Introduction
Lung cancer was responsible for the majority of
cancer-associated mortalities in China in 2014, with risk factors
attributed to tobacco or asbestos exposure, indoor air pollution,
carcinogenic products and genetic susceptibility (1). Lung cancer can be divided into non-small
cell lung cancer (NSCLC) and small cell lung cancer (SCLC); 85% of
cases are NSCLC (2). The major
histological subtypes of NSCLC include adenocarcinoma, squamous
cell carcinoma (SCC) and large cell carcinoma (3), with distinct phenotypes. SCC is the
second most prevalent (~30%) among cases of NSCLC. Despite the
recent advances of targeted therapy in lung adenocarcinoma,
platinum doublet chemotherapy remains the cornerstone treatment in
metastatic SCC of the lung (4).
Arsenic trioxide (ATO) has previously been used as a
traditional Chinese medicine; synergistic combination of ATO with
all-trans retinoic acid (ATRA) has been demonstrated to lower the
relapse rate during treatment of acute promyelocytic leukaemia
(APL) (5). ATO activates the caspase
signalling pathway, decreases the mitochondrial membrane potential
and promotes the production of reaction oxygen species (ROS),
leading to apoptosis (6). In
addition, ATO induces apoptosis by downregulating B-cell lymphoma-2
(Bcl-2) and inducing G2/M cell cycle arrest (7).
The anticancer activity of ATO has been reported in
other solid tumours (8). In addition,
a previous study from our group reported the in vitro and
in vivo activity of ATO in SCLC (9). Given the limited therapeutic options in
advanced lung SCC, the aim of the present study was to investigate
the role of ATO in treatment of SCC using in vitro cell line
and in vivo tumour xenograft models.
Materials and methods
Cell lines and reagents
A panel of 4 squamous cell lung carcinoma cell lines
was obtained from American Type Culture Collection (Manassas, VA,
USA). SK-MES-1 and SW900 cells were cultured in Eagle's Minimum
Essential Medium (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) and Leibovitz's L-15 medium (Gibco; Thermo Fisher
Scientific, Inc.), respectively. H520 and H2170 cells were cultured
in RMPI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.). All
media were supplemented with 10% foetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.). Cells were incubated at 37°C in a
humidified atmosphere supplied with 5% CO2. ATO
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was dissolved in
1.65 M NaOH in a final concentration of 2.5 mM.
Cell viability assay
MTT assay was performed as previously described
(10). SK-MES-1 (2,000 cells/well),
SW900 (20,000 cells/well), H520 (10,000 cells/well) or H2170
(20,000 cells/well) cells were seeded in 96-well plate overnight at
37°C. Cells were incubated for 72 h with ATO solution (0.625, 1.25,
2.5, 5, 10 and 20 µM) and untreated cells served as a control.
Cells were further incubated at 37°C for 2 h following addition of
20 µl of MTT solution (0.25 mg/ml final concentration) (USB
Corporation, Cleveland, OH, USA). All solutions were then removed
and 50 µl dimethyl sulfoxide was added to solubilize the formazan
crystals. Optical density was measured at 570 nm using a FLUOstar
OPTIMA micro-plate reader (BMG Labtech GmbH, Ortenberg, Germany).
All experiments were performed ≥3 times.
Annexin binding assay
Annexin binding assay was performed using an annexin
V-phycoerythrin (PE)/7-aminoactinomycin D (7-AAD) apoptosis
detection kit (BD Biosciences, Franklin Lakes, NJ, USA) (9). Cells were treated for 72 h with ATO
(1.25, 2.5 and 5 µM) and untreated cells served as a control. Cells
(1×106) were collected, washed with PBS and re-suspended
in binding buffer. Cells were then stained for 15 min at room
temperature in darkness with 300 µl binding buffer containing 5 µl
of annexin V-PE and 5 µl of 7-AAD. The excitation/emission (Ex/Em)
of PE-annexin V and 7-AAD were 488/578 and 546/647 nm,
respectively. Signals were detected using a flow cytometer
(Cytomics FC500 with CXP software version 1.0, Beckman Coulter,
Inc., Brea, CA, USA) using Fluorescence Light (FL), FL2 and FL4
channels. A total of 10,000 events/sample were recorded. The
percentage of apoptotic cells (PE+/7-AAD- and PE+/7-AAD+) was
calculated. The experiments were performed in triplicate.
Mitochondrial membrane depolarization
detection
JC-1 (Sigma-Aldrich; Merck KGaA) staining was
performed as previously described (11). Cells were treated for 72 h with ATO
(1.25, 2.5 and 5 µM) and untreated cells served as a control. Cells
(1×106) were collected, washed with PBS and stained at
37°C for 15 min in darkness with JC-1 (2 µM final concentration) in
plain medium. Signals were detected using a flow cytometer
(Cytomics FC500 with CXP software version 1.0, Beckman Coulter,
Inc.) with FL1 and FL2 channels. A total of 10,000 events/sample
were captured. The experiments were performed in triplicate.
Cell cycle analysis
Cells were treated for 72 h at the beginning of
experiment with ATO (1.25, 2.5 and 5 µM) and untreated cells served
as a control. Cells were fixed with 70% cold ethanol and stored at
−20°C. Cells were incubated at 37°C in the dark with plain medium
containing RNase A (Thermo Fisher Scientific, Inc.) and propidium
iodide (Sigma-Aldrich). Signals were detected using a flow
cytometer (Cytomics FC500 with CXP software version 1.0, Beckman
Coulter, Inc.) with FL3 channel.
Western blot analysis
Western blot analysis was performed as previously
reported (11). Cells
(1×107) were collected and lysed for 1 h on ice with
RIPA buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM
Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate,
2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM
Na3VO4 and 1 µg/ml leupeptin) containing a
protease inhibitor cocktail. Tissue samples were lysed for 1 h on
ice with T-PER® Tissue Protein Extraction Reagent
(Thermo Fisher Scientific, Inc.) containing a protease inhibitor
cocktail. Protein concentration was detected using a Bradford assay
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Protein (25–100
µg) was then mixed with a 5X sample buffer (0.1 M Tris-HCl, pH 6.8,
50% glycerol, 10% SDS, 5% β-mercaptoethanol and 0.05% bromophenol
blue) and boiled for 5 min. Proteins were separated using SDS-PAGE
(10–15%). Proteins were then transferred to nitrocellulose blotting
membranes (GE Healthcare Life Sciences, Little Chalfont, UK). The
membrane was blocked at 4°C for 1 h with 5% blotting-grade blocker
(Bio-Rad Laboratories, Inc.) and further incubated at 4°C overnight
with primary antibody (Cell Signaling Technology, Inc., Danvers,
MA, USA; all in 1:1,000) [rabbit anti-human Bcl-2 antagonist/killer
protein (Bak) (cat no. 3814), rabbit anti-human Bcl-2 (cat no.
2872), rabbit anti-human cleaved caspase 3 (cat no. 9661), rabbit
anti-human E2F transcription factor 1 (E2F1; cat no. 3742), rabbit
anti-human poly ADP-ribose polymerase (PARP; cat no. 9542), rabbit
anti-human ribonucleotide reductase M1 (RRM1; cat no. 8637), rabbit
anti-human thymidylate synthase (TYMS; cat no. 5449) and rabbit
anti-human X-linked inhibitor of apoptosis protein (XIAP; cat no.
2042)]. The membrane was washed at 4°C for 10 min with
Tris-Buffered Saline and Tween 20 (TBST) 3 times. The membrane was
then incubated at 4°C for 90 min with corresponding horseradish
peroxidase-conjugated secondary antibody (anti-mouse IgG, cat no.
7076, 1:1,000 or anti-rabbit IgG, cat no. 7074, 1:1,000; both from
Cell Signaling Technology, Inc.). Protein expression was detected
using Amersham™ ECL™ Western Blotting Detection Reagents (GE
Healthcare Life Sciences, catalogue no. RPN2106). Relative protein
expression was normalized with mouse anti-human β-actin
(Sigma-Aldrich, cat no. A1978, 1:1,000). Band intensities were
analysed by GelQuant version 1.8.2 (BioSystematica, Llandysul,
UK).
Tumour growth inhibition in vivo
A total of 24 BALB/cA-nude mice (female,
4–6-week-old, 10–14 g, BALB/cAnN-nu, Charles River Laboratories,
Wilmington, USA) were obtained and kept in 12/12 h light/dark cycle
with temperature (16–26°C) and humidity (30–70%) control and ad
libitum diet was provided. The animal protocol (approval
reference number: 2860-12) was approved by the Animal Ethics
Committee of The University of Hong Kong. SW900 cells
(107) were re-suspended in 100 µl phosphate buffered
saline and mixed with 100 µl ice-cold highly concentrated Matrigel
matrix (BD Biosciences) prior to subcutaneous injection at the back
of the nude mice. The mice were randomized into 3 groups (n=8) when
the tumour volume reached 100 mm3. PBS and ATO (3.75 and
7.5 mg/kg) were injected intraperitoneally and daily in control and
treatment groups, respectively. Their general condition and body
weight were monitored daily. Tumour size was measured on alternate
days using digital calipers and calculated using the formula
[volume=1/2 × length × width × height] (12). The experimental endpoint was reached
when the length of tumour exceeded 17 mm in the control group.
Tumours were collected for western blot analysis and hematoxylin
and eosin staining.
Hematoxylin and eosin staining
Tissues were fixed overnight at 4°C in PBS 10%
formaldehyde. Tissues were cut into 5 µm sections. The slides were
deparaffinized by immersion in xylene (Merck KGaA). Subsequently,
tissue sections were rehydrated with 100, 95 and 75% ethanol
(Sigma-Aldrich; Merck KGaA) and distilled water (5 min for each
step). Nuclei were stained for 2 min at room temperature with
hematoxylin (Sigma-Aldrich; Merck KGaA) and cytoplasm was stained
for 30 sec at room temperature with eosin (BD Biosciences) to
visualize cellular structures. Slides were immersed in water to
stop staining. For the dehydrating process, sections were
dehydrated in 75, 95 and 100% ethanol and xylene (5 min for each
step). Slides were mounted with Histofluid (Marienfeld-Superior,
Germany). Images were captured using a Nikon Ni-U fluorescence
microscope (Nikon, Tokyo, Japan) equipped with a camera/detector
Diagnostic Instrument RT3 Slider (Meyer Instruments, Houston, USA).
Pictures were captures at ×200 magnification using CFI Plan Fluor
DLL 20X objective (Nikon). Images were obtained using NIS-Elements
Basic Research software (Laboratory Imaging, Prague, Czech
Republic).
Statistical analysis
Data from three individual experiments are presented
as the mean ± standard deviation. Comparison between groups was
performed using Student's two-tailed t-test using Prism version
5.01 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
ATO treatment decreases SK-MES-1 and
SW900 cell viability
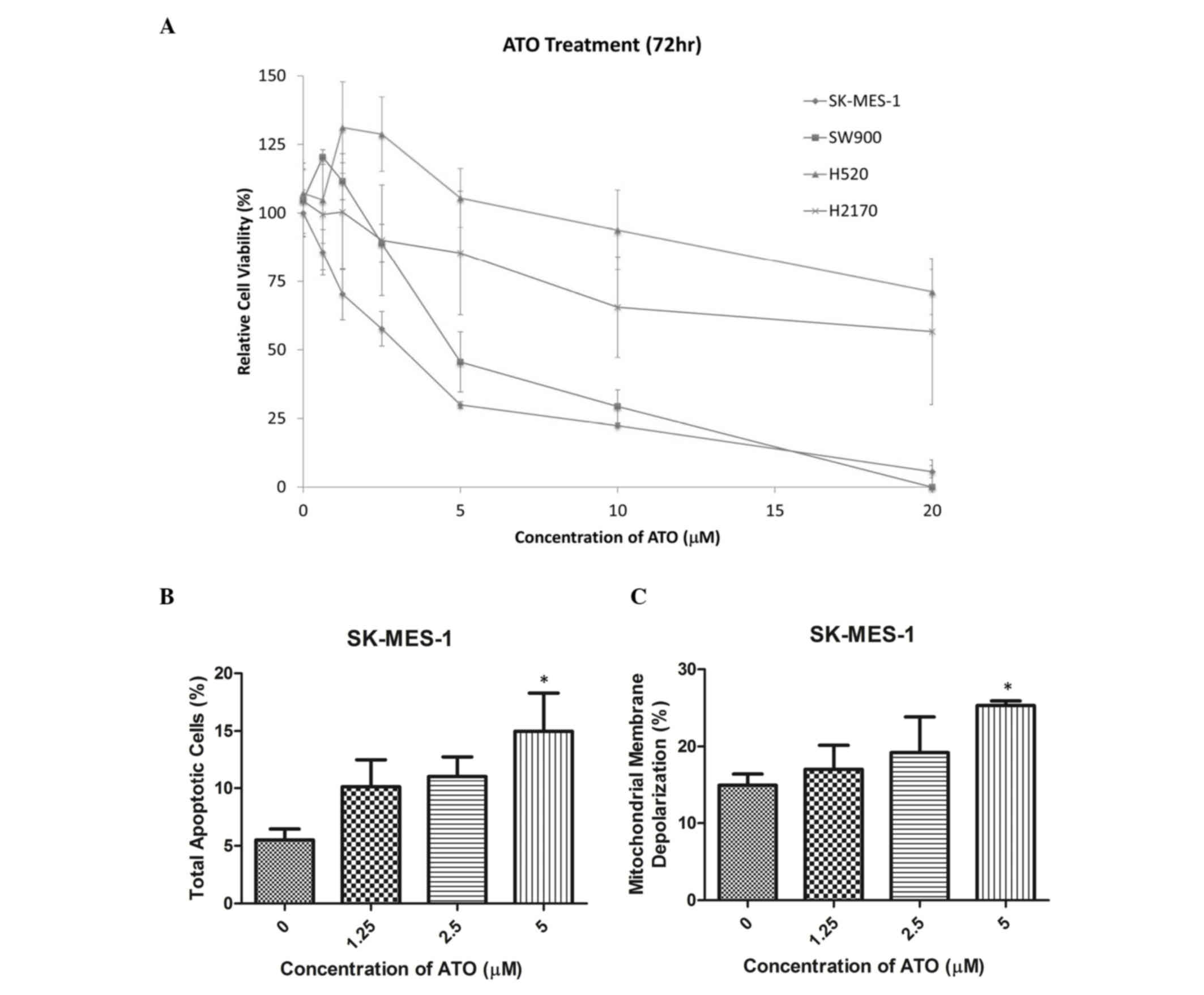
Cell viability was reduced by ATO in a
dose-dependent manner in SK-MES-1 and SW900 cells (Fig. 1A). The half-maximal inhibitory
concentration (IC50) values (72 h following ATO
treatment) in SK-MES-1 and SW900 cells were 2.5 and 5 µM,
respectively, while those in H520 and H2170 cells were >10 µM.
SK-MES-1 and SW900 cells were selected for further experiments, as
their IC50 values were within clinically reachable
concentrations of ATO (13). In
SK-MES-1 cells, ATO treatment markedly increased the percentage of
late apoptotic cells in a dose-dependent manner, as detected using
an annexin V-PE/7-AAD assay. A concentration of 5 µM ATO
significantly increased the percentage of late apoptotic cells
compared with the control group (15 vs. 5%; P<0.05; Fig. 1B). In addition, treatment with 5 µM
ATO significantly increased the percentage of SK-MES-1 cells with
mitochondrial membrane depolarization compared with the control
group (26 vs. 12%; P<0.05; Fig.
1C). Cell cycle analysis using propidium iodide staining
demonstrated that ATO treatment induced G2/M arrest in
SK-MES-1 and SW900 cells (Tables I
and II).
 | Table I.The percentage of cells in different
phases of the cell cycle following ATO treatment in SK-MES-1
cells. |
Table I.
The percentage of cells in different
phases of the cell cycle following ATO treatment in SK-MES-1
cells.
|
| Cell cycle phase
distribution, % |
|---|
|
|
|
|---|
| ATO dose |
Sub-G1 | G1 | S | G2/M |
|---|
| 0 µM | 9.9±4.6 | 55.1±4.0 | 14.2±2.1 | 20.9±2.8 |
| 1.25 µM | 11.3±4.3 | 48.2±1.2a | 16.6±3.6 | 23.9±2.0 |
| 2.5 µM | 12.7±3.5 | 44.7±2.8a | 15.5±5.6 | 27.1±0.9a |
| 5 µM | 10.6±5.3 | 36.4±1.1b | 19.0±1.3 | 34.1±5.1a |
| Table II.The percentage of cells in different
phases of the cell cycle following ATO treatment in SW900
cells. |
Table II.
The percentage of cells in different
phases of the cell cycle following ATO treatment in SW900
cells.
|
| Cell cycle phase
distribution, % |
|---|
|
|
|
|---|
| ATO dose |
Sub-G1 | G1 | S |
G2/M |
|---|
| 0 µM | 3.1±1.5 | 56.8±1.9 | 10.4±1.7 | 29.7±1.3 |
| 1.25 µM | 2.3±0.9 | 47.3±5.6 | 11.4±0.5 | 39.0±5.9 |
| 2.5 µM | 2.3±0.6 |
44.0±1.8a | 11.6±0.2 |
42.1±2.6a |
| 5 µM | 1.9±0.3 |
40.8±5.6b | 11.3±1.2 |
46.0±5.4b |
Expression of apoptosis and DNA
replication-associated proteins following treatment with ATO
Following treatment with 5 µM ATO, expression of
XIAP and Bcl-2 was significantly downregulated in SW900 and
SK-MES-1 cells, respectively, compared with the control groups
(P<0.01 and P<0.05; Fig. 2A and
B, respectively). Bak was significantly upregulated in SW900
cells compared with the control group (P<0.05; Fig. 2C) and expression of cleaved caspase 3
was significantly increased in SK-MES-1 and SW900 cells compared
with the control groups (P<0.001 and P<0.05, respectively;
Fig. 2D). A significant increase in
cleaved PARP expression was observed in SK-MES-1 cells compared
with the control group (P<0.01; Fig.
2E). ATO (1.25, 2.5 and 5 µM) treatment significantly
downregulated the expression of E2F1 compared with the control
group in SK-MES-1 (P<0.05, P<0.001 and P<0.001,
respectively; Fig. 2F). RRM1
expression was significantly decreased compared with the control
group in SK-MES-1 cells (2.5 and 5 µM ATO; P<0.05 and P<0.01,
respectively; Fig. 2G) and TYMS
expression was significantly decreased in SK-MES-1 cells treated
with 5 µM ATO compared with the control group (P<0.05; Fig. 2H).
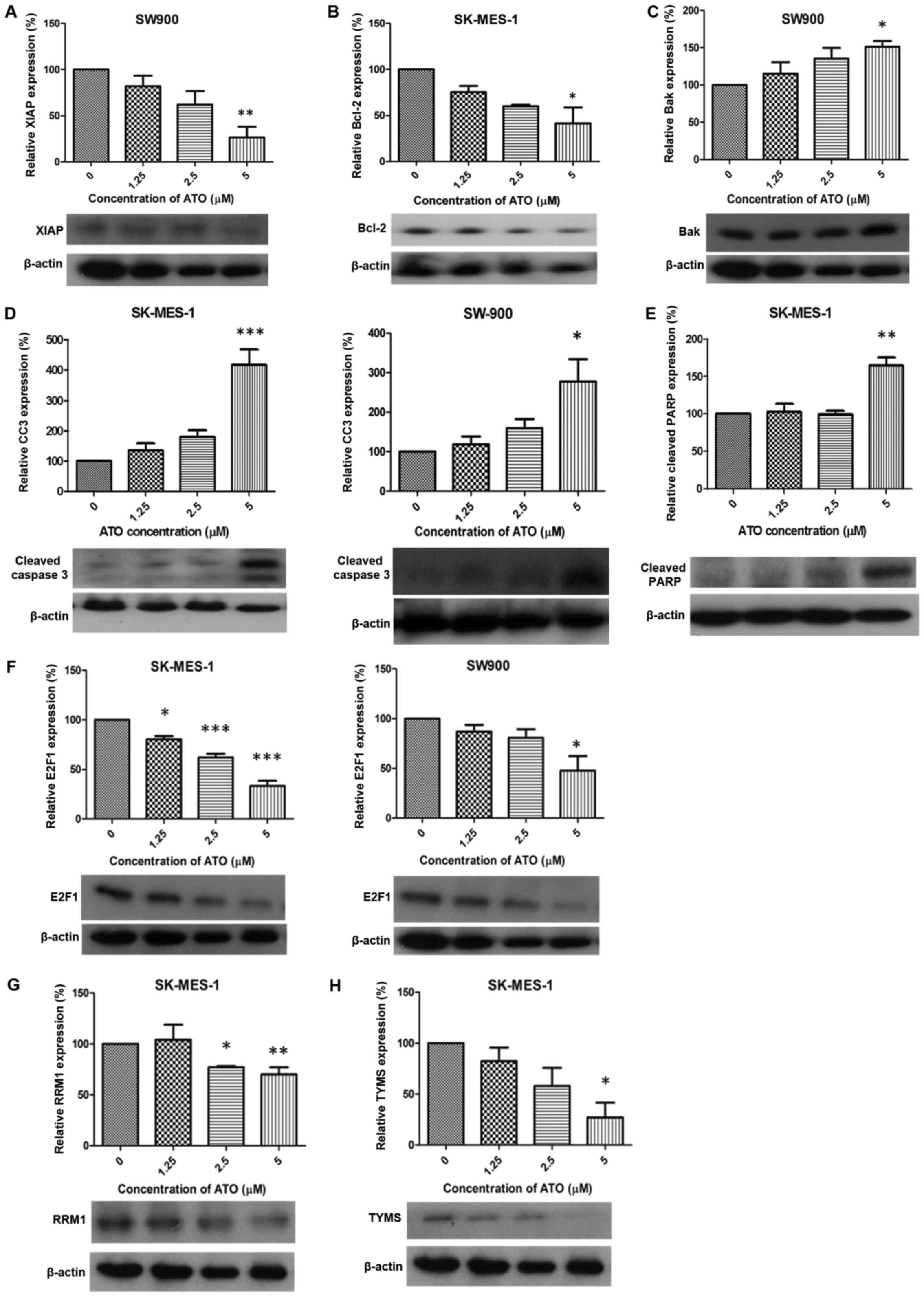 | Figure 2.Alteration of protein expression
following ATO treatment. ATO induced (A) XIAP downregulation
(SW900), (B) Bcl-2 suppression (SK-MES-1), (C) Bak upregulation
(SW900), (D) CC3 elevation (SK-MES-1 and SW900), (E) cleaved PARP
upregulation (SK-MES-1), (F) E2F1 suppression (SK-MES-1 and SW900),
(G) RRM1 downregulation (SK-MES-1) and (H) TYMS suppression
(SK-MES-1). Results were measured in triplicate experiments.
*P<0.05, **P<0.01 and ***P<0.001 compared with the control
group. XIAP, X-linked inhibitor of apoptosis; Bcl-2, apoptosis
regulator Bcl-2; Bak, Bcl-2 antagonist/killer protein; CC3, cleaved
caspase 3; PARP, poly ADP-ribose polymerase; RRM1, ribonucleotide
reductase M1; TYMS, thymidylate synthase. |
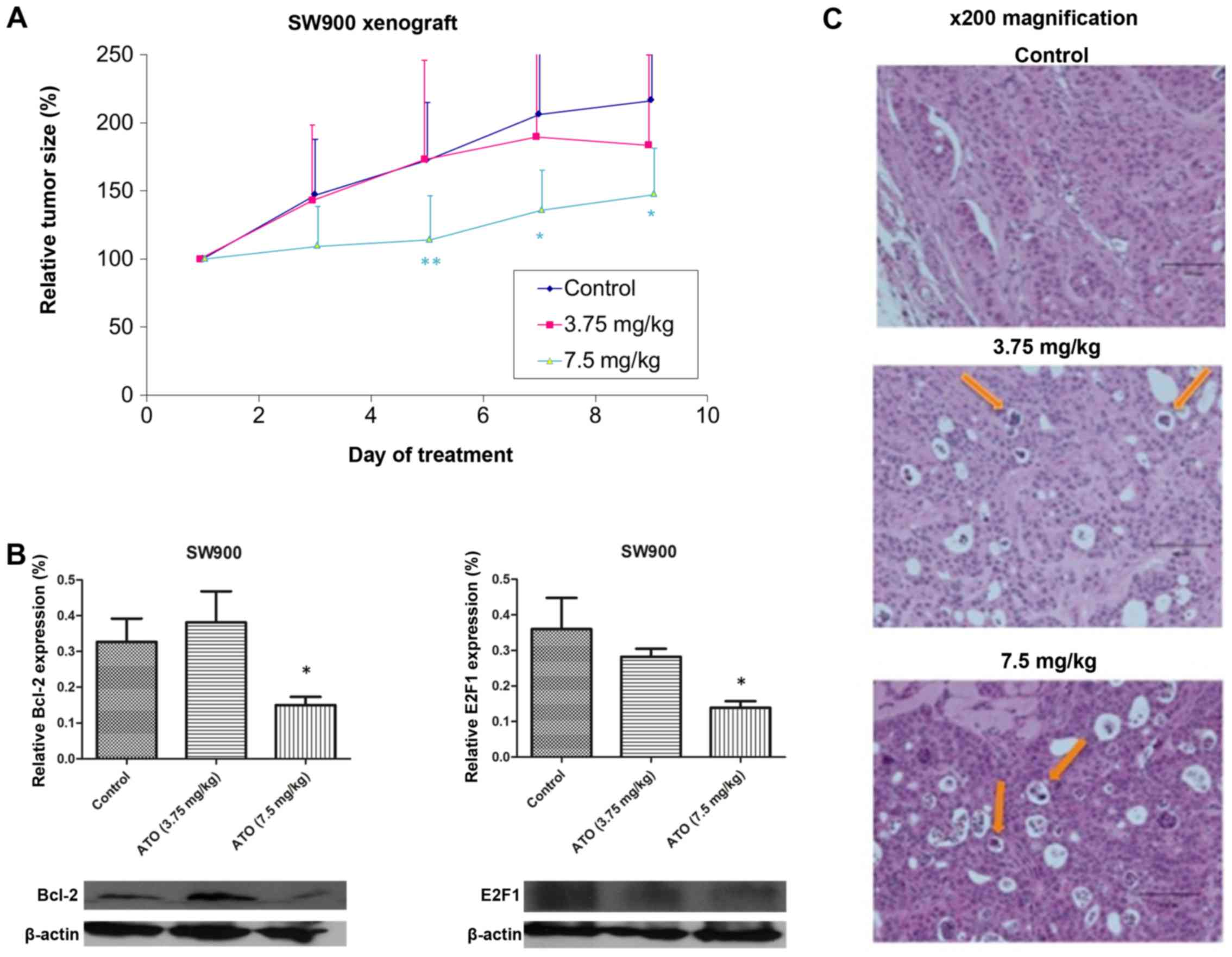
In vivo effect of ATO in SW900
xenograft model
In order to assess the effect of ATO on tumour
growth in vivo, mice were subcutaneously injected with SW900
cells, tumours were allowed to grow to ~100 mm3, then
the mice were treated daily with PBS (control) or ATO (3.75 or 7.5
mg/kg). No significant differences in baseline tumour volume were
observed between each group (Fig.
3A). The tumour size in control group of the SW900 xenograft
model reached the humane endpoint on day 9, when mice were
sacrificed and tumours were collected. Only high-dose ATO (7.5
mg/kg) significantly suppressed tumour growth compared with the
control group (P=0.0425; Fig. 3A).
There were no evident toxic effects due to ATO treatment. Western
blot analysis demonstrated significant downregulation of Bcl-2 and
E2F1 expression in tumour samples following 7.5 mg/kg ATO treatment
(P<0.05; Fig. 3B). Hematoxylin and
eosin staining revealed more apoptotic bodies in the 7.5 mg/kg ATO
group compared with the 3.75 mg/kg and control groups (Fig. 3D).
Discussion
Data from the present study demonstrated that ATO
significantly decreases the viability of the SK-MES-1 and SW900 SCC
cell lines within clinically achievable concentrations (13). ATO induced G2/M phase cell
cycle arrest and activated apoptosis by upregulating a number of
pro-apoptotic proteins. In addition, proteins associated with DNA
replication and repair, including E2F1 and TYMS were downregulated
following ATO treatment. Furthermore, ATO treatment significantly
reduced tumour growth in vivo in a SW900 xenograft
model.
ATO is a traditional Chinese medicine that has been
used clinically as an anti-cancer agent in APL, with or without
combined use of all-trans retinoic acid (14). ATO inhibits cell growth in solid
tumours other than non-haematological carcinoma, including
colorectal carcinoma (15). ATO
induces apoptosis in lung adenocarcinoma (11) and SCLC in vitro and in
vivo (9). However, the effect of
ATO in SCC remains unknown.
Phosphatidylserine externalization is a recognition
ligand for phagocytes to detect apoptotic cells (16). Phosphatidylserine externalization has
been observed in H841 SCLC cells (9)
and H23 lung adenocarcinoma cells (10). Mitochondrial membrane depolarization
is another indicator of apoptosis; it has been demonstrated that
ATO can induce mitochondrial membrane depolarization in lung cancer
cells (11). DNA damage is typically
sensed at the G1/S checkpoint or G2/M
checkpoint, leading to either DNA repair or cell apoptosis. ATO
treatment has been demonstrated to induce G2 arrest in
Calu-6 cells (1–3 µM) (17).
XIAP belongs to the inhibitor of apoptosis protein
family, and inhibits apoptosis through activation of the caspase
activation pathway via caspase 3, 7 and 9 (18). A previous report demonstrated that
expression of XIAP was decreased following ATO treatment in SCLC
H841 cells (9). Bcl-2 is responsible
for the release of apoptotic inducing factor and cytochrome
c from the mitochondria, which leads to apoptosis (19). Bcl-2 has been revealed to be
downregulated following ATO treatment in lung adenocarcinoma
(11) and SCLC (9). Bak is a pro-apoptotic member of the
Bcl-2 family of proteins (20) and
has been demonstrated to be upregulated in ATO-treated lung
adenocarcinoma cell lines, an effect mediated by truncation of
BH3-interacting domain death agonist (11).
PARP is a nuclear protein that facilitates DNA
repair when cells are undergoing genomic DNA damage from
environmental stress (21). PARP is
one of the major substrates for executor caspase 3, which
represents a hallmark of apoptosis (22). Activation of caspase 3 was observed in
lung adenocarcinoma (23) and SCLC
(9). Caspase-dependent apoptosis can
be divided into the intrinsic (mitochondria-mediated) and extrinsic
(death receptor-mediated) pathways (24). Data from the present study
demonstrated that in ATO-treated SK-MES-1 cells, downregulation of
Bcl-2 was accompanied by mitochondrial membrane depolarization.
Cleavage of caspase-3 and an increase in PARP expression indicated
that the intrinsic pathway was activated by ATO in SK-MES-1 cells.
By contrast, in ATO-treated SW900 cells, upregulation of Bak did
not induce mitochondrial membrane depolarization. Suppression of
XIAP and activation of caspase-3 suggested that the extrinsic
pathway could be the predominant pathway in this cell type.
Therefore, ATO could induce apoptosis via different pathways that
were cell-line dependent.
E2F1 is a transcription factor belonging to the E2F
family of proteins, which is involved in the G1 to S
phase transition and cancer cell proliferation (25). High E2F1 gene expression in NSCLC has
been associated with more aggressive phenotype of tumour cells
(25). In addition, ATO has
previously been demonstrated to downregulate E2F1 and its
associated genes, including cyclin A2, c-myc and S-phase kinase
associated protein 2 in lung adenocarcinoma (11).
TYMS is a key enzyme for the biosynthesis of
thymidylate and is involved in DNA synthesis (26). High tumoral TYMS expression has been
associated with poor clinical outcomes (25). In addition, ATO has been demonstrated
to downregulate TYMS protein and mRNA expression, leading to tumour
growth inhibition in lung adenocarcinoma cell lines (11).
RRM1 encodes the regulatory subunit of
ribonucleotide reductase and acts on ribonucleoside diphosphates
that are required for deoxynucleotide production (27). A previous clinical study demonstrated
that patients with higher tumoral RRM1 expression exhibited a lower
survival rate when treated with gemcitabine-based therapy (28). ATO downregulated RRM1 expression in
lung adenocarcinoma, which may cause inhibition of DNA synthesis
(11).
In conclusion, the results from the present study
indicate that ATO decreases the viability of SCC SK-MES-1 and SW900
cells within clinically achievable concentrations, partially
through apoptosis and inhibition of cell proliferation.
Furthermore, ATO induced apoptosis via the intrinsic pathway in
SK-MES-1 cells and the extrinsic pathway in SW900 cells. The
antitumor effects of ATO were confirmed using a xenograft
model.
References
|
1
|
Hu J, Qian GS and Bai CX: Lung Cancer
Study Group of Chinese Thoracic S and Chinese Alliance Against Lung
Cancer Expert Group: Chinese consensus on early diagnosis of
primary lung cancer (2014 version). Cancer. 121:(Suppl 17).
S3157–S3164. 2015. View Article : Google Scholar
|
|
2
|
Ho JC, Tam TC and Lam SK: Salvage therapy
beyond targeted therapy in lung adenocarcinoma. Semin Respir Crit
Care Med. 34:837–844. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
West H, Harpole D and Travis W: Histologic
considerations for individualized systemic therapy approaches for
the management of non-small cell lung cancer. Chest. 136:1112–1118.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ang YL, Tan HL and Soo RA: Best practice
in the treatment of advanced squamous cell lung cancer. Ther Adv
Respir Dis. 9:224–235. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ravandi F, Estey E, Jones D, Faderl S,
O'Brien S, Fiorentino J, Pierce S, Blamble D, Estrov Z, Wierda W,
et al: Effective treatment of acute promyelocytic leukemia with
all-trans-retinoic acid, arsenic trioxide, and gemtuzumab
ozogamicin. J Clin Oncol. 27:504–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rojewski MT, Körper S and Schrezenmeier H:
Arsenic trioxide therapy in acute promyelocytic leukemia and
beyond: From bench to bedside. Leuk Lymphoma. 45:2387–2401. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park JW, Choi YJ, Jang MA, Baek SH, Lim
JH, Passaniti T and Kwon TK: Arsenic trioxide induces G2/M growth
arrest and apoptosis after caspase-3 activation and bcl-2
phosphorylation in promonocytic U937 cells. Biochem Biophys Res
Commun. 286:726–734. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Evens AM, Tallman MS and Gartenhaus RB:
The potential of arsenic trioxide in the treatment of malignant
disease: Past, present, and future. Leuk Res. 28:891–900. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng CY, Lam SK, Li YY and Ho JC: Arsenic
trioxide-induced cytotoxicity in small cell lung cancer via altered
redox homeostasis and mitochondrial integrity. Int J Oncol.
46:1067–1078. 2015.PubMed/NCBI
|
|
10
|
Lam SK, Mak JC, Zheng CY, Li YY, Kwong YL
and Ho JC: Downregulation of thymidylate synthase with arsenic
trioxide in lung adenocarcinoma. Int J Oncol. 44:2093–2102.
2014.PubMed/NCBI
|
|
11
|
Lam SK, Li YY, Zheng CY, Leung LL and Ho
JC: E2F1 downregulation by arsenic trioxide in lung adenocarcinoma.
Int J Oncol. 45:2033–2043. 2014.PubMed/NCBI
|
|
12
|
Euhus DM, Hudd C, LaRegina MC and Johnson
FE: Tumor measurement in the nude mouse. J Surg Oncol. 31:229–234.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ardalan B, Subbarayan PR, Ramos Y,
Gonzalez M, Fernandez A, Mezentsev D, Reis I, Duncan R, Podolsky L,
Lee K, et al: A phase I study of 5-fluorouracil/leucovorin and
arsenic trioxide for patients with refractory/relapsed colorectal
carcinoma. Clin Cancer Res. 16:3019–3027. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Au WY, Kumana CR, Lee HK, Lin SY, Liu H,
Yeung DY, Lau JS and Kwong YL: Oral arsenic trioxide-based
maintenance regimens for first complete remission of acute
promyelocytic leukemia: A 10-year follow-up study. Blood.
118:6535–6543. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakagawa Y, Akao Y, Morikawa H, Hirata I,
Katsu K, Naoe T, Ohishi N and Yagi K: Arsenic trioxide-induced
apoptosis through oxidative stress in cells of colon cancer cell
lines. Life Sci. 70:2253–2269. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han YH, Kim SZ, Kim SH and Park WH:
Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a
G2 arrest of the cell cycle and apoptosis accompanied with the
depletion of GSH. Cancer Lett. 270:40–55. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Silke J, Kratina T, Ekert PG, Pakusch M
and Vaux DL: Unlike Diablo/smac, Grim promotes global
ubiquitination and specific degradation of X chromosome-linked
inhibitor of apoptosis (XIAP) and neither cause apoptosis. J Biol
Chem. 279:4313–4321. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seite P, Hillion J, d'Agay MF, Gaulard P,
Cazals D, Badoux F, Berger R and Larsen CJ: BCL2 gene activation
and protein expression in follicular lymphoma: A report on 64
cases. Leukemia. 7:410–417. 1993.PubMed/NCBI
|
|
20
|
Chao DT and Korsmeyer SJ: BCL-2 family:
Regulators of cell death. Annu Rev Immunol. 16:395–419. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Satoh MS and Lindahl T: Role of poly
(ADP-ribose) formation in DNA repair. Nature. 356:356–358. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Han YH, Kim SZ, Kim SH and Park WH:
Arsenic trioxide inhibits growth of As4.1 juxtaglomerular cells via
cell cycle arrest and caspase-independent apoptosis. Am J Physiol
Renal Physiol. 293:F511–F520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim HR, Kim EJ, Yang SH, Jeong ET, Park C,
Kim SJ, Youn MJ, So HS and Park R: Combination treatment with
arsenic trioxide and sulindac augments their apoptotic potential in
lung cancer cells through activation of caspase cascade and
mitochondrial dysfunction. Int J Oncol. 28:1401–1408.
2006.PubMed/NCBI
|
|
24
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang CL, Liu D, Nakano J, Yokomise H,
Ueno M, Kadota K and Wada H: E2F1 overexpression correlates with
thymidylate synthase and survivin gene expressions and tumor
proliferation in non small-cell lung cancer. Clin Cancer Res.
13:6938–6946. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rustum YM, Harstrick A, Cao S, Vanhoefer
U, Yin MB, Wilke H and Seeber S: Thymidylate synthase inhibitors in
cancer therapy: Direct and indirect inhibitors. J Clin Oncol.
15:389–400. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Boukovinas I, Papadaki C, Mendez P, Taron
M, Mavroudis D, Koutsopoulos A, Sanchez-Ronco M, Sanchez JJ,
Trypaki M, Staphopoulos E, et al: Tumor BRCA1, RRM1 and RRM2 mRNA
expression levels and clinical response to first-line gemcitabine
plus docetaxel in non-small-cell lung cancer patients. PLoS One.
3:e36952008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rosell R, Danenberg KD, Alberola V, Bepler
G, Sanchez JJ, Camps C, Provencio M, Isla D, Taron M, Diz P, et al:
Ribonucleotide reductase messenger RNA expression and survival in
gemcitabine/cisplatin-treated advanced non-small cell lung cancer
patients. Clin Cancer Res. 10:1318–1325. 2004. View Article : Google Scholar : PubMed/NCBI
|