Introduction
v-Myc avian myelocytomatosis viral oncogene
neuroblastoma-derived homolog (MYCN), also known as nMYC, was first
described in 1983 as a gene homologous with vMYC, but is distinct
from the classical v-myc avian myelocytomatosis viral oncogene
homolog (cMYC) proto-oncogene and is highly amplified in high-risk
neuroblastoma (1). Although nMYC and
cMYC genes share high levels of homology in their coding regions
and their protein products, they vary widely in their tissue
expression profiles (2). According to
the International Neuroblastoma Risk Group, the most established
risk factors for high-risk neuroblastoma are age, histology, grade
of tumor differentiation, chromosome 11q status, DNA ploidy and
nMYC amplification status (3). nMYC
amplification is observed in ~25% of neuroblastoma cases and is
associated with poor prognosis and is also considered the
best-known genetic predictor for neuroblastoma risk (4). Additionally, nMYC amplification status
is also used as a diagnostic tool to determine the prognostic
outcomes of neuroblastoma treatments (5).
Radiation therapy is one of the standard therapies
used for the treatment of recurrent high-risk neuroblastoma.
Radiation therapy has led to significant success and a previous
study has illustrated that patients treated using 21 to 24 Gy
radiation at the local tumor sites resulted in 94% local control,
66% event-free survival and 86% overall survival, making it a vital
treatment strategy for the treatment of neuroblastoma (6). However, a significant proportion of
recipients continue to exhibit recurrence. Radiation affects cells
primarily by inducing DNA damage and cells have evolved repair
mechanisms to counter this damage. Numerous mechanisms of radiation
resistance have evolved, which include activation of heat-shock
proteins and an alteration of response signaling pathways that
promote cell survival (7).
In order to determine whether neuroblastoma cells
that are capable of surviving high doses of radiation exhibit
changes in expression of nMYC, the effect of low-dose (5 Gy) and
high-dose (25 Gy) radiation on neuroblastoma cells was studied. The
results demonstrate that chronic but not acute radiation results in
the loss of nMYC mRNA and protein expression and loss of nMYC gene
copy number. A further unidentified mechanism by which
neuroblastoma cells may adapt to radiation by actively suppressing
genomic copy number was also demonstrated.
Materials and methods
Cell lines and culture conditions
The neuroblastoma cell line SK-N-BE (2) was obtained from the American Type
Culture Collection (Manassas, VA, USA) and the NB1691 cell line was
provided by Dr Peter Houghton (St. Jude Children's Research
Hospital, Memphis, TN, USA). Cells were cultured in RPMI medium
with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C
in a humidified atmosphere containing 5% CO2. The two
cell lines were screened to be free of Mycoplasma
contamination using a polymerase chain reaction (PCR)
Mycoplasma detection kit (ABM, Inc., Richmond, BC, Canada).
Human umbilical vein endothelial cells (HUVECs) were obtained from
Thermo Fisher Scientific, Inc. (Waltham, MA, USA) and cultured in
Medium 200 supplemented with 1X low-serum growth medium. Low
passage number (3) HUVECs were used
for the tube formation assay.
Antibodies
Antibodies were obtained from the following sources:
cMYC (cat. no. 13987s), nMYC (cat. no. 9405s) and anti-mouse IgG
(cat. no. 7076s) from Cell Signaling Technologies, Inc. (Danvers,
MA, USA), β-actin (cat. no. sc47778) and anti-rabbit IgG (cat. no.
sc2030) from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Radiation treatment
Radiation was performed using the RS2000 radiator
(Rad Source Technologies, Inc., Boca Raton, FL, USA). For acute
radiation, a single dose of 5 Gy was administered and cells were
processed for all the experiments. Chronic radiation treatment is
discussed as follows.
Generation of radiation-resistant
cells
SK-N-BE (2) and
NB-1691 cells were treated with 5 Gy radiation and returned to the
CO2 incubator and grown at 37°C in a humidified
atmosphere. Radiosensitive cells died following between 3 and 5
days and the surviving cells grew as colonies. These colonies were
then carefully expanded until they became confluent. These cells
were then radiated again with 5 Gy and the cycle was continued
until the cells were treated with a cumulative dosage of 25 Gy. The
surviving cells were designated as chronic radiated cells and used
for additional experiments. This was performed to recapitulate the
clinical radiation dosage regime. Radiation-resistant cells were
termed chronic radiation cells.
Colony formation assay
Cells were counted, and 1,000 cells were plated in
60-mm plates. The plates were incubated at 37°C for 2 weeks with
the medium changed every 3 days. The cells were then fixed with
acetic acid/methanol and stained with 0.5% crystal violet for 2 h.
The plates were de-stained with water, air-dried and imaged. The
assay was performed in triplicate.
Cell viability assay
A total of 1,000 cells were plated in 96-well plates
and incubated for 24, 48, 72 and 96 h at 37°C. MTT was added to a
final concentration of 0.5 mg/ml and incubated for an additional 2
h at 37°C. The reaction was stopped by adding 100 µl dimethyl
sulfoxide for 30 min and the absorbance was read at 550 nm using a
spectrophotometer. Untreated wild-type cells served as the control
group. A total of 12 replicates were included for each time point
and the mean percentage proliferation was plotted.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from cells using TRIzol
(Thermo Fisher Scientific, Inc.) and converted into cDNA using an
iScript cDNA synthesis kit (Bio-Rad Laboratories, Inc., Hercules,
CA, USA), according to the manufacturer's protocol. Amplification
of cDNA was performed using iTaq Universal SYBR-Green Supermix
(Bio-Rad Laboratories, Inc.) using primers targeting cMYC and nMYC,
and normalized against hypoxanthine-guanine
phosphoribosyltransferase (HPRT). The primer sequences used were as
follows: nMYC forward, 5′-CACAAGGCCCTCAGTACCTC-3′ and reverse,
5′-ACCACGTCGATTTCTTCCTC-3′; cMYC forward,
5′-CGTCTCCACACATCAGCACAA-3′ and reverse,
5′-CACTGTCCAACTTGACCCTCTTG-3′; HPRT forward,
5′-TGACACTGGCAAAACAATGCA-3′ and reverse
5′-GGTCCTTTTCACCAGCAAGCT-3′. The thermocycling conditions were as
follows: Initial denaturation at 95°C for 30 sec, followed by 40
cycles of 95°C for 5 sec and 60°C for 30 sec. A total of 4
replicates/sample were performed. mRNA levels were quantified using
the 2−ΔΔCq method and plotted as arbitrary units
according to standard protocols on Microsoft Excel (Microsoft
Corporation, Redmond, WA, USA) (8).
Western blotting
Whole cell lysates were prepared by treating cells
with M-PER mammalian protein extraction reagent (Thermo Fisher
Scientific, Inc.) and protein concentration was measured using
Pierce 660 nm protein assay reagent (Thermo Fisher Scientific,
Inc.). Equal concentrations of protein (40 µg) were separated by
SDS-PAGE (10% gels) and transferred onto polyvinylidene fluoride
membranes. Blots were blocked with 5% skimmed milk in PBS
containing Tween-20 (PBST) for 1 h at room temperature, probed with
appropriate primary antibody (1:1,000 dilution) in PBST overnight
at 4°C and secondary antibodies (1:5,000 dilution) for 1 h at room
temperature. Protein bands were visualized using Pierce enhanced
chemiluminescence western blot substrate (Thermo Fisher Scientific,
Inc.), and β-actin was used as a loading control. Western blot
analysis was performed to confirm RT-PCR results.
Confocal microscopy
Control, acute and chronic radiation-treated cells
were plated in 8-well chamber slides. After 24 h, cells were fixed
in formaldehyde for 10 min and permeabilized with 0.1% Igepal-300.
The slides were blocked with 1% BSA in PBST for 30 min and probed
with primary antibody (cMYC or nMYC) for 1 h at room temperature.
The slides were washed and treated with appropriate fluorescent
secondary antibody for 30 min. Nuclear staining was performed using
propidium iodide (BioSure®, Grass Valley, CA, USA) and
slides were mounted using Vectashield mounting medium (Vector
Laboratories, Inc., Burlingame, CA, USA). Images were captured
using a confocal microscope (Olympus Corporation, Center Valley,
PA, USA).
nMYC gene amplification/copy number
analysis
Copy number analysis was performed using the
comparative Cq method as previously described (9). Genomic DNA was isolated from the control
and the treated cells using the DNA mini kit (Qiagen, Inc.,
Valencia, CA, USA) and amplification was performed using iTaq
Universal SYBR-Green Supermix (Bio-Rad Laboratories, Inc.). The
thermal cycling conditions maintained were as follows: Initial
denaturation at 95°C for 30 sec, followed by 40 cycles of 95°C for
5 sec and 60°C for 30 sec. A total of four replicates were included
for each sample, and the copy number was calculated by normalizing
against a calibrator DNA sample that possessed a disomic copy
number of genes (control genomic DNA, Applied Biosystems; Thermo
Fisher Scientific, Inc.). The mean copy number of two reference
genes, B-cell maturation antigen (BCMA) and syndecan 4 (SDC4), was
used for normalization. The copy number of nMYC was calculated
using the formula:.
2–ΔΔCq=(1+E)–ΔCqgene(1+E)–ΔCqreference
where E represents efficiency, set at 0.95; ∆Cq gene
represents the difference in Cq value between test sample and
calibrator sample for the target gene; ∆Cq reference represents the
difference in Cq value between test sample and calibrator sample
for the reference gene.
The primer sequences used were as follows: nMYC
forward, 5′-CGCAAAAGCCACCTCTCATTA-3′; reverse,
5′-TCCAGCAGATGCCACATAAGG-3′; BCMA forward,
5′-CGACTCTGACCATTGCTTTCC-3′ and reverse, 5′-AAGCAGCTGGCAGGCTCTT-3′
and SDC4 forward, 5′-CAGGGTCTGGGAGCCAAGT-3′ and reverse,
5′-GCACAGTGCTGGACATTGACA-3′.
Fluorescent in-situ hybridization
(FISH)
FISH was performed in cells using the nMYC
amplification probe (red) spanning the nMYC gene in chromosome 2
position 2p24.3 (Cytocell, Cambridge, UK). Control and treated
cells were plated in 8-well chamber slides. After 24 h, the cells
were fixed using methanol: acetic acid for 5 min, denatured by
heating at 75°C for 5 min and hybridized with probe at 37°C
overnight according to the manufacturer's protocol. The slides were
washed, mounted and visualized using a confocal microscope. A
control probe (green) targeting lymphoid nuclear protein related to
AF4 gene at chromosome 2 position 2q11.2 was included in the probe
mix and served as an internal control for chromosome number.
Conditioned medium collection
NB-1691 and SK-N-BE (2) wild-type, acute and chronic radiation
treated cells were plated in 60 mm cell culture plates and grown in
RPMI 1640 medium (cat. no. MT10040CV; Thermo Fisher Scientific
Inc.) with 10% FBS and 1% penicillin streptomycin. When the cells
reached ~80% confluence, they were washed with RPMI 1640 medium
without FBS and antibiotics, resuspended in 2 ml serum-free medium
and incubated overnight in a humidified 37°C incubator. The
conditioned medium was collected and debris removed by
centrifugation at 800 × g for 5 min at room temperature. The
supernatant was separated and used for subsequent angiogenesis
assays.
In vitro angiogenesis assay
The angiogenesis assay was performed using the HUVEC
tube formation assay. HUVECs were first stained with a
cell-permeant fluorescent dye, calcein acetoxymethyl ester (AM)
(Thermo Fisher Scientific, Inc.) at a concentration of 2 µg/ml and
incubated at 37°C in the dark for at least 30 min. A total of 40 µl
of Geltrex basement membrane matrix (Thermo Fisher Scientific,
Inc.) was coated on 96-well plates and allowed to solidify for 30
min. HUVECs were trypsinized and resuspended in SFM. Cells were
plated at a concentration of 20,000 cells/well in 100 µl
appropriate conditioned medium. Cells with complete medium were
used as a positive control and cells with SFM were used as negative
control. The plate was incubated at 37°C in a humidified incubator
for 3 h. Tube formation was visualized using an inverted
fluorescence microscope at ×10 magnification.
In vitro invasion assay
Invasion was performed using the Oris Pro 96-well
invasion assay kit (Platypus Technologies, Madison, WI, USA) as
described below. A total of 20,000 calcein AM-treated cells were
plated in collagen I-coated 96-well plates and incubated for 1 h in
a humidified 37°C incubator. The cells were overlaid with collagen
I and incubated for an additional 1 h in a humidified 37°C
incubator. Following polymerization, 100 µl complete medium was
added to all the wells. Pre-invasion images were captured using an
inverted fluorescent microscope at ×10 magnification. The plate was
then incubated at 37°C for 48 h. After 48 h, post-invasion images
were also captured.
Statistical analysis
All statistical analysis was performed using
GraphPad Prism (version 6.0; GraphPad Software, Inc., La Jolla, CA,
USA). Student's t-tests were used to compare significance between
study groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
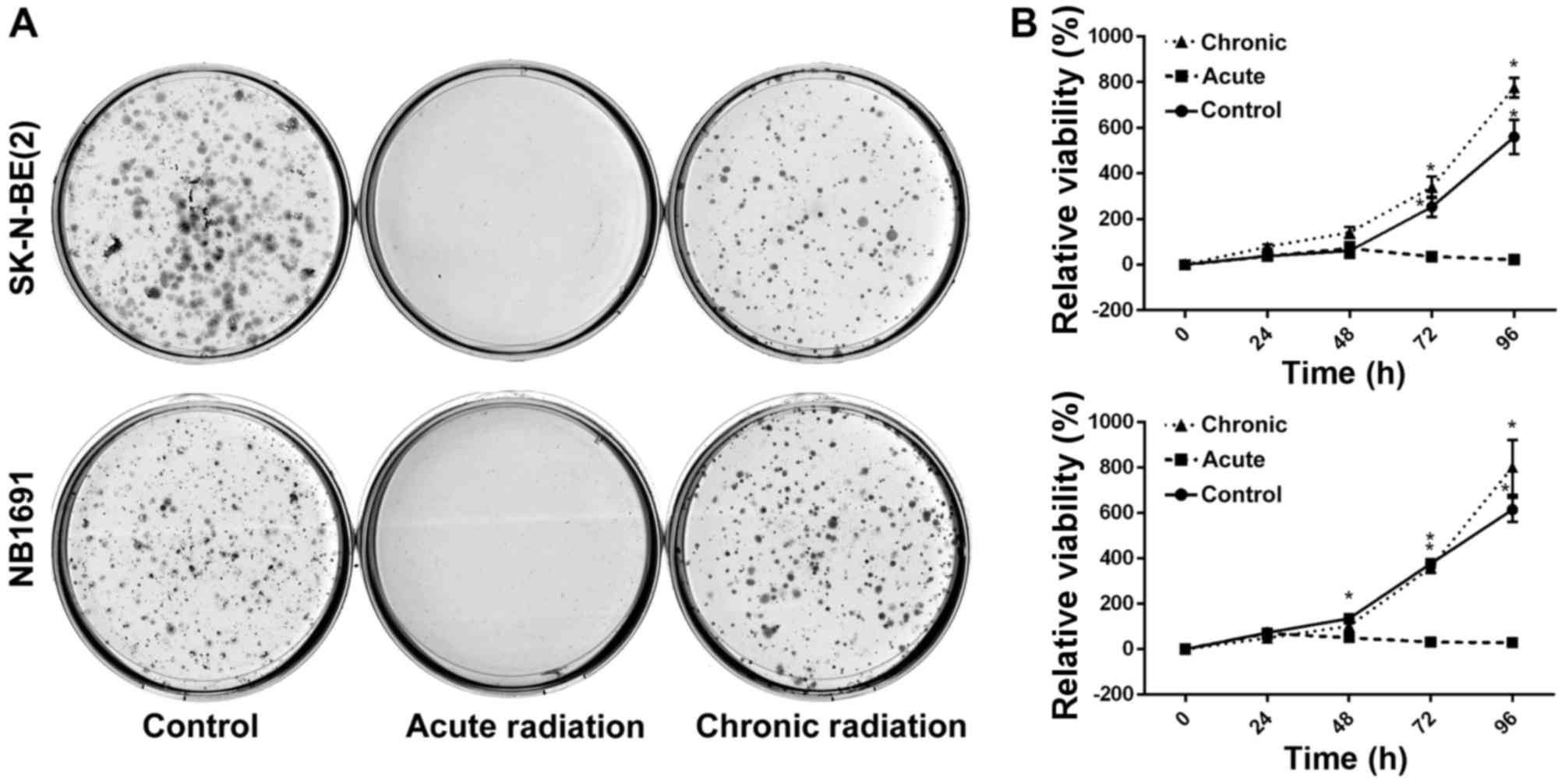
Chronic radiation increases cell
viability and survival
To test the effect of radiation on neuroblastoma
cells, NB1691 and SK-N-BE (2) cells
were exposed to acute radiation (single 5 Gy dose) and chronic
radiation (5 doses of 5 Gy each, cumulative dosage of 25 Gy). It
was revealed that cells resistant to chronic radiation were able to
form colonies, whereas control cells that were exposed to acute
radiation (5 Gy) exhibited little to no anchorage-dependent
proliferation (Fig. 1A).
Additionally, to determine whether cells exposed to acute radiation
and resistant cells exposed to chronic radiation exhibited any
change in proliferative potential, an MTT assay was performed. It
was observed that over a period of 96 h, the proliferative
potential of radiation-resistant cells was similar, if not greater,
compared with un-radiated controls, whereas cells exposed to acute
radiation demonstrated little to no proliferation (Fig. 1B).
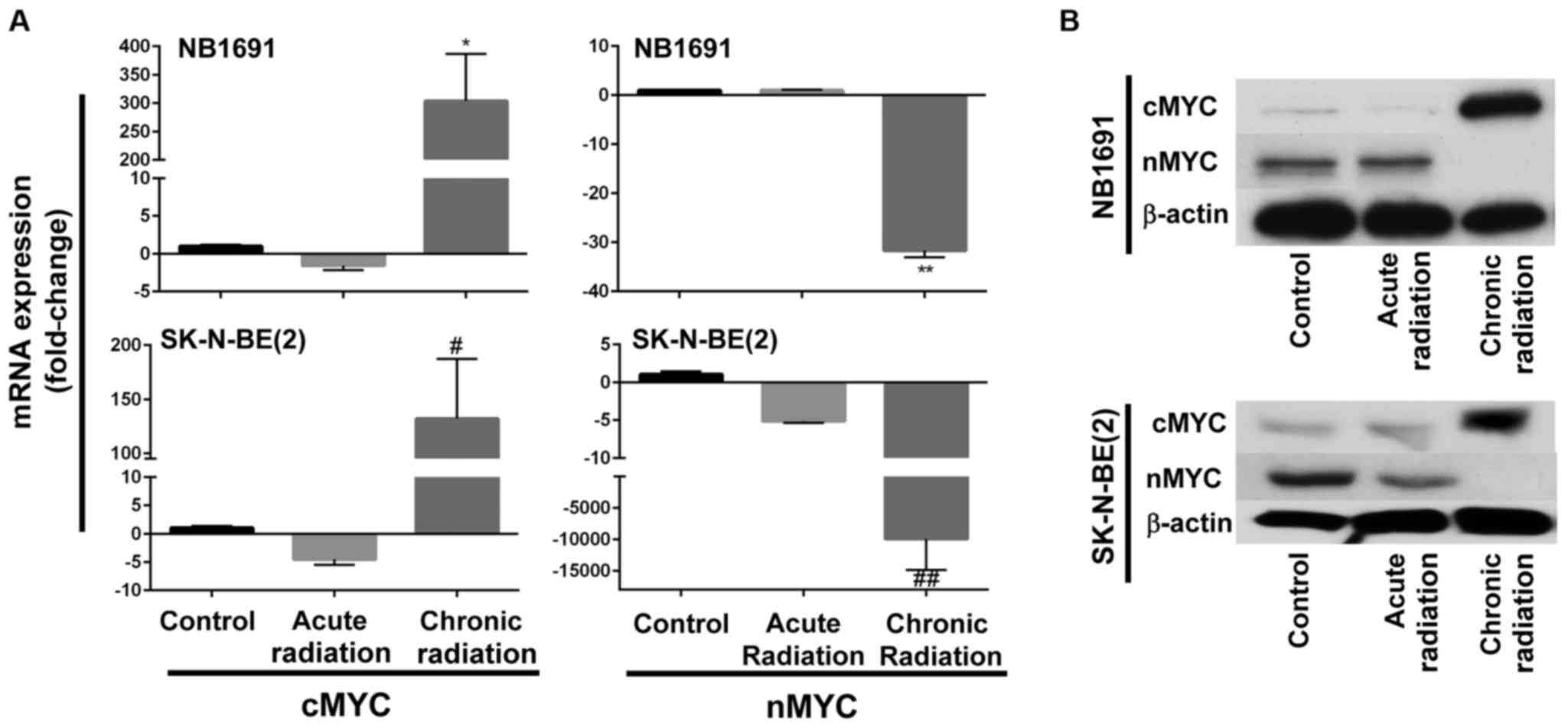
Chronic radiation induces loss of nMYC
expression
Fig. 1B indicates that
neuroblastoma cells exposed to chronic radiation exhibited high
proliferation rates when compared with cells exposed to acute
radiation. It is known that increased expression of nMYC is
associated with increased proliferation (10). Therefore, in an attempt to determine
whether chronic radiation influenced the expression levels of nMYC
expression, RT-qPCR analysis was used to determine the expression
levels of nMYC and cMYC. It was observed that radiation-resistant
(chronic) cells exhibited a significant loss of nMYC mRNA
expression in the NB1691 and SK-N-BE (2) cell lines. Additionally, expression
levels of cMYC mRNA were increased in the two NB1691 and SK-N-BE
(2) cells exposed to chronic
radiation (Fig. 2A). This change was
additionally corroborated and validated with mRNA expression levels
with protein expression levels of nMYC and cMYC. It was observed
that a decrease in nMYC mRNA corresponded to a decrease in nMYC
protein levels and an increase in cMYC mRNA levels corresponded to
an increase in cMYC protein levels in NB1691 and SK-N-BE (2) cells exposed to chronic radiation
(Fig. 2B).
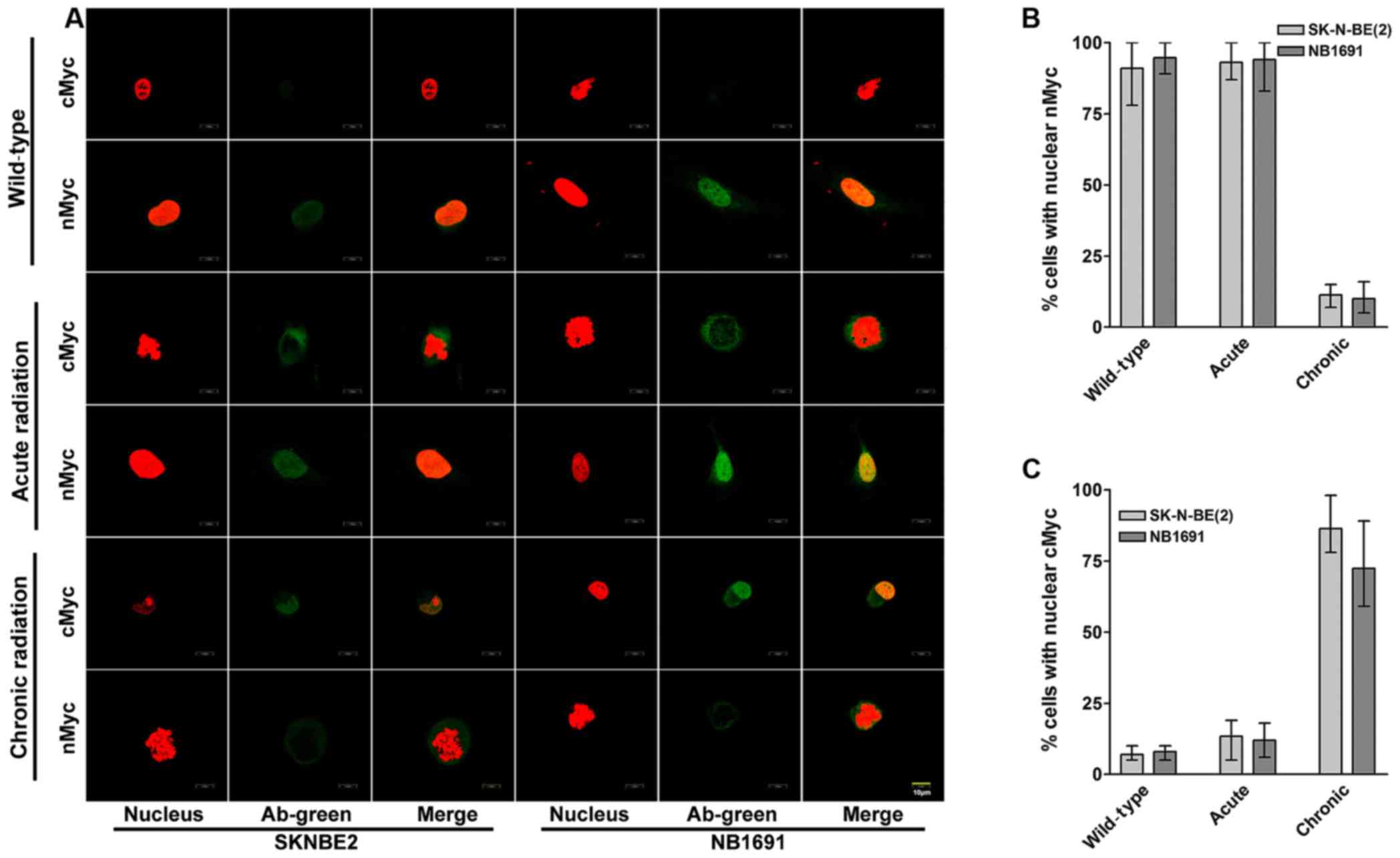
cMYC localizes in the nucleus of
chronic radiation cells
To study the cellular localization of cMYC and nMYC
proteins in acute and chronic radiation cells, confocal microscopy
was performed for cMYC and nMYC in the NB1691 and SK-N-BE (2) cell lines. It was observed that in
controls and acute radiation cells, high expression of nMYC was
predominantly exhibited in the nucleus, whereas lower expression of
cMYC was exhibited in the cytoplasm. Notably, chronic radiation
cells demonstrated a significant loss of nMYC expression in the
nucleus, which was compensated by a corresponding increase in the
expression of cMYC (Fig. 3A).
Quantitative analysis demonstrated that only 11% of SK-N-BE
(2) cells and 10% of NB-1691 cells
expressed nMYC in the nucleus of chronic radiation cells compared
with 80% in control SK-N-BE (2) and
84% in NB-1691 control cells (Fig.
3B). In contrast, following treatment with chronic radiation,
cMYC expression was observed in the nucleus of 86% SK-N-BE
(2) cells and 72% NB-1691 cells
compared with 7 and 8% in their respective controls (Fig. 3C).
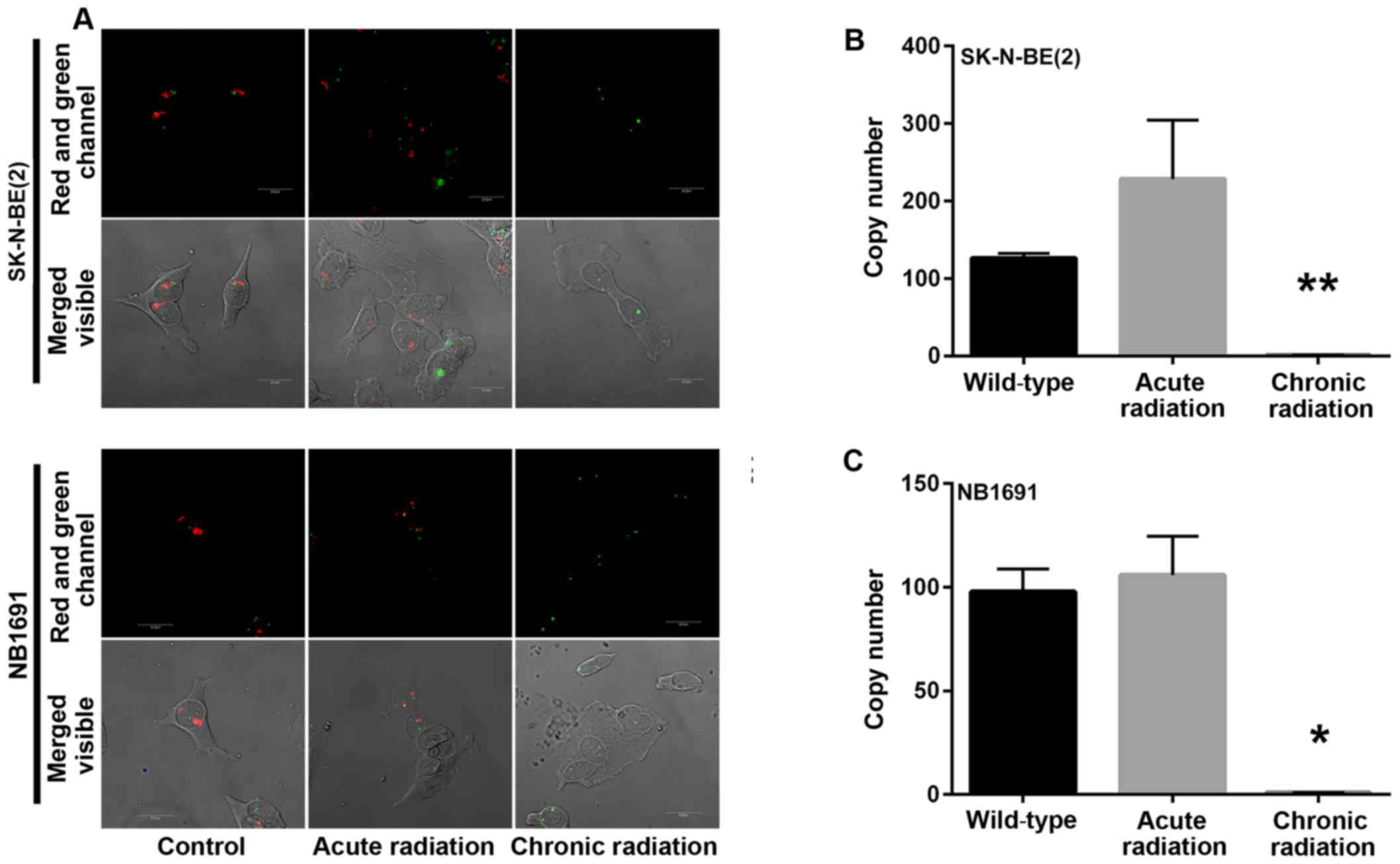
Chronic radiation induces loss of MYCN
gene amplification
Amplification of the nMYC gene in neuroblastoma has
been identified to be markedly associated with advanced disease
stage (5). A mechanism by which
radiation kills tumor cells is by DNA damage. In the present study,
initial experiments demonstrated a loss of nMYC mRNA and protein
expression in cells that were exposed to chronic radiation, the
effect of radiation on nMYC gene amplification using FISH and qPCR
based gene copy number analysis was examined. From the FISH
analysis, it was observed that the nMYC gene copy number was
significantly decreased in chronic radiation cells when compared
with controls or acute radiation cells (Fig. 4A). The FISH results were confirmed by
a qPCR-based copy number analysis that also demonstrated that
chronic radiation resulted in significant loss of nMYC copy number
in NB1691 and SK-N-BE (2) cells
(Fig. 4B).
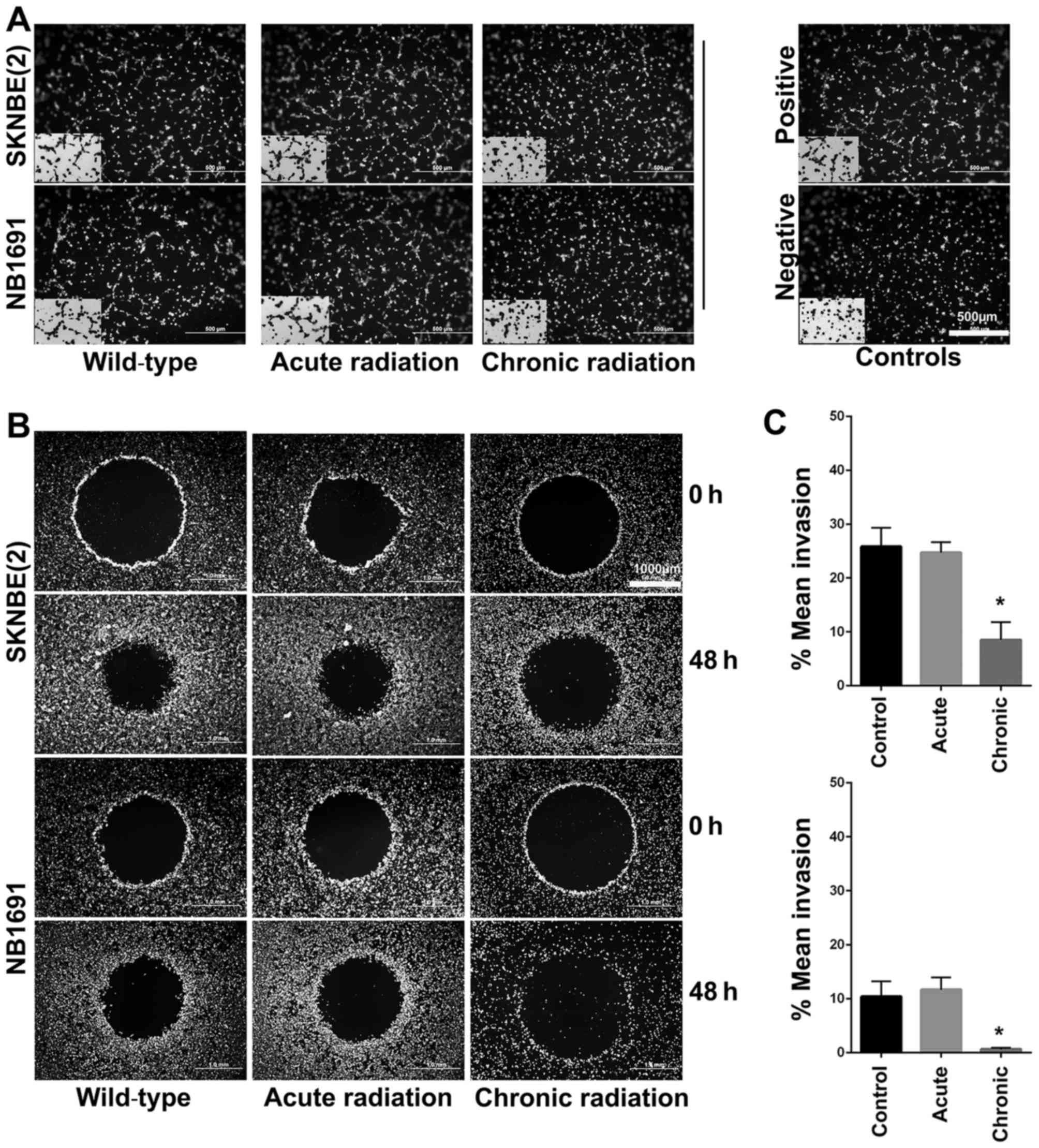
Chronic radiation inhibits
angiogenesis and invasion in neuroblastoma cells
Using the well-established HUVEC tube formation
assay, the effect of radiation on neuroblastoma cells was
investigated. The results revealed that chronic, but not acute,
radiation resulted in significant inhibition of secretion of
pro-angiogenic factors (Fig. 5A). The
results also demonstrated that cells treated with chronic but not
acute radiation exhibited decreased invasive potential (Fig. 5B and C).
Discussion
In the present study, the effect of acute and
chronic radiation on high-risk neuroblastoma cells was analyzed. As
expected, the majority of cells treated with acute radiation did
not survive and failed to proliferate efficiently, as seen by the
colony formation and viability assays. In order to simulate
biological and clinical radiation treatment modalities, the few
cells that survived acute radiation were selected and clonally
expanded. These radiation-resistant cells were additionally treated
with multiple cycles of 5 Gy radiation for a total of 25 Gy, and
clonally expanded. Notably, these cells that survived chronic
radiation demonstrated biological characteristics similar to those
of the control untreated cells. These chronic radiation cells were
able to form individual colonies and the un-radiated controls.
Additionally, the results also demonstrated that these cells
regained their proliferation potential, and were also proliferating
at a higher rate compared with the controls. It is known that in
response to radiation, several pathways are activated that target
DNA repair, including cell cycle regulation, proliferation and
apoptosis and other pathways (11).
Since nMYC has been implicated in the aggressive
behavior of neuroblastoma, the present study sought to explore the
expression levels of nMYC at the mRNA and protein levels. It was
revealed that chronic radiation resulted in the loss of nMYC mRNA
and protein expression, and that this was accompanied by a
corresponding increase in the expression of cMYC. There are no
concrete reports on the distinct roles of cMYC and nMYC, and nMYC
and cMYC regulate the same genes involved in the cell cycle,
proliferation and death, and may partially compensate for the
functions of each other (4). In
murine development, nMYC has been demonstrated to successfully
replace cMYC functions (12).
However, in a previous study with embryonic stem cells, cMYC
upregulation failed to compensate for nMYC functions (13). Additionally, it is known that in
neuroblastoma there is an inverse association between nMYC and cMYC
expression (14). High nMYC
expression due to nMYC amplification is known to suppress cMYC
expression in neuroblastoma cells (14), thereby indicating that nMYC function
may be compensated for by cMYC.
To additionally explore the role of nMYC/cMYC, the
present study sought to determine their subcellular localization.
It was observed that cMYC was localized in the nucleus of SK-N-BE
(2) and NB-1691 cells in response to
chronic radiation, which was accompanied by a loss of nMYC
expression. A previous study indicated that cMYC was primarily
localized to the nucleus in patients with Burkitt lymphoma, whereas
in patients with diffuse large B-cell lymphomas, it was observed
predominantly in the cytoplasm (15).
It has already been demonstrated that cMYC protein possesses two
nuclear localization signal regions (16). Following nuclear localization, it has
been identified that cMYC binds to its partner Max in promyelocytic
leukemia nuclear bodies and fibrillarin (17,18). The
present study was able to identify low levels of cMYC expression in
the cytoplasm of cells treated with acute radiation. The hypothesis
of the present study is that radiation upregulates the expression
of cMYC protein in the cytoplasm. However, high levels of nMYC
present in the nucleus prevent nuclear translocation of cMYC.
During chronic radiation, loss of nMYC favors nuclear import of
cMYC, which in turn activates the pathways needed for survival and
proliferation.
The mechanism by which chronic radiation results in
the loss of nMYC but not cMYC remains unknown. Radiation has been
demonstrated to result in a significant decrease in amplified gene
copy number in transformed human cell lines (17). This nMYC loss may have occurred either
due to the sustained DNA damage to the cells or the loss of key DNA
repair mechanisms. The underlying molecular mechanism for the
specificity of this loss and gain remains to be assessed. The
results of the present study are vital as there are very few
studies that have documented changes in gene copy number following
exposure to radiation, and this has a major impact on the future of
radiation therapy.
nMYC amplification has been associated with
increased angiogenesis and metastasis in children with
neuroblastoma (19). In the present
study, chronic radiation cells demonstrated decreased angiogenic
and invasive potential. It is suggested that this may be a result
of the loss of key signaling pathways regulated by nMYC. The
results of the present study also support the view that certain
cancer cells adapt to treatment pressure by switching on genes that
aid dormancy. Such dormant cells with poor metastatic ability have
been demonstrated to exist in neuroblastoma (20). It is hypothesized that these cells may
act as precursors for metastases.
In the present study, certain important differences
between acute and chronic radiation treatment of nMYC amplified
neuroblastoma have been highlighted. It has been demonstrated that
cells treated with chronic radiation, but not acute radiation, were
able to form colonies and proliferate efficiently. Contrary to
previous hypotheses, the results of the present study suggest that
there is severe loss of expression of nMYC mRNA and protein in
response to chronic radiation, and is compensated for by the
nuclear localization of cMYC. The results of the present study also
revealed that chronic radiation induces copy number changes in nMYC
and, to the best of our knowledge, the present study provides the
first experimental demonstration that indicates that nMYC gene copy
number is severely altered following chronic radiation. Additional
studies examining the exact mechanisms that result in the loss of
nMYC gene copy number, and factors that drive the cells to survive,
will lead the design of improved treatment strategies and the
overcoming of radiation resistance.
Acknowledgements
The present study was supported by the National
Cancer Institute (grant no. R01CA147792).
References
|
1
|
Kohl NE, Kanda N, Schreck RR, Bruns G,
Latt SA, Gilbert F and Alt FW: Transposition and amplification of
oncogene-related sequences in human neuroblastomas. Cell.
35:359–367. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kohl NE, Legouy E, DePinho RA, Nisen PD,
Smith RK, Gee CE and Alt FW: Human N-myc is closely related in
organization and nucleotide sequence to c-myc. Nature. 319:73–77.
1986. View
Article : Google Scholar
|
|
3
|
Cohn SL, Pearson AD, London WB, Monclair
T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, et
al: The international neuroblastoma risk group (INRG)
classification system: An INRG task force report. J Clin Oncol.
27:289–297. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang M and Weiss WA: Neuroblastoma and
MYCN. Cold Spring Harb Perspect Med. 3(a014415)2013.PubMed/NCBI
|
|
5
|
Brodeur GM, Seeger RC, Schwab M, Varmus HE
and Bishop JM: Amplification of N-myc in untreated human
neuroblastomas correlates with advanced disease stage. Science.
224:1121–1124. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gatcombe HG, Marcus RB Jr, Katzenstein HM,
Tighiouart M and Esiashvili N: Excellent local control from
radiation therapy for high-risk neuroblastoma. Int J Radiat Oncol
Biol Phys. 74:1549–1554. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morgan MA and Lawrence TS: Molecular
pathways: Overcoming radiation resistance by targeting DNA damage
response pathways. Clin Cancer Res. 21:2898–2904. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
De Preter K, Speleman F, Combaret V, Lunec
J, Laureys G, Eussen BH, Francotte N, Board J, Pearson AD, De Paepe
A, et al: Quantification of MYCN, DDX1, and NAG gene copy number in
neuroblastoma using a real-time quantitative PCR assay. Mod Pathol.
15:159–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schweigerer L, Breit S, Wenzel A,
Tsunamoto K, Ludwig R and Schwab M: Augmented MYCN expression
advances the malignant phenotype of human neuroblastoma cells:
Evidence for induction of autocrine growth factor activity. Cancer
Res. 50:4411–4416. 1990.PubMed/NCBI
|
|
11
|
Maier P, Hartmann L, Wenz F and Herskind
C: Cellular Pathways in Response to Ionizing Radiation and Their
Targetability for Tumor Radiosensitization. Int J Mol Sci.
17:1022016. View Article : Google Scholar
|
|
12
|
Malynn BA, de Alboran IM, O'Hagan RC,
Bronson R, Davidson L, DePinho RA and Alt FW: N-myc can
functionally replace c-myc in murine development, cellular growth,
and differentiation. Genes Dev. 14:1390–1399. 2000.PubMed/NCBI
|
|
13
|
Stanton BR, Perkins AS, Tessarollo L,
Sassoon DA and Parada LF: Loss of N-myc function results in
embryonic lethality and failure of the epithelial component of the
embryo to develop. Genes Dev. 6:2235–2247. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Breit S and Schwab M: Suppression of MYC
by high expression of NMYC in human neuroblastoma cells. J Neurosci
Res. 24:21–28. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ruzinova MB, Caron T and Rodig SJ: Altered
subcellular localization of c-myc protein identifies aggressive
B-cell lymphomas harboring a c-MYC translocation. Am J Surg Pathol.
34:882–891. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dang CV and Lee WM: Identification of the
human c-myc protein nuclear translocation signal. Mol Cell Biol.
8:4048–4054. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Soldani C, Bottone MG, Biggiogera M,
Alpini C, Scovassi AI, Martin T and Pellicciari C: Nuclear
localization of phosphorylated c-myc protein in human tumor cells.
Eur J Histochem. 46:377–380. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smith KP, Byron M, O'Connell BC, Tam R,
Schorl C, Guney I, Hall LL, Agrawal P, Sedivy JM and Lawrence JB:
C-myc localization within the nucleus: Evidence for association
with the PML nuclear body. J Cell Biochem. 93:1282–1296. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Meitar D, Crawford SE, Rademaker AW and
Cohn SL: Tumor angiogenesis correlates with metastatic disease,
N-myc amplification, and poor outcome in human neuroblastoma. J
Clin Oncol. 14:405–414. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Edry Botzer L, Maman S, Sagi-Assif O,
Meshel T, Nevo I, Bäuerle T, Yron I and Witz IP: Lung-residing
metastatic and dormant neuroblastoma cells. Am J Pathol.
179:524–536. 2011. View Article : Google Scholar : PubMed/NCBI
|