Introduction
Renal cell carcinoma (RCC) is a common form of
urological malignancy. It accounts for 2.6% of adult malignancies
globally, and its incidence has steadily risen over the past decade
(1–3).
According to the Heidelberg classification system, the histological
subtypes of RCC include chromophobe, clear cell, papillary, and
unclassified carcinomas (2). The most
common subtype of RCC is clear cell RCC (ccRCC), accounting for
~82% of RCCs (3).
Patients with RCC display a high metastatic index,
with one third of patients presenting with metastatic disease at
initial diagnosis or following nephrectomy (4). Therapeutic approaches for RCC are
multifaceted and include surgery, immunomodulatory therapy,
radiotherapy and targeted therapy (1,5). Despite
the widespread use of multimodal treatments, patients with RCC
generally have poor prognoses (4).
RCC is considered to be a radioresistant tumor, and the molecular
mechanisms underlying this radiation resistance remain largely
unknown. This resistance to current therapies severely limits the
clinical management of RCC in patients. Thus, radiotherapy is used
mostly for palliation of metastases or local tumor growth (6).
Recent research has identified a rare subpopulation
of cancer stem cells, otherwise known as tumor-initiating cells
(TICs), in malignant tumors (including RCC) that may confer
resistance to radiotherapy (7,8). A number
of studies have isolated pools of TICs from RCC cell lines and
tumor specimens (9–14). RCC TICs appear to be responsible for
malignancy progression, relapse and metastasis (8–13). Zhong
et al (12) have demonstrated
that stem cell-like mammospheres from the RCC cell line SK-RC-42
exhibited greater resistance to irradiation than monolayers.
Furthermore, several genetic and cellular adaptations within TICs
may confer resistance to radiation. These adaptations include
efficient DNA repair, free radical scavenging, upregulated cell
cycle control, relative quiescence cell cycle kinetics and specific
interactions with the stromal microenvironment (15).
TIC-mediated radiation resistance has been reported
in various tumors; however, the correlation between radiation
resistance and TICs in RCC remains elusive. The present study aims
to investigate the role of TICs in radiation resistance and
describe the molecular characteristics of RCC TICs.
Materials and methods
Isolation of primary RCC cells from
human ccRCC tumors
Tumor specimens were obtained from patients at the
Henan Provincial People's Hospital and the People's Hospital of
Zhengzhou University (Zhengzhou, China). All patients gave informed
consent for their tumor samples to be used. The present study was
approved by the Internal Review and the Ethics Boards of Henan
Provincial People's Hospital and the People's Hospital of Zhengzhou
University. Tumor samples were isolated from a 47-year-old male
patient with ccRCC during radical nephrectomy. Fresh tumors were
minced, suspended in Dulbecco's Modified Eagle's Medium/nutrient
mixture F-12 (DMEM/F12; Invitrogen, Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), and mixed with 300 U/ml collagenase I
(Invitrogen, Thermo Fisher Scientific Inc.) and hyaluronidase
(Calbiochem; EMD Millipore, Billerica, MA, USA), followed by
overnight incubation at 37°C in 5% CO2. Enzymatically
disaggregated suspensions were filtered using a 40 µm cell strainer
and washed twice with phosphate buffered saline (PBS), and red
blood cells were lysed with ammonium chloride lysing buffer. The
resulting single tumor cells were cultured in DMEM/F12 supplemented
with 10% fetal bovine serum (FBS; Hyclone; GE Healthcare Life
Sciences, Logan, UT, USA) at 37°C in a humidified atmosphere
containing 5% CO2.
Radiation
Cells were irradiated at room temperature using a
60Co laboratory irradiator (Beijing Normal University,
Beijing, China) at a dose rate of 1 Gy/min. The cultured cells were
irradiated with a single dose of 3 Gy. For fractionated radiation,
cells were either irradiated for 2–3 consecutive days, or
sham-irradiated (controls). Irradiated and sham-irradiated cells
were cultured for an additional 48 h and used in subsequent
experiments.
Sphere formation assay
Cells were plated at 1×104/well in
ultra-low-attachment 6-well plates and grown in serum-free
DMEM/F12, supplemented with 20 ng/ml epidermal growth factor, 10
ng/ml human recombinant basic fibroblast growth factor-basic, and
1% B27 supplement (all from Invitrogen; Thermo Fisher Scientific,
Inc.). The medium was changed every 2 days. Following 10 days in
culture, colonies containing >20 cells were counted. To evaluate
cell self-renewal ability, mammospheres were digested with 0.15%
trypsin to be reseeded at 5×103/well.
Side population analysis
Side population (SP) analysis was performed as
described by Goodell et al (16) with slight modifications. Briefly, the
cells were suspended at a density of 1×106 cells/ml in
pre-warmed DMEM/F12, supplemented with 2% FBS (Invitrogen; Thermo
Fisher Scientific, Inc.) and 10 mmol/l
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES). This
was followed by incubation with 5 mg/ml Hoechst 33342 (Invitrogen;
Thermo Fisher Scientific, Inc.) with or without 50 µM verapamil
(Sigma-Aldrich, St. Louis, MO, USA), an ABC transporter inhibitor,
in the dark at 37°C for 90 min with interval mixing. Following
staining, cells were washed twice with ice-cold PBS and resuspended
in cold PBS. Flow cytometry analysis was subsequently performed
using FACSAria II (Becton Dickinson; BD Biosciences, San Jose, CA,
USA). Hoechst 33342 was stimulated using a 355 nm UV laser and
detected using a 450/BP50 filter for blue fluorescence and 660/BP50
filter for red fluorescence.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was obtained from cells using RNAiso Plus
(Takara Biotechnology Co., Ltd. Dalian, China), and reverse
transcription was performed according to Takara's protocol. qPCR
was performed using a SYBR-Green I Master Mix kit (Takara
Biotechnology Co., Ltd.) on the Bio-Rad IQ5 Real-Time-PCR reaction
system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
relative amounts of mRNA were calculated from the comparative
threshold cycle values using glyceraldehyde 3-phosphate
dehydrogenase as a reference gene (all primers depicted in Table I). PCR was carried out with the
following cycling conditions: 95°C for 2 min followed by 38 cycles
of amplification (denaturation at 95°C for 15 sec, annealing at
58°C for 20 sec and extension at 72°C for 30 sec).
 | Table I.Primer sequences used in RT-qPCR. |
Table I.
Primer sequences used in RT-qPCR.
| Gene | Sense and anti-sense
(5′ to 3′) | Product size
(bp) |
|---|
| ATM |
CCAAGTATGTAACCAACAATAGAAGAAG | 81 |
|
|
TGGATCCAGCTATTTGGTTTGA |
|
| ATR |
TGTCTCTACTCTTCACGGCATGTT | 82 |
|
|
AAGAGGTCCACATGTCCGTGTT |
|
| Bmi1 |
AAATGCTGGAGAACTGGAAAG | 124 |
|
|
CTGTGGATGAGGAGACTGC |
|
| Chk1 |
TTGGATAAACAGGGAAGTGAACAC | 108 |
|
|
GGTGAATATAGTGCTGCTATGTTGACA |
|
| Chk2 |
CCCAAGGCTCCTCCTCACA | 81 |
|
|
AGTGAGAGGACTGGCTGGAGTT |
|
| GAPDH |
AATTGAGCCCGCAGCCTCCC | 153 |
|
|
CCAGGCGCCCAATACGACCA |
|
| Nanog |
ATTCAGGACAGCCCTGATTCTTC | 76 |
|
|
TTTTTGCGACACTCTTCTCTGC |
|
| Oct4 |
GTGGAGAGCAACTCCGATG | 86 |
|
|
TGCTCCAGCTTCTCCTTCTC |
|
| Sox2 |
CGAGTGGAAACTTTTGTCGGA | 74 |
|
|
TGTGCAGCGCTCGCAG |
|
Tumorigenicity assay and in vivo
micro-positron emission tomogtaphy (PET) imaging
The study was approved by the Ethics Committee of
Zhengzhou University (Zhengzhou, China). Briefly, 1×105
cells suspended in 100 µl Matrigel (BD Biosciences) were injected
subcutaneously into the left flank region of 4-week-old male
NOD/SCID mice obtained from the Institute of Laboratory Animal
Science, Peking University Health Science Center (Beijing, China).
Tumor growth was monitored for 4 weeks following transplantation
using a MOSAIC animal PET scanner (Philips Medical Systems, Inc.,
Bothell, WA, USA). For microPET imaging, the mice were subjected to
fasting for 10 h prior to injection with fluorodeoxyglucose
(18F-FDG), but were allowed free access to water. Mice
were anesthetized intraperitoneally with 100 mg/kg pentobarbital
(Sigma-Aldrich), and then injected intravenously with ~3.7 MBq
18F-FDG (Department of Nuclear Medicine, Peking
University First Hospital, Beijing, China). To quantify the data,
the area density of formed tumors was calculated using Gel-Pro
Analyzer software ver. 3.0 (Media Cybernetics, Inc., Rockville, MD,
USA).
Clone formation assay
For each experiment, a total of 600–1,000 cells were
plated in 25 cm2 flasks in triplicate and cultured in
DMEM/F12 (supplemented with 10% FBS). To evaluate the DNA damage
checkpoint response, primary ccRCC cells were cultured for 24 h
before incubation with 100 nM AZD7762 (AstraZeneca R&D, Boston,
MA, USA) for 1 h prior to irradiation and 24 h following radiation.
The cells were cultured for an additional 9 days and subsequently
fixed and stained with 0.5% crystal violet. Colonies containing
>50 cells were counted. The clone formation efficiency was
calculated as the ratio of the clone number to the seeded cell
number.
Cell cycle analysis
Irradiated and sham-irradiated cells were pretreated
with or without 100 nM AZD7762 (AstraZeneca R&D) for 1 h. The
cells were harvested and washed with PBS 24 h following radiation,
fixed in 70% ethanol for 30 min at 4°C, and stained with PBS
containing 40 µg/ml RNaseA and 10 µg/ml propidium iodine, in the
dark for 30 min. Cell cycle distribution was measured using the
FACSCalibur™ flow cytometer (BD Biosciences), and data were
analyzed using the BD CellQuest™ software ver. 3.1 (BD
Biosciences).
Statistical analysis
Data are expressed as means ± SEM (n=3). The
differences between SP cells were analyzed using Student's t-test.
Statistical analyses were performed using SPSS software, ver. 17
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Increased self-renewal capacity and
expression of stemness genes following fractionated radiation
treatment
The intrinsic or acquired resistance of RCC cells to
current therapeutic strategies severely limits their efficacy, and
most patients with RCC succumb to the disease following the failure
of current treatments. However, the molecular mechanisms
contributing to the radioresistance of RCC tumors remain largely
unknown. Therefore, the present study investigated whether TICs are
enriched after receiving clinical fractions of radiation. Freshly
isolated primary ccRCC cells were irradiated as follows: i) A
single dose of 3 Gy on day 3 (R1); ii) 2 daily doses of 3 Gy on day
2 (R2); or iii) 3 daily doses of 3 Gy on day 1 (R3). Corresponding
controls (CTR) were sham-irradiated on day 3. Following
irradiation, the cells were incubated for an additional 48 h to
simulate a typical weekend treatment gap.
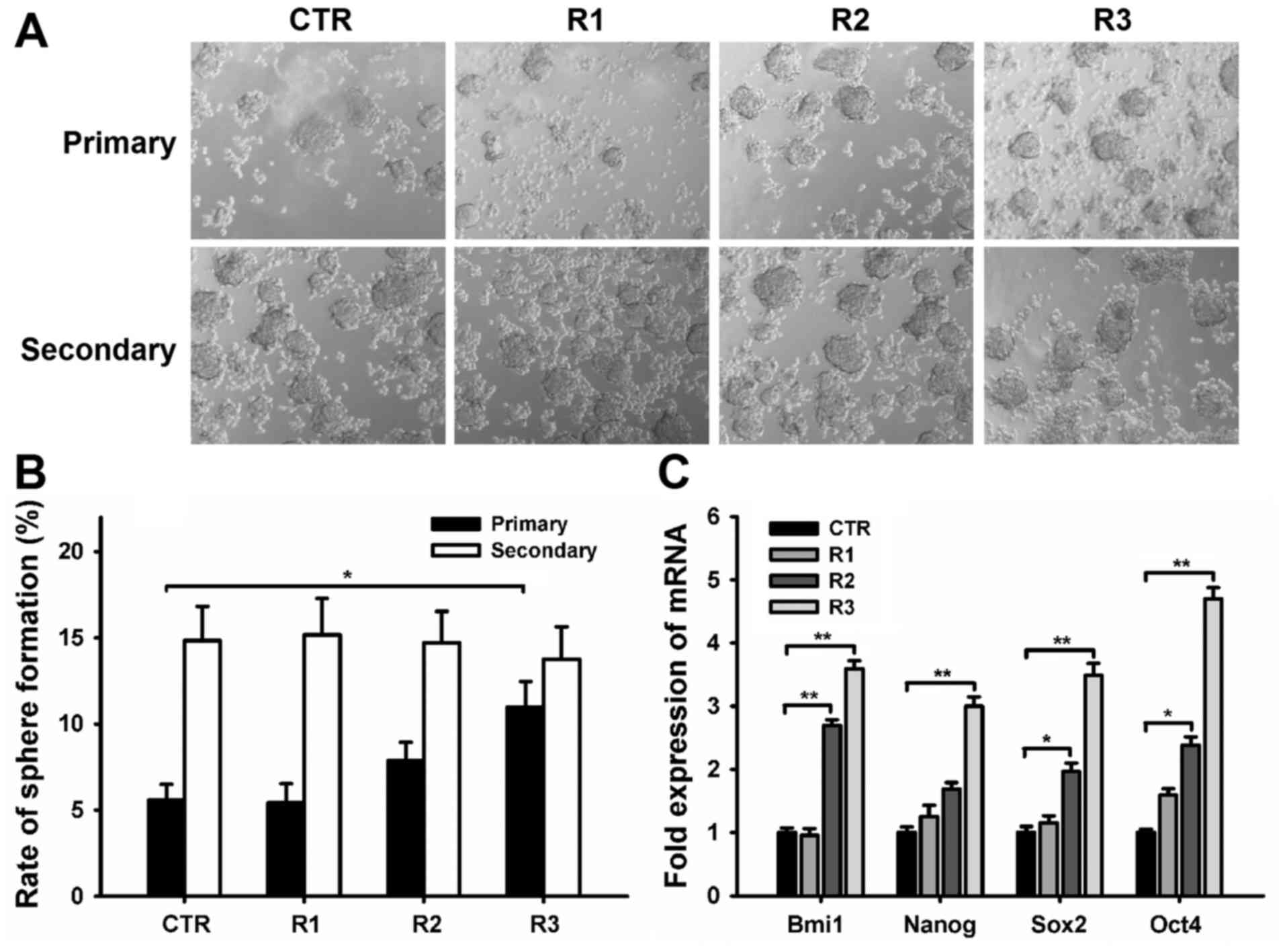
Sphere formation is a well-described characteristic
of TICs, reflecting their potential for self-renewal (17). Thus, the capacity for sphere formation
by primary ccRCC cells following multiple rounds of fractionated
radiation was evaluated in non-adherent and serum-starved medium.
Mammospheres containing >20 cells were counted following 10 days
in culture (Fig. 1A). The rate of
sphere formation in cells from the CTR, R1, R2, and R3 groups were
5.59±0.90, 5.42±1.12, 7.86±1.07, and 10.98±2.47% respectively
(Fig. 1B). The R3 group exhibited a
~2 fold higher frequency of mammosphere formation than that of the
CTR group (Fig. 1B). The self-renewal
capacity of primary formed mammospheres was analyzed among the four
groups by dissociating the mammospheres into single cells, and
reculturing them in tumor sphere medium. The rate of sphere
formation in secondary mammospheres was 14.84±1.98% in the CTR,
15.18±2.10% in R1, 14.71±1.82% in R2, and 13.76±1.88% in R3
(Fig. 1B). The secondary frequencies
of spherical colony formation were similar among all the groups,
indicating that capacity for self-renewal was retained after
irradiation.
TICs display conserved stem and progenitor cell
phenotypes (7,18); therefore, the expression of embryonic
stem cell (ES)-associated genes was evaluated to confirm the
stemness phenotype of cells subjected to fractionated radiation.
RT-qPCR results demonstrated that the R2 and R3 groups expressed ES
marker genes, such as Bmi1, Nanog, Sox 2 and Oct4, more highly than
CTR (Fig. 1C).
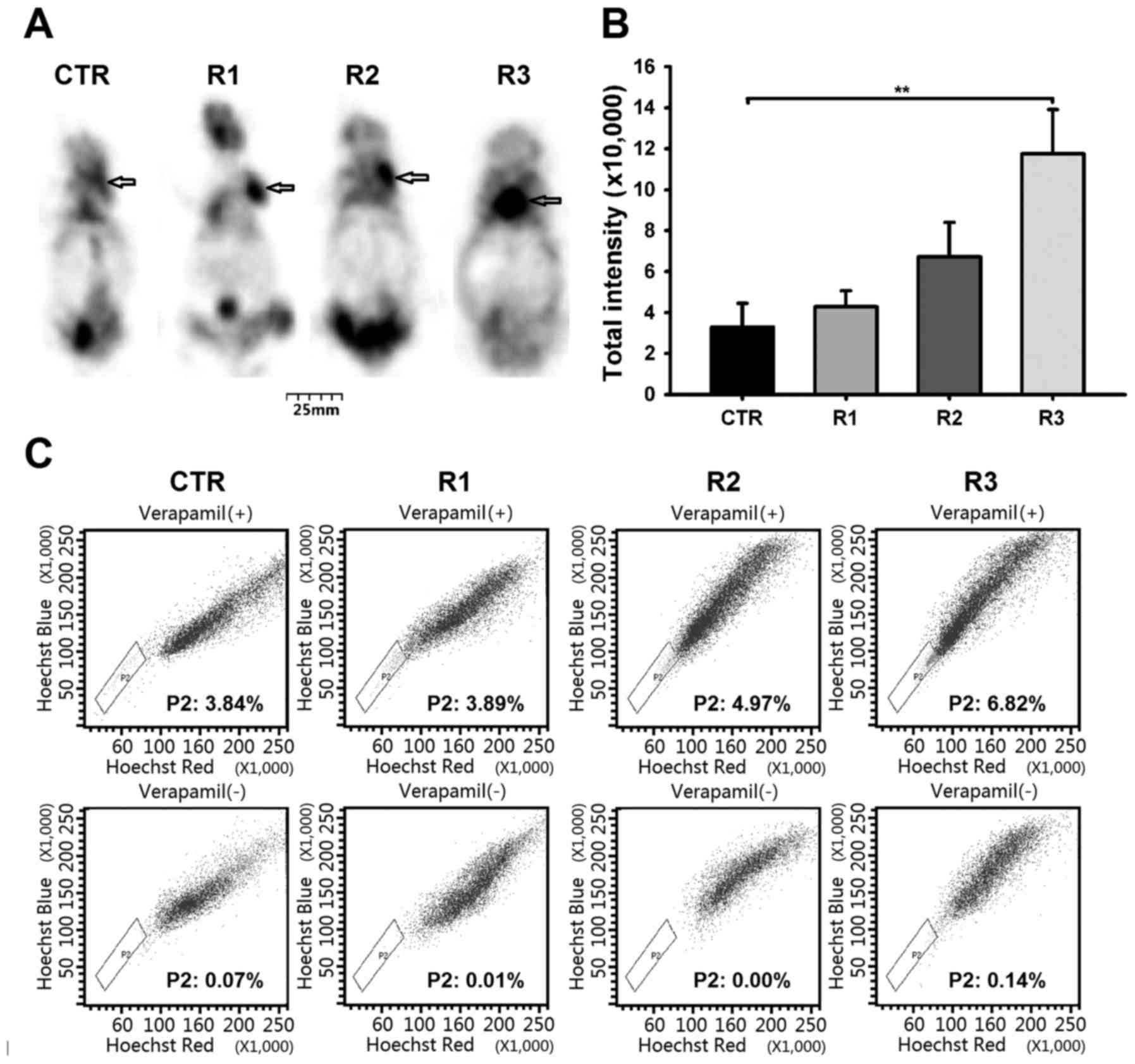
Fractionated irradiated cells exhibit
high tumorigenicity and contain a larger SP
Tumorigenic capacity of TICs was determined by
injecting immunocompromised mice with RCC cells, using a widely
accepted assay (19,20). To determine whether irradiated cells
exhibit higher tumorigenicity, the cells were injected
subcutaneously into the left flank region of NOD/SCID mice. The
Tumor growth of mice injected with cells from each group (CTR, R1,
R2, and R3) was monitored at 4 weeks post-transplantation (Fig. 2A, indicated by arrow). The total
densities of formed tumors were 3.29±1.16 in CTR, 4.28±0.79 in R1,
6.72±1.68 in R2, and 11.75±2.16 in R3 (×10,000; Fig. 2B). The total tumor densities formed in
the R3 group were significantly higher than those formed in the CTR
group (**P<0.01).
Previous studies have demonstrated that SPs within
different cancer cells are less differentiated and have
characteristics of stem/progenitor cells (7,16,21). In addition, it has been confirmed that
SP cells in RCCs exhibit TIC characteristics (8,9,22). Fig. 2C
presents the fraction of SP cells observed within each group. These
were 3.84, 3.89, 4.97 and 6.82% in the CTR, R1, R2, and R3 groups,
respectively. The frequency of SP cells increased from 3.84% in the
CTR group to 6.82% in the R3 group (P<0.05; Fig. 2C), demonstrating enrichment of the SP
fraction that occurred following fractionated radiation
treatment.
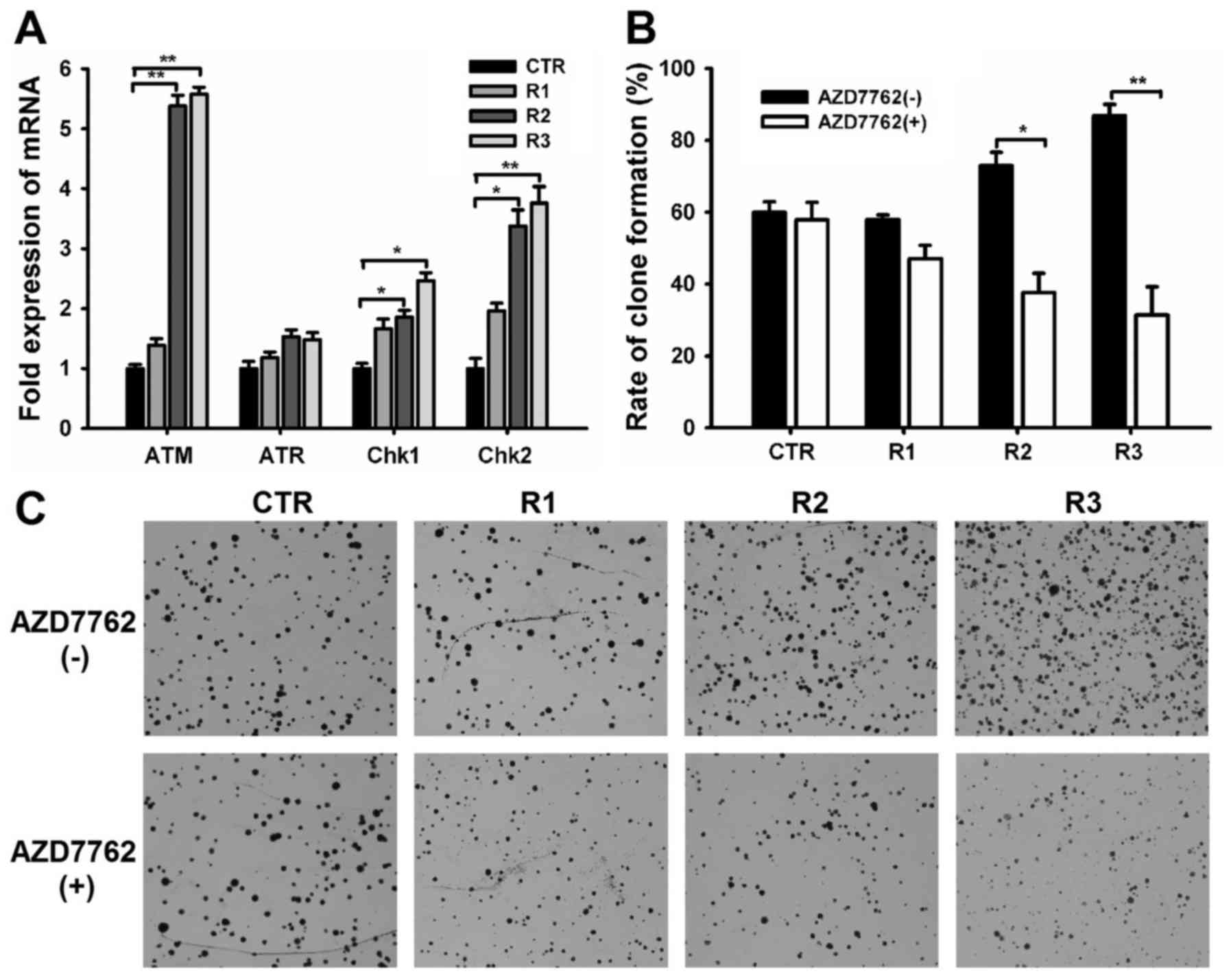
Upregulation of DNA damage checkpoint
genes after fractionated radiation
The TIC subpopulation was enriched following
fractionated radiation, suggesting that TICs within primary ccRCC
mediate resistance to ionizing radiation. DNA damage leads to
radiation-induced cell lethality; therefore the DNA damage
checkpoint response serves an essential role in cellular
radiosensitivity (23,24). To determine the role of the DNA damage
checkpoint response in TIC radioresistance, the present study
examined the expression of DNA damage checkpoint-associated genes
including ataxia-telangiectasia-mutated serine/threonine kinase
(ATM), ATM and Rad3-related serine/threonine kinase (ATR),
checkpoint kinase 1 (Chk1), and Chk2. The expression levels of ATM,
Chk1, and Chk2 were significantly higher in cells subjected to
fractionated irradiation than in sham-irradiated cells (Fig. 3A), however, no significant difference
in ATR gene expression was observed among the four groups. These
data indicate that fractionated irradiated cells may activate
checkpoint responses to a greater extent than sham-irradiated
cells, suggesting that the radiation resistance of enriched TICs is
due to increased checkpoint activation.
A novel checkpoint kinase inhibitor, AZD7762, was
used to confirm the above hypothesis. Results from the clonogenic
survival assay demonstrated that 100 nM AZD7762 exerted minimal
cytotoxicity in the CTR group but yielded radiation enhancement
effects that overcome the resistance of irradiated cells,
significantly reducing the rate of clone formation in R2 and R3
cells (Fig. 3B and C; P<0.05).
These data confirm that the preferential checkpoint response in
radiation-enriched TICs is associated with cellular resistance to
radiation.
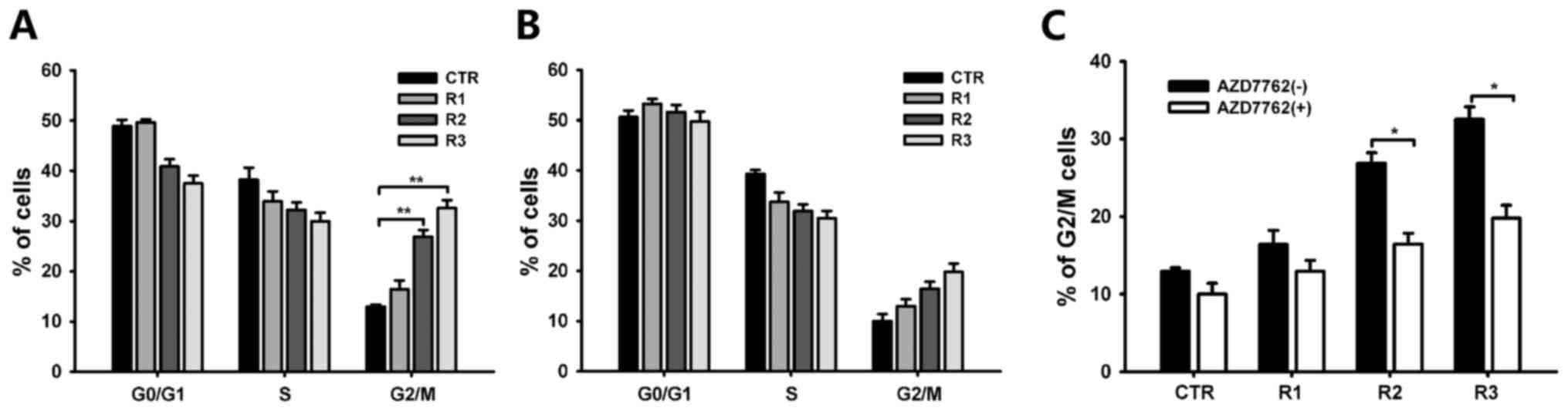
Arrest of fractionated radiated cells
in G2/M phase
Checkpoint activation induces cell-cycle arrest in
order to repair damaged DNA. Therefore, the cell cycle distribution
among the four groups was analyzed. Cell cycle analysis revealed
induction of G2/M arrest following irradiation with 3×3 Gy; from
12.94±0.47 (CTR) to 32.57±1.58% (R3 group; Fig. 4A). This arrest was abolished by
AZD7762 administration. AZD7762 specifically abrogated the G2
checkpoint as evidenced by an increased G1 cell population in
groups R2 and 3 (Fig. 4B and C).
These results indicate that cells exposed to fractionated radiation
were arrested in the G2/M phase, due to the induction of the DNA
damage checkpoint response, possibly via Chk1 and Chk2.
Discussion
In this study, a TIC-like subpopulation was enriched
fol-lowing exposure to fractionated radiation. This subpopulation
displayed preferential activation of DNA damage
checkpoint-associated genes, resulting in cell cycle arrest to
repair damaged DNA.
TICs are known to mediate tumor cell resistance to
current therapeutic options, and serve as candidate biological
targets for overcoming resistance to conventional therapy. Previous
studies have isolated and identified a TIC-like subpopulation of
cells from RCC (9–14) using approaches based on the notion
that TICs have conserved stem and progenitor cell functions and
phenotypes (7,25). However, the associations among the
different TIC populations isolated remain unclear. Methods of
isolating TICs may not be the same for each type of tumor cell
(7,26). For example, the sphere-forming assay
is a relatively simple yet robust method for isolating and
expanding stem cell populations; however, there is scant definitive
information regarding what type of cancer cells are propagated
under these conditions (26). Surface
marker-based cell sorting is a widely used method, however, its
frequency is highly variable even among the same type of cancer
cell. Therefore, TICs may exhibit heterogeneity and display several
common markers (7).
The current study demonstrated the enrichment of
TIC-like cells following their exposure to fractionated radiation.
The cells that survived displayed TIC features, including an
increased propensity to form mammospheres (Fig. 1), tumorigenicity (Fig. 2), and heightened clonogenic efficiency
(Fig. 3), compared with
sham-irradiated cells. The surviving cells also expressed higher
levels of ES-associated genes, such as Bmi1, Nanog, Sox2, and Oct4
(Fig. 1). Furthermore, the results
indicated that the SP was enriched during fractionated irradiation
(Fig. 2). Therefore, the results of
the current study provides direct evidence that TIC cells are
present in radiation resistant ccRCC cells.
The secondary frequency of spherical colonies
indicated that fractionated irradiation had no significant effects
on the self-renewing capacity of surviving ccRCC cells (Fig. 1), suggesting that fractionated
radiation may be a valuable method of enriching TIC subpopulations.
This may be useful for future investigations into radiation
resistance. The heterogenicity of the TIC population was retained
to a large extent in this selection model, compared with the
traditional cell sorting isolation method, often based on surface
markers.
Accumulating evidence suggests that TIC resistance
to ionizing radiation may arise from enhanced DNA repair,
quiescence propensity, mechanisms of free-radical scavenging,
upregulated cell cycle control and specific interaction with the
stromal microenvironment (15,27). The
biological efficacy of ionizing radiation is dependent mainly on
its ability to stimulate DNA lesions. DNA damage may induce cell
cycle arrest to preserve DNA integrity, through the DNA damage
checkpoint response (23,24). In response to DNA damage, two critical
genes, ATM and ATR, initiate cell cycle arrest via Chk1 and Chk2
(28). ATM activation is generally
considered to be a response to DNA lesions induced by irradiation,
whereas ATR activation is primarily sensitive to UV damage. The
ATM/ATR-Chk signaling pathway triggers G2/M phase arrest through
the inhibition of cyclin-dependant protein kinase, allowing damaged
DNA to undergo repair (28,29). The ability to repair DNA damage is
essential to cellular survival, as DNA lesions may induce apoptosis
and senescence (24,30).
The present study demonstrated that the expression
of ATM, Chk1, and Chk2 (but not ATR) were all significantly
upregulated following fractionated irradiation exposure, and the
irradiated cells were arrested in G2/M phase (Figs. 3 and 4).
Thus, the survival of TIC-like cells following fractionated
radiation may be partly attributed to heightened checkpoint
activation, which mediates G2/M arrest. This was confirmed by the
fact that AZD7762 (a novel checkpoint kinase inhibitor) abrogated
the G2/M phase arrest and induced ccRCC cell sensitivity to
irradiation (Figs. 3 and 4).
In conclusion, the present study demonstrated that
TIC-like cells were enriched after fractionated radiation. This
enriched subpopulation of cells within ccRCC tumors may contribute
to ccRCC radioresistance by activating the DNA damage checkpoint
response and arresting the cell cycle within the G2/M phase, thus
facilitating the repair of damaged DNA. Therefore, the DNA damage
checkpoint signal pathway may be a potential therapeutic target for
overcoming ccRCC resistance to radiotherapy.
Acknowledgements
The present study was supported by the State Key
Clinical Specialty Construction Project of China (grant no.
2013-544).
References
|
1
|
Rini BI, Campbell SC and Escudier B: Renal
cell carcinoma. Lancet. 373:1119–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ljungberg B, Hanbury DC, Kuczyk MA,
Merseburger AS, Mulders PF, Patard JJ and Sinescu IC: European
Association of Urology Guideline Group for renal cell carcinoma:
Renal cell carcinoma guideline. Eur Urol. 51:1502–1510. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Flanigan RC, Campbell SC, Clark JI and
Picken MM: Metastatic renal cell carcinoma. Curr Treat Options
Oncol. 4:385–390. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Albiges L, Choueiri T, Escudier B, Galsky
M, George D, Hofmann F, Lam T, Motzer R, Mulders P, Porta C, et al:
A systematic review of sequencing and combinations of systemic
therapy in metastatic renal cancer. Eur Urol. 67:100–110. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
De Meerleer G, Khoo V, Escudier B, Joniau
S, Bossi A, Ost P, Briganti A, Fonteyne V, Van Vulpen M, Lumen N,
et al: Radiotherapy for renal-cell carcinoma. Lancet Oncol.
15:e170–e177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao YJ, Li B, Wu XY, Cui J and Han JK:
Thyroid tumor-initiating cells: Increasing evidence and
opportunities for anticancer therapy (Review). Oncol Rep.
31:1035–1042. 2014.PubMed/NCBI
|
|
8
|
Bussolati B, Dekel B, Azzarone B and
Camussi G: Human renal cancer stem cells. Cancer Lett. 338:141–146.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang B, Huang YJ, Yao ZJ, Chen X, Guo SJ,
Mao XP, Wang DH, Chen JX and Qiu SP: Cancer stem cell-like side
population cells in clear cell renal cell carcinoma cell line 769P.
PLoS One. 8:e682932013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ueda K, Ogasawara S, Akiba J, Nakayama M,
Todoroki K, Ueda K, Sanada S, Suekane S, Noguchi M, Matsuoka K and
Yano H: Aldehyde dehydrogenase 1 identifies cells with cancer stem
cell-like properties in a human renal cell carcinoma cell line.
PLoS One. 8:e754632013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bussolati B, Bruno S, Grange C, Ferrando U
and Camussi G: Identification of a tumor-initiating stem cell
population in human renal carcinomas. FASEB J. 22:3696–3705. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhong Y, Guan K, Guo S, Zhou C, Wang D, Ma
W, Zhang Y, Li C and Zhang S: Spheres derived from the human
SK-RC-42 renal cell carcinoma cell line are enriched in cancer stem
cells. Cancer Lett. 299:150–160. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Galleggiante V, Rutigliano M, Sallustio F,
Ribatti D, Ditonno P, Bettocchi C, Selvaggi FP, Lucarelli G and
Battaglia M: CTR2 identifies a population of cancer cells with stem
cell-like features in patients with clear cell renal cell
carcinoma. J Urol. 192:1831–1841. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lucarelli G, Galleggiante V, Rutigliano M,
Vavallo A, Ditonno P and Battaglia M: Isolation and
characterization of cancer stem cells in renal cell carcinoma.
Urologia. 82:46–53. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pajonk F, Vlashi E and McBride WH:
Radiation resistance of cancer stem cells: The 4 R's of
radiobiology revisited. Stem Cells. 28:639–648. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Goodell MA, Brose K, Paradis G, Conner AS
and Mulligan RC: Isolation and functional properties of murine
hematopoietic stem cells that are replicating in vivo. J Exp Med.
183:1797–1806. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pastrana E, Silva-Vargas V and Doetsch F:
Eyes wide open: A critical review of sphere-formation as an assay
for stem cells. Cell Stem Cell. 8:486–498. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang P, Zhang Y, Mao L, Zhang Z and Chen
W: Side population in oral squamous cell carcinoma possesses tumor
stem cell phenotypes. Cancer Lett. 277:227–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Welte Y, Adjaye J, Lehrach HR and
Regenbrecht CR: Cancer stem cells in solid tumors: Elusive or
illusive? Cell Commun Signal. 8:62010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Clarke MF, Dick JE, Dirks PB, Eaves CJ,
Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM: Cancer
stem cells-perspectives on current status and future directions:
AACR workshop on cancer stem cells. Cancer Res. 66:9339–9344. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Golebiewska A, Brons NH, Bjerkvig R and
Niclou SP: Critical appraisal of the side population assay in stem
cell and cancer stem cell research. Cell Stem Cell. 8:136–147.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nishizawa S, Hirohashi Y, Torigoe T,
Takahashi A, Tamura Y, Mori T, Kanaseki T, Kamiguchi K, Asanuma H,
Morita R, et al: HSP DNAJB8 controls tumor-initiating ability in
renal cancer stem-like cells. Cancer Res. 72:2844–2854. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K
and Linn S: Molecular mechanisms of mammalian DNA repair and the
DNA damage checkpoints. Annu Rev Biochem. 73:39–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Carr AM: DNA structure dependent
checkpoints as regulators of DNA repair. DNA Repair (Amst).
1:983–994. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dalerba P, Cho RW and Clarke MF: Cancer
stem cells: Models and concepts. Annu Rev Med. 58:267–284. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Visvader JE and Lindeman GJ: Cancer stem
cells in solid tumours: Accumulating evidence and unresolved
questions. Nat Rev Cancer. 8:755–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moncharmont C, Levy A, Gilormini M,
Bertrand G, Chargari C, Alphonse G, Ardail D, Rodriguez-Lafrasse C
and Magné N: Targeting a cornerstone of radiation resistance:
Cancer stem cell. Cancer Lett. 322:139–147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Smith J, Tho LM, Xu N and Gillespie DA:
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and
cancer. Adv Cancer Res. 108:73–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Enomoto M, Goto H, Tomono Y, Kasahara K,
Tsujimura K, Kiyono T and Inagaki M: Novel positive feedback loop
between Cdk1 and Chk1 in the nucleus during G2/M transition. J Biol
Chem. 284:34223–34230. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou BB and Elledge SJ: The DNA damage
response: Putting checkpoints in perspective. Nature. 408:433–439.
2000. View
Article : Google Scholar : PubMed/NCBI
|