05 or
<0.01 was considered to indicate a statistically significant
difference.
Results
DCA-induced autophagy in TE-1
cells
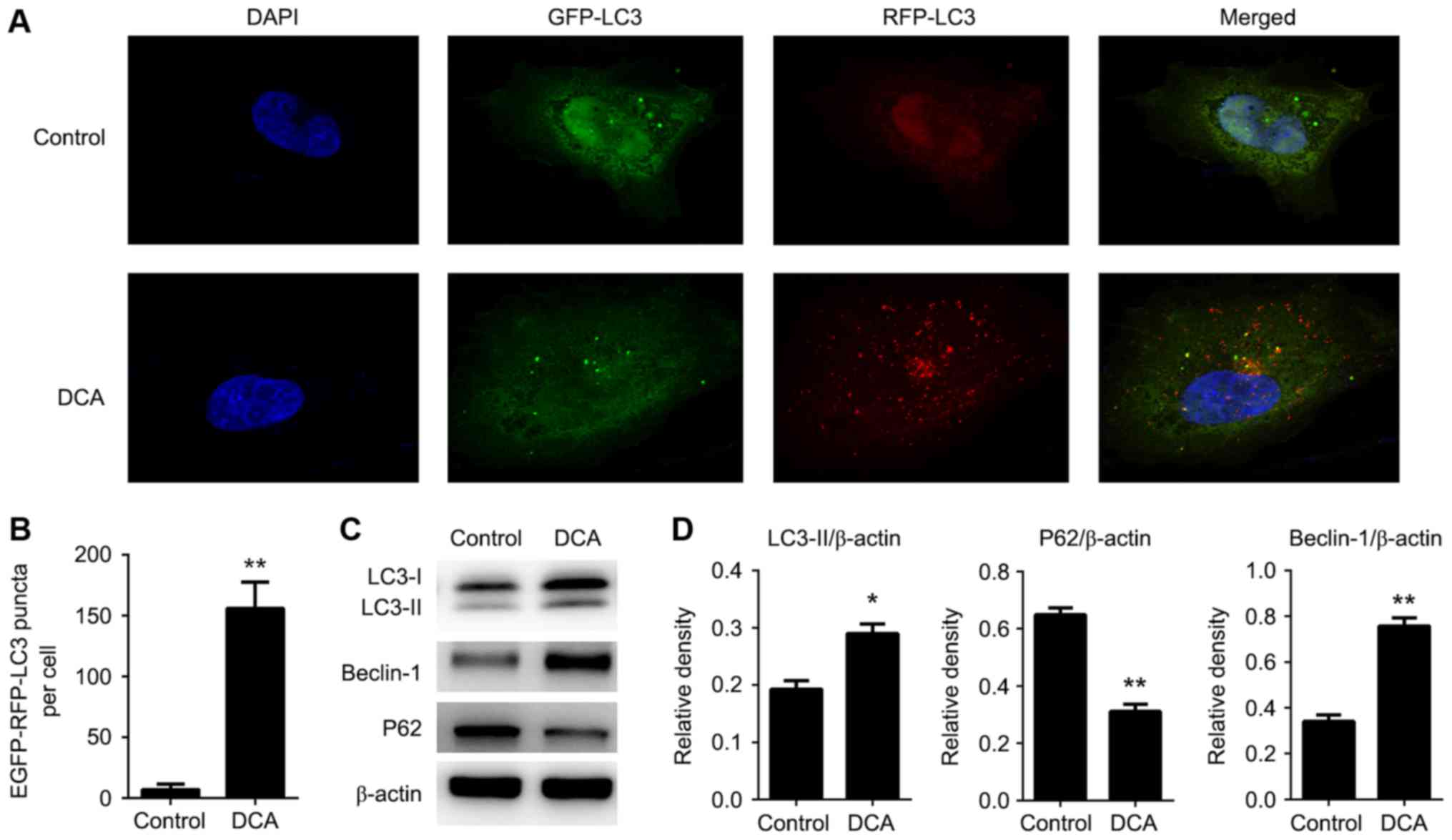
Autophagy is a protective mechanism against
extracellular stress. To investigate the role of DCA in autophagy
regulation of TE-1 cell, we examined the localization of LC3 using
an immunofluorescence assay. We utilized adenovirus encoding a
tandem GFP-RFP-LC3 construct to confirm autophagy induction
(14). The basis of this function is
that the neutral autophagosome and acidic autolysosome showed
different pH sensitivity. The GFP-LC3 degrades, while the RFP-LC3
maintains the puncta when autophagosome fuses with the lysosome to
form autolysosomes. After infection with this adenovirus, we
observed the successful introduction of LC3 proteins upon treatment
with DCA relative to the control (Fig.
1A-B). Moreover, to examine whether autophagy-related proteins
are altered in response to DCA treatment, western blot analysis was
performed. The protein levels of LC3-II, P62 (SQSTM-1) and Beclin-1
were examined. We observed a significant decrease in the levels of
P62 following DCA treatment relative to control conditions.
Moreover, the ratio of the classical autophagy pathway proteins,
LC3-II and Beclin-1/β-actin, was also markedly augmented (Fig. 1C-D). Taken together, these results
indicate that the autophagic flux which occurs in TE-1 cells is
enhanced by DCA treatment.
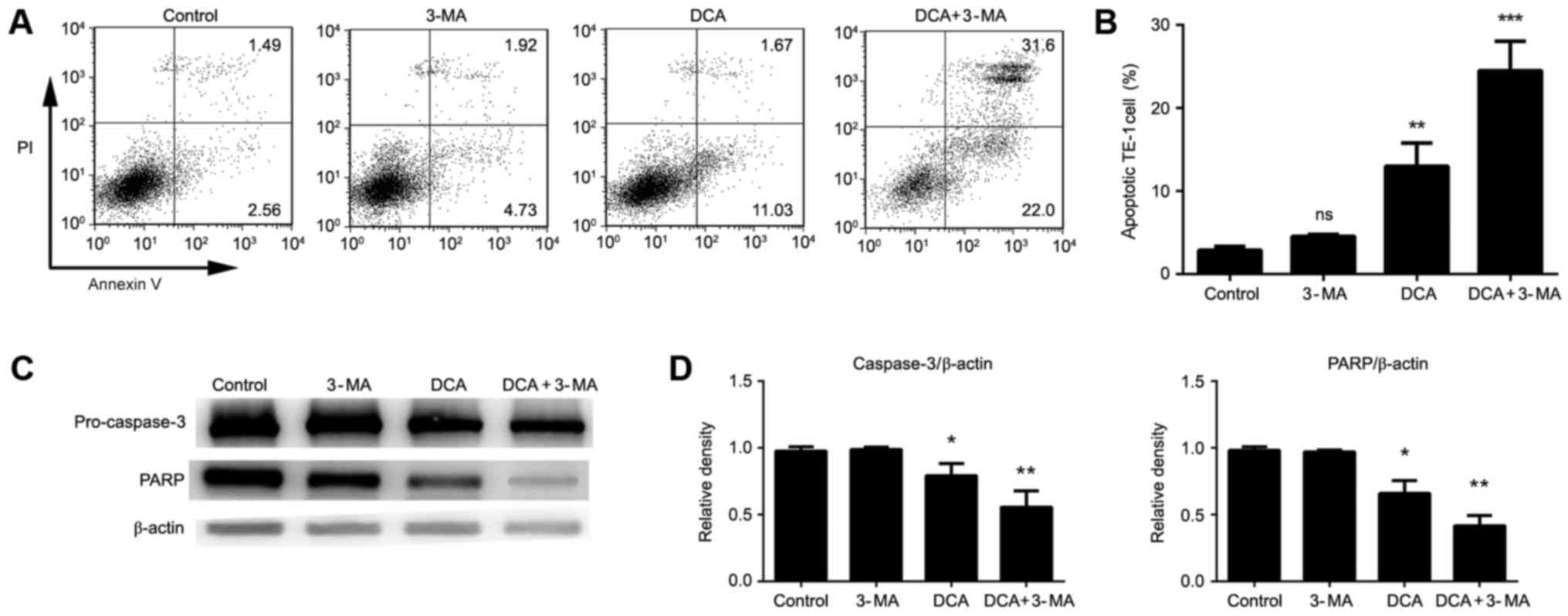
Inhibition of DCA-induced autophagy
facilitates apoptosis of TE-1 cells
The induction of autophagy is considered to be a
protection behavior against apoptosis in cancer cells (15). To examine the influence of DCA
treatment on TE-1 apoptosis, cells were treated with DCA for 24 h,
and flow cytometry was performed. Our results show that lower dose
DCA treatment did not induce a significant level of apoptosis in
TE-1 cells compared with the control. 3-MA, a widely used autophagy
inhibitor, was used to investigate the functional role of autophagy
in response to DCA treatment. Briefly, TE-1 cells were treated
alone or in combination with DCA and 3-MA for 24 h, and the levels
of apoptosis were measured. Interestingly, DCA-induced apoptosis
was markedly enhanced via combined treatment with 3-MA and DCA
compared to either treatment alone or the control group (Fig. 2A-B). Subsequently, the critical
apoptosis-related proteins PARP and Pro-caspase-3 were measured by
western blot analysis and were found to be significantly reduced
upon treatment with DCA and 3-MA (Fig.
2C-D). These results indicated that the antitumor effect of DCA
might be strengthened when the level of autophagy in tumor cells is
inhibited.
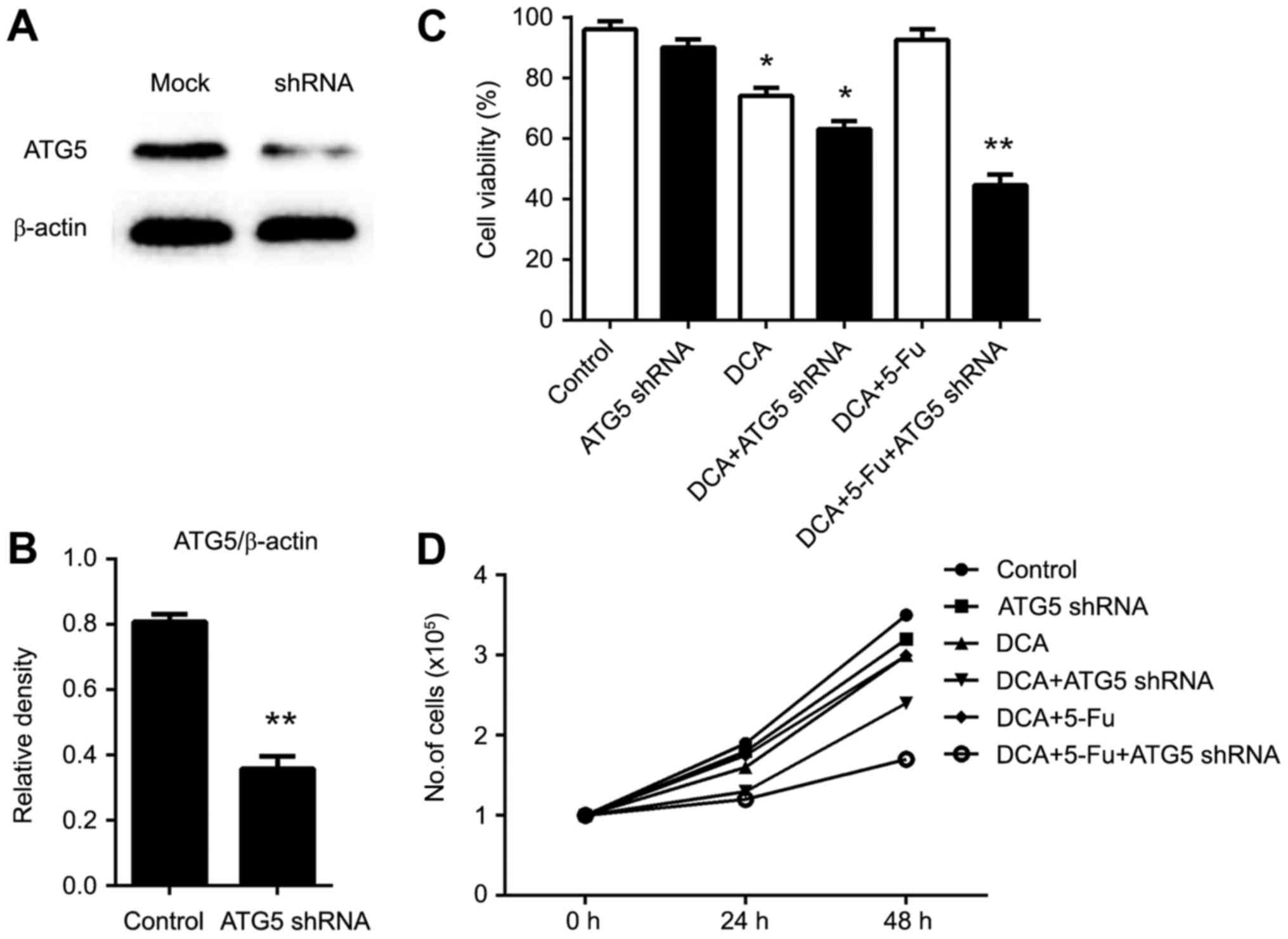
RNA interference of Atg5 enhanced the
drug sensitivity of DCA and 5-fluorouracil (5-FU)
5-fluorouracil (5-FU) is a commonly used antitumor
chemotherapeutic, and combination treatment with DCA may achieve
better outcomes (16). Induction of
autophagy in tumor cells is considered one of the primary
mechanisms of chemoresistance (17).
To investigate whether the inhibition of autophagy could improve
the effects of combination chemotherapy, we used Atg5 siRNA to
inhibit autophagy. The interference of Atg5 mRNA was confirmed by
western blot (Fig. 3A and B). As
shown in Fig. 3C-D, cell number and
viability were significantly reduced in cells knocked down for Atg5
in the DCA- and 5-FU-treated conditions. In addition, apoptosis was
markedly increased in cells treated with the DCA and 5-FU
combination treatment, indicating that inhibition of Atg5
expression could enhance drug sensitivity.
DCA-induced autophagy occurs via
inhibition of the AKT-mTOR signaling pathway
It is known that the AKT/mTOR pathway is a key
negative regulator of autophagy (17). Here, we examined whether DCA treatment
is involved in the regulation of the Akt-mTOR pathway. The
phosphorylation status of both Akt and mTOR was significantly
decreased in DCA-treated cells compared with the control. These
findings suggested that DCA induced autophagy by inhibition of the
AKT/mTOR pathway in TE-1 cells (Fig.
4A-B). To investigate whether DCA played an identical role in
other cells, we examined the proliferation and the level of
autophagy-related proteins in relation to DCA treatment in TE-8
cells, which correspond to moderately differentiated esophageal
squamous carcinoma. The cell autophagic activity was increased
during DCA treatment, and the cell viability was comparable to that
of TE-1 cells and was inhibited significantly upon treatment with
DCA, 5-FU and ATG5 shRNA. To gain insight into the mechanism of
DCA-induced autophagic activity in TE-8 cells, we examined the
AKT-mTOR signaling pathway by western blot assay. Our results
revealed that DCA treatment enhanced the dephosphorylation of AKT
and mTOR, which indicated that DCA-induced autophagy through the
AKT-mTOR signaling pathway was pervasive in tumor cells (Fig. 4C-E).
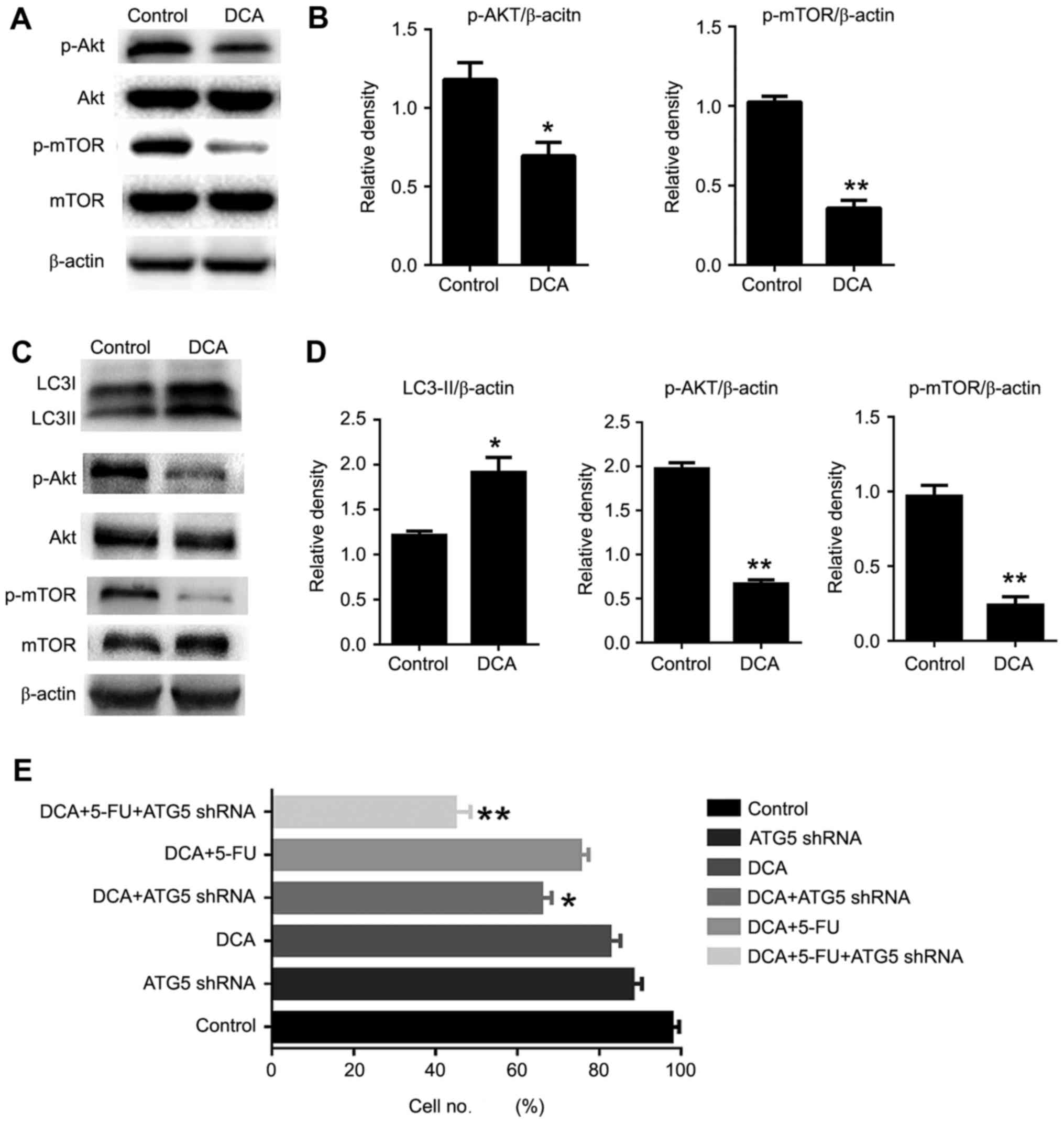 | Figure 4.Akt-mTOR pathway was involved in
DCA-induced autophagy in TE-1 cells. (A) Cells were treated with
DCA (25 mM) for 24 h; the expression of Akt, p-Akt, mTOR, p-mTOR
was analyzed by western blot. (B) Comparisons of the intensities of
proteins were statistically estimated and are presented as the mean
± SD for 3 independent experiments (ns, not significant;
**P<0.01 vs. control). (C) TE-8 cells were treated with DCA (25
mM) for 24 h; the expression of LC3, Akt, p-Akt, mTOR, p-mTOR was
analyzed by western blot analysis. (D) Comparisons of the
intensities of autophagic proteins were statistically estimated and
are presented as the mean ± SD for 3 independent experiments (n.s,
not significant; **P<0.01 vs. control). (E) TE-8 cells were
infected with ATG5-knockdown plasmid for 12 h, then treated with
DCA or 5 µg/ml 5-FU for 24 h, and cell proliferation was analyzed.
Data are represented as the mean ± SD for 3 independent
experiments. **P<0.01; ns, not significant. |
Discussion
DCA, as an inhibitor of the 3-PDPK1-4 family, has
been reported to inhibit tumor proliferation by reversing the
bio-energetic profile in a variety of cancer models (18–20). DCA
inhibits the activity of pyruvate dehydrogenase kinase (PDK) and
subsequently increases the activity of pyruvate PDH, which reverses
mitochondrial dysfunction and reactivates mitochondria-dependent
apoptosis (10,19,21).
Autophagy is a self-protective cellular mechanism that provides
energy for cells. Previous studies have demonstrated that
protective autophagy induced by chemotherapy and radiotherapy can
resist cell apoptosis (8,9). However, the internal interaction between
autophagy and DCA was unknown in esophageal squamous carcinoma
cells.
In the present study, we found that DCA induced
autophagy in human esophageal squamous carcinoma cells with minimal
apoptosis and induced high levels of autophagy-related proteins.
Moreover, autophagy inhibition by Atg5 siRNA or 3-MA treatment
significantly improved DCA-induced apoptosis and drug sensitivity
in TE-1 cells. These results suggest that autophagy plays a
protective role in tumor cells in response to DCA treatment. Hence,
low-concentration DCA treatment in conjunction with autophagy
inhibitors may exert antitumor activity.
The Akt-mTOR signaling is an important negative
pathway that regulates autophagy. In the present study, we
demonstrated that the phosphorylation of Akt and mTOR was inhibited
by DCA treatment, indicating that DCA-induced autophagy occurs by
inhibition of Akt-mTOR signaling. Lin et al reported that
DCA induced autophagy was accompanied by ROS production and mTOR
inhibition, reduced lactate excretion, reduced kPL and
increased NAD+/NADH ratio in colorectal carcinoma cells,
which was consistent with our findings (18).
In conclusion, DCA inhibits glycolysis and reduces
lactate accumulation, which destroys the acidified tumor
microenvironment. DCA treatment resulted in significant apoptosis
in human colon cancer cells, but apoptosis was decreased upon
treatment with DCA under hypoxia (22), which may suggest that a high level of
autophagy occurs in hypoxic conditions. In the present study, we
found that DCA treatment increased autophagy-related protein
production and inhibited the mTOR pathway in esophageal squamous
carcinoma cancer cells. These results demonstrate that autophagy
plays a protective role in DCA treatment, and further studies are
required to verify these results under hypoxia.
References
|
1
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gupta VK, Scheunemann L, Eisenberg T,
Mertel S, Bhukel A, Koemans TS, Kramer JM, Liu KS, Schroeder S,
Stunnenberg HG, et al: Restoring polyamines protects from
age-induced memory impairment in an autophagy-dependent manner. Nat
Neurosci. 16:1453–1460. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Denton D, Nicolson S and Kumar S: Cell
death by autophagy: Facts and apparent artifacts. Cell Death
Differ. 19:87–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Teng RJ, Du J, Welak S, Guan T, Eis A, Shi
Y and Konduri GG: Cross talk between NADPH oxidase and autophagy in
pulmonary artery endothelial cells with intrauterine persistent
pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol.
302:L651–L663. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baehrecke EH: Autophagy: Dual roles in
life and death? Nat Rev Mol Cell Biol. 6:505–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fan QW and Weiss WA: Autophagy and Akt
promote survival in glioma. Autophagy. 7:536–538. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang K, Liu R, Li J, Mao J, Lei Y, Wu J,
Zeng J, Zhang T, Wu H, Chen L, et al: Quercetin induces protective
autophagy in gastric cancer cells: Involvement of Akt-mTOR- and
hypoxia-induced factor 1α-mediated signaling. Autophagy. 7:966–978.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cheng P, Ni Z, Dai X, Wang B, Ding W, Rae
Smith A, Xu L, Wu D, He F and Lian J: The novel BH-3 mimetic
apogossypolone induces Beclin-1- and ROS-mediated autophagy in
human hepatocellular carcinoma [corrected] cells. Cell Death Dis.
4:e4892013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin JF, Tsai TF, Liao PC, Lin YH, Lin YC,
Chen HE, Chou KY and Hwang TI: Benzyl isothiocyanate induces
protective autophagy in human prostate cancer cells via inhibition
of mTOR signaling. Carcinogenesis. 34:406–414. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bonnet S, Archer SL, Allalunis-Turner J,
Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta
L, Bonnet S, et al: A mitochondria-K+ channel axis is suppressed in
cancer and its normalization promotes apoptosis and inhibits cancer
growth. Cancer Cell. 11:37–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cao W, Yacoub S, Shiverick KT, Namiki K,
Sakai Y, Porvasnik S, Urbanek C and Rosser CJ: Dichloroacetate
(DCA) sensitizes both wild-type and over expressing Bcl-2 prostate
cancer cells in vitro to radiation. Prostate. 68:1223–1231. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun RC, Fadia M, Dahlstrom JE, Parish CR,
Board PG and Blackburn AC: Reversal of the glycolytic phenotype by
dichloroacetate inhibits metastatic breast cancer cell growth in
vitro and in vivo. Breast Cancer Res Treat. 120:253–260. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun W, Zhou S, Chang SS, McFate T, Verma A
and Califano JA: Mitochondrial mutations contribute to HIF1alpha
accumulation via increased reactive oxygen species and up-regulated
pyruvate dehydrogenease kinase 2 in head and neck squamous cell
carcinoma. Clin Cancer Res. 15:476–484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yuan K, Huang C, Fox J, Laturnus D,
Carlson E, Zhang B, Yin Q, Gao H and Wu M: Autophagy plays an
essential role in the clearance of Pseudomonas aeruginosa by
alveolar macrophages. J Cell Sci. 125:507–515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang X, Overholtzer M and Thompson CB:
Autophagy in cellular metabolism and cancer. J Clin Invest.
125:47–54. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tong J, Xie G, He J, Li J, Pan F and Liang
H: Synergistic antitumor effect of dichloroacetate in combination
with 5-fluorouracil in colorectal cancer. J Biomed Biotechnol.
2011:7405642011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rubinsztein DC, Codogno P and Levine B:
Autophagy modulation as a potential therapeutic target for diverse
diseases. Nat Rev Drug Discov. 11:709–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin G, Hill DK, Andrejeva G, Boult JK,
Troy H, Fong AC, Orton MR, Panek R, Parkes HG, Jafar M, et al:
Dichloroacetate induces autophagy in colorectal cancer cells and
tumours. Br J Cancer. 111:375–385. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Michelakis ED, Webster L and Mackey JR:
Dichloroacetate (DCA) as a potential metabolic-targeting therapy
for cancer. Br J Cancer. 99:989–994. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Papandreou I, Goliasova T and Denko NC:
Anticancer drugs that target metabolism: Is dichloroacetate the new
paradigm? Int J Cancer. 128:1001–1008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Michelakis ED, Sutendra G, Dromparis P,
Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR,
Fulton D, et al: Metabolic modulation of glioblastoma with
dichloroacetate. Sci Transl Med. 2:31ra342010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shahrzad S, Lacombe K, Adamcic U, Minhas K
and Coomber BL: Sodium dichloroacetate (DCA) reduces apoptosis in
colorectal tumor hypoxia. Cancer Lett. 297:75–83. 2010. View Article : Google Scholar : PubMed/NCBI
|