Introduction
Glioblastoma is one of the most common types of
malignant tumors and exhibits a poor prognosis (1). Due to the high invasiveness and
heterogeneity, glioma has been demonstrated to exhibit resistance
to traditional treatment, including surgery, irradiation and
chemotherapy (1). The average
survival time for patients with glioblastoma has marginally changed
over the previous years and remains at ~8–12 months (2). Therefore, there is an urgent requirement
to develop novel therapy strategies in order to treat this
malignant neoplasm.
Temozolomide (TMZ) is an orally administered
alkylating agent with promising antitumor activity and ability to
cross the brain-blood barrier (2).
Randomized clinical trials have been performed to demonstrate that
TMZ-based standard chemotherapy may significantly improve survival
time and quality of life for patients with glioblastoma (3). The mechanism of action of TMZ is
attributed to cell apoptosis due to induction of DNA O6-methyl
guanine. However, treatment with TMZ alone is insensitive to
specific patients who highly express the O6-methylguanine-DNA
methyltransferase (MGMT). MGMT acts as a transferase and removes
the cytotoxic O6-alkylguanine DNA adducts induced by TMZ (4). Previous studies have revealed that
phosphatidylinositol 3-kinase (PI3K)/AKT/mechanistic target of
rapamycin signaling pathway over-activation is a common event in
glioma and its downstream targets, including p53, nuclear factor
(NF)-κB. p53 and NF-κB contribute to chemotherapy resistance by
mediating the transcription of a number of genes, including MGMT
(5).
NVP-BKM120, an oral highly selective pan-class I
PI3K inhibitor, has been used in previous studies to treat various
solid types of cancer and demonstrated an ability to induce cancer
cell apoptosis and reverse chemoresistance. In phase I clinical
trials, NVP-BKM120 was demonstrated to suppress tumor growth and
proliferation at tolerated doses in various types of solid cancer
(6).
In the present study, the authors hypothesized that
targeting the PI3K/AKT signaling pathway by BKM120 in combination
with TMZ may be a promising strategy for the treatment of glioma.
Furthermore, the present study investigated the possible mechanism
underlying the PI3K/AKT signaling pathway and drug resistance.
Materials and methods
Tumor cell line
To investigate the TMZ-resistance mechanism of
glioma cells, the present study used the C6 glioma cell line, a
MGMT-positive cell line (Type Culture Collection of the Chinese
Academy of Sciences, Shanghai, China). In the present study, glioma
cells were cultured in Dulbecco's modified Eagle's medium
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C in an atmosphere containing 5%
CO2 for 24 or 48 h.
Reagents
TMZ was supplied by Tasly Pharmaceutical Co., Ltd.
(Tianjin, China). The PI3K inhibitor, BKM120, was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Stock solutions of
TMZ and BKM120 (TMZ, 120 mM; BKM120, 3 mM) were made by dissolving
in dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck KGaA). For TMZ
treatment solutions, the DMSO concentrations were limited to <1%
(v/v), and the vehicle controls were added at the same
concentration as DMSO. The terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay cell death kit was purchased
from Roche Diagnostics (Indianapolis, IN, USA). The Cell Counting
Kit-8 (CCK-8) and Hoechst 33342 were supplied by Beyotime Institute
of Biotechnology (Haimen, China). All primary antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA),
including rabbit anti-rat antibodies against PI3K (cat. no. 4257,
1:1,000 dilution), phosphorylated (p)-Akt (Ser473) (cat.
no. 4060, 1:1,000 dilution), NF-κB p65 (cat. no. 3033, 1:1,000
dilution), pS6 (cat. no. 2215S, 1:1,000 dilution), cleaved
caspase-3 (cat. no. 9664, 1:1,000 dilution), Bax (cat. no. 14796,
1:1,000 dilution) and GAPDH (cat. no. 5174, 1:1,000 dilution,), and
goat anti-rabbit horseradish peroxidase (HRP)-conjugated
immunoglobulin G (IgG) secondary antibody (cat. no. 31460, 1:1,000
dilution; Pierce; Thermo Fisher Scientific, Inc.).
Cell viability assay
Cell viability and coefficient of drug interactions
(CDIs) were determined by CCK-8 assay. The cells were seeded with a
density of 5×103 cells/well in 96-well plates and
incubated, at 37°C, in complete medium (DMEM with 20% FBS) with
serial dilutions of TMZ and/or BKM120 for 24 h. Tumor cells were
divided into four groups (TMZ alone, BKM120 alone, TMZ and BKM120
combined, vehicle control). In the TMZ treatment group, TMZ was
added to the culture medium at final concentrations of 300, 600,
900 and 1,200 µM. In the BKM120 treatment groups, BKM120 was added
to culture medium at final concentrations of 300, 600, 1,500 and
3,000 nM. In the combination treatment group, BKM120 (300 nM) was
added to culture mediums in combination with various concentrations
of TMZ (300, 600, 900 and 1,200 µM). Following incubation at 37°C
for 12, 24 and 48 h, CCK-8 solution was added into each well with a
final concentration of 20 µl/well. Following incubation for 4 h,
absorbance was determined at 490 nm using a microplate reader. Cell
viabilities and CDIs were evaluated using the data from the
independent CCK-8 assay results. The IC50 value was
defined as the mean drug concentration required for inhibiting 50%
of cell viability compared with the vehicle controls.
Hoechst 33342 staining
Briefly, C6 glioma cells were plated in 6-well
plates with a density of 1×105 cells/well and treated
with TMZ (300 µM) and/or BKM120 (300 nM) at 37°C for 24 h. The
cells were washed in PBS three times and incubated in Hoechst 33342
solution (10 µg/ml) for 30 min at 4°C. Finally, fluorescence
microscopy (magnification, ×40) was preformed to observe the
nuclear changes of C6 glioma cells. For each treatment group,
≥1,000 cells were analyzed in triplicate.
TUNEL staining
Following treatment with drugs (TMZ, 300 µM; BKM120,
300 nM) for 24 h and washing with PBS three times, the cells were
incubated with stationary liquid (4% paraformaldehyde) for 30 min,
and subsequently treated with 3% hydrogen peroxide for 10 min to
block endogenous peroxides at room temperature. Following washing
with PBS, 0.1% Triton was added to perforate the cells for 10 min
at room temperature. Subsequently, the slides were incubated with
terminal deoxynucleotidyl transferase enzyme for 1 h at room
temperature. Gold antifade mountant with DAPI (P36935; Invitrogen;
Thermo Fisher Scientific, Inc.) was used to mount the slides,
according to the manufacturer's instructions. Images were captured
using a microscope camera system (BX63; Olympus, Tokyo, Japan)
(magnification, ×40).
Western blot analysis
Following drug treating with TMZ (300 µM) and/or
BKM120 (300 nM) for 24 h at 37°C, proteins were extracted using a
cell lysis buffer (PBS, 1% Triton, 1% SDS, protease inhibitor
cocktail III (539134; Merck KGaA), phosphatase inhibitor cocktail
III (k276-1EA; BioVision, Inc., Milpitas, CA, USA). The proteins
were centrifuged for 10 min at 10,000 × g at 4°C and protein was
determined using the bicinchoninic assay method. The proteins (10
µg/lane) were separated using 12% SDS-PAGE (15% gel for cleaved
caspase-3) and subsequently transferred to nitrocellulose
membranes. Nitrocellulose membranes were blocked with 5% milk for 2
h at 37°C and subsequently incubated with primary antibodies
against PI3K, pAkt (Ser473), pS6, cleaved caspase-3, Bax
and GAPDH at 4°C overnight. The membranes were subsequently washed
with Tris-buffered saline supplemented with 1% Tween-20 three times
and incubated with goat anti-rabbit immunoglobulin G at 37°C for 1
h. In addition, the enhanced chemiluminescence western blotting
detection system (Medicare X-Ray-Processor 102; Kodak, Rochester,
NY, USA) was used to visualize antibody complexes. Finally, protein
densities were measured using ImageJ software (version 2.0;
National Institutes of Health, Bethesda, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
For RT-qPCR, C6 glioma cells were collected
following treatment with TMZ (300 µM) and/or BKM120 (300 nM) for 24
h, and subsequently ≥1 mg total RNA was extracted from individual
samples using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
All RNAs were reverse transcribed into cDNA using the Revert Aid
First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Inc.)
for RT-PCR. cDNA was added to a total volume of 20 µl. qPCR was
performed using the SYBR Premix Ex Taq™ II kit (Takara
Biotechnology, Co., Ltd., Dalian, China) for MGMT and β-actin. The
2−ΔΔCq method (7) was used
to determine the relative mRNA expression levels of MGMT, with
β-actin mRNA as an endogenous control. All experiments were
repeated three times.
The temperature protocol for qPCR was as follows: i)
50°C for 2 min, 1 cycle; ii) 95°C for 10 min, 1 cycle; iii) 95°C
for 15 sec, 60°C for 30 sec and 72°C for 30 sec, 40 cycles; and iv)
72°C for 10 min, 1 cycle. The temperature protocol for reverse
transcription was as follows: i) 25°C for 5 min; ii) 42°C for 30
min; and iii) 85°C for 5 min. Primer sequences used were as
follows: MGMT mRNA forward, 5′-AGGAGCGATGAGGAGCAATC-3′ and reverse,
5′-CAGACTAACCCTCGACCGAG-3′; β-actin mRNA forward,
5′-GAGACCTTCAACACCCCAGC-3′ and reverse,
5′-CCCTTTAGCACGCACTGTA-3′.
Statistical analysis
Each analysis was repeated for ≥3 independent
experiments. Statistical analysis of results was performed using
SPSS software version 21 (IBM Corp., Armonk, NY, USA). All data
were tested for significance by unpaired Student's t-test or
one-way analysis of variance (ANOVA). Tukey's test and Dunnett's
correction were performed following ANOVA. P<0.05 was considered
to indicate a statistically significant difference. All data are
presented as the mean ± standard deviation.
Results
Synergistic cytotoxicity from
combination of BKM120 and TMZ in C6 glioma cells
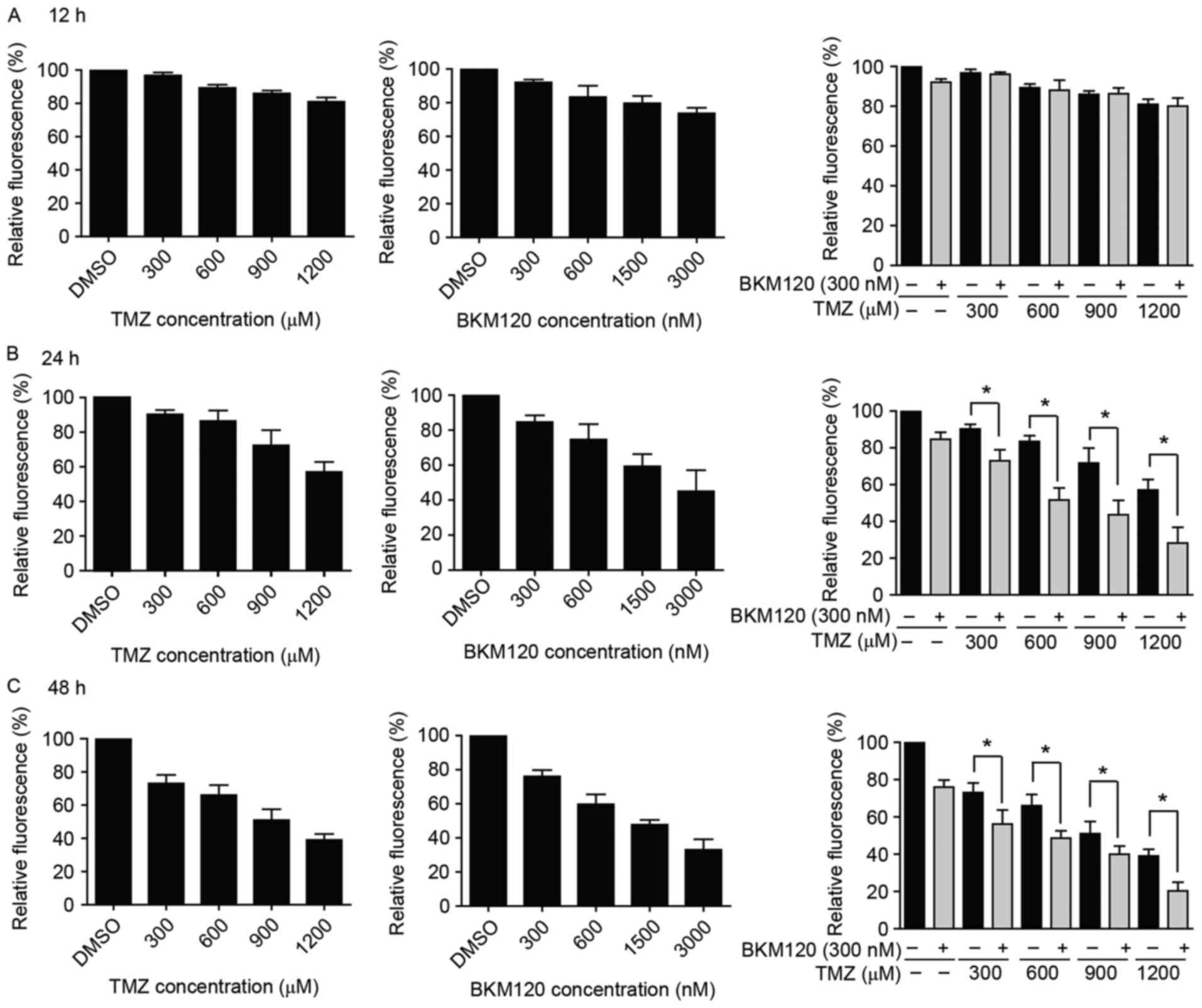
According to the hypothesis of the present study
(Fig. 1A and B), the present study
verified the PI3K-induced inhibition of BKM120 in C6 glioma cells
(Fig. 1C) and subsequently performed
CCK-8 analysis to detect the synergistic effect of BKM120 and TMZ.
C6 glioma cells were treated alone with increasing doses of TMZ or
BKM120 for 12, 24 and 48 h. There were decreased cell viabilities
at 24 and 48 h, compared with that observed at 12 h (Fig. 2A). The IC50 of TMZ (24 h,
1,137.67±271.53 µM; 48 h, 969.09±269.61 µM) and BKM120 (24 h,
4.78±1.63 µM; 48 h, 2.20±0.64 µM) achieved a high value, which
suggested that C6 glioma cells are insensitive to TMZ or BKM120
monotherapy.
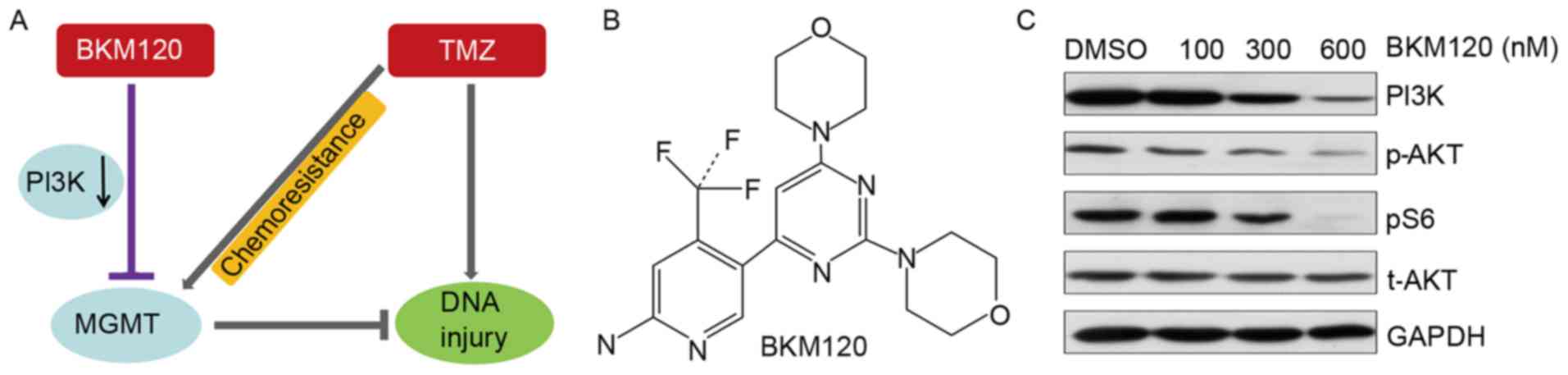 | Figure 1.(A) Hypothesized mechanism of action
of the BKM120 and TMZ combination treatment. (B) Chemical structure
of BKM120. (C) C6 glioma cells were treated with increased doses of
BKM120 (100, 300 and 600 nM) and DMSO for 24 h, respectively. The
expression of PI3K, p-Akt and pS6 was reduced in BKM120 treated C6
cells. TMZ, temozolomide; DMSO, dimethyl sulfoxide; MGMT,
O6-methylguanine-DNA methyltransferase; PI3K, phosphatidylinositol
3-kinase; p, phosphorylated; Akt, protein kinase B; t, total
protein. |
However, when cells were treated with the
combination treatment, which comprised a low concentration of
BKM120 (300 nM) and various concentrations of TMZ (300, 600, 900
and 1,200 µM) for 24 and 48 h, the IC50 of TMZ was
significantly decreased (24 h, 1,137.67±271.53 µM to 346.67±134.53
µM; 48 h, 969.09±269.61 to 794.75±314.09 µM; P<0.05, unpaired
Student's t-test; Fig. 2B and C). The
CDI values were close to 0.7, which indicated that BKM120 increased
the toxicity of TMZ (Table I).
Furthermore, the present study confirmed that the inhibition of
PI3K was induced by BKM120 in C6 glioma cells, and the expression
levels of its downstream targets (p-AKT, pS6) were also decreased
(Fig. 1C).
 | Table I.Coefficient of drug interaction of
combination treatment with TMZ and BKM120. |
Table I.
Coefficient of drug interaction of
combination treatment with TMZ and BKM120.
| TMZ, µM | BKM120, nM | CDI |
|---|
| 300 | 300 | 0.81±0.05 |
| 600 | 300 | 0.73±0.08 |
| 900 | 300 | 0.65±0.06 |
| 1,200 | 300 | 0.81±0.03 |
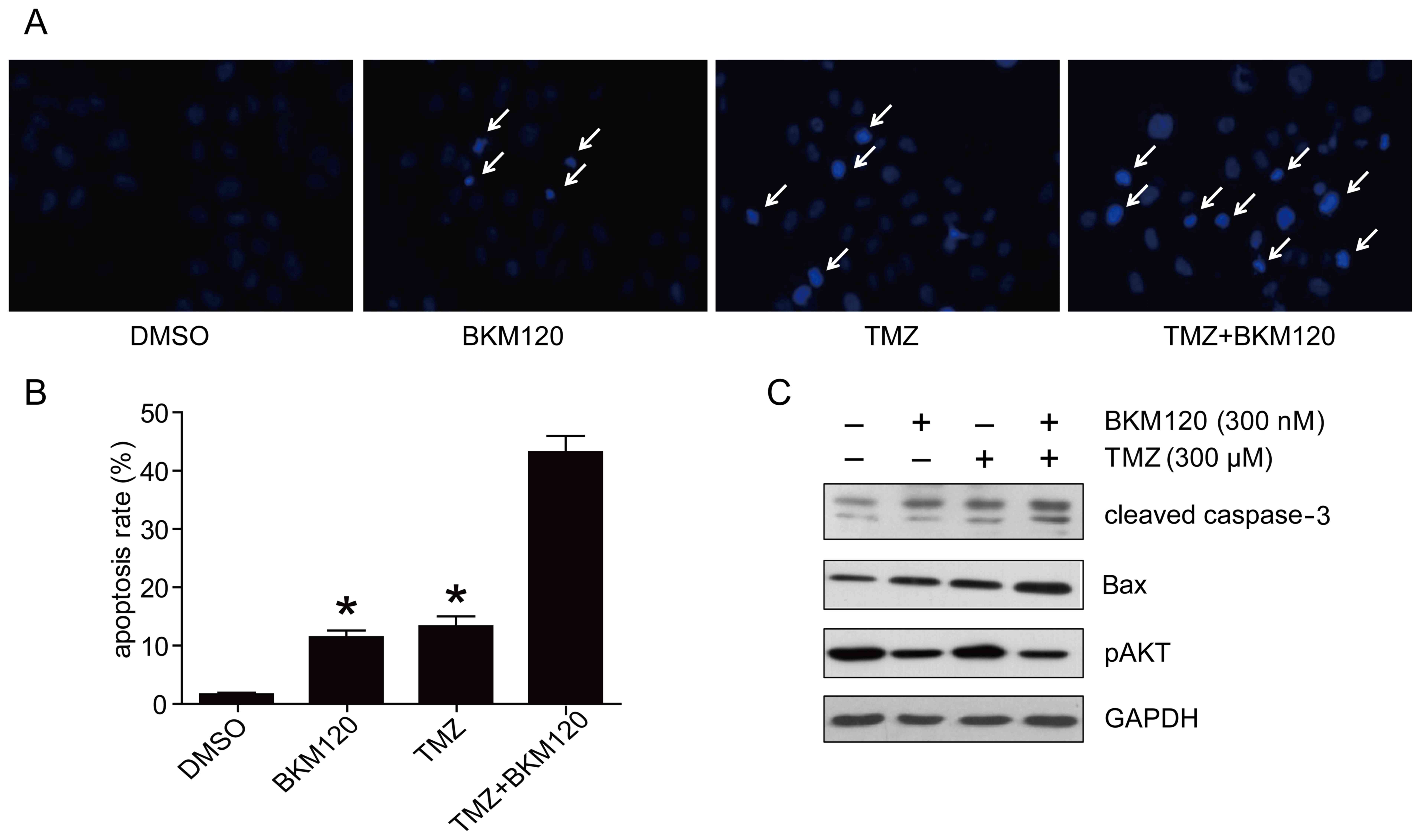
BKM120 increases TMZ-induced apoptosis
of C6 glioma cells
Hoechst 33342 staining assay was performed to
determine the level of apoptosis in cells following monotherapy or
combination therapy. In the combination treatment group, C6 glioma
cells exhibited more nuclear apoptotic changes, including nuclear
condensation and aggregation (Fig.
3A). The rate of Hoechst 33342-positive cells in the
combination treatment group was significantly increased, compared
with that of the TMZ group or the BKM120 group (44.06±7.28 vs.
17.68±3.04% and 13.37±2.23%, respectively; P<0.05, one-way
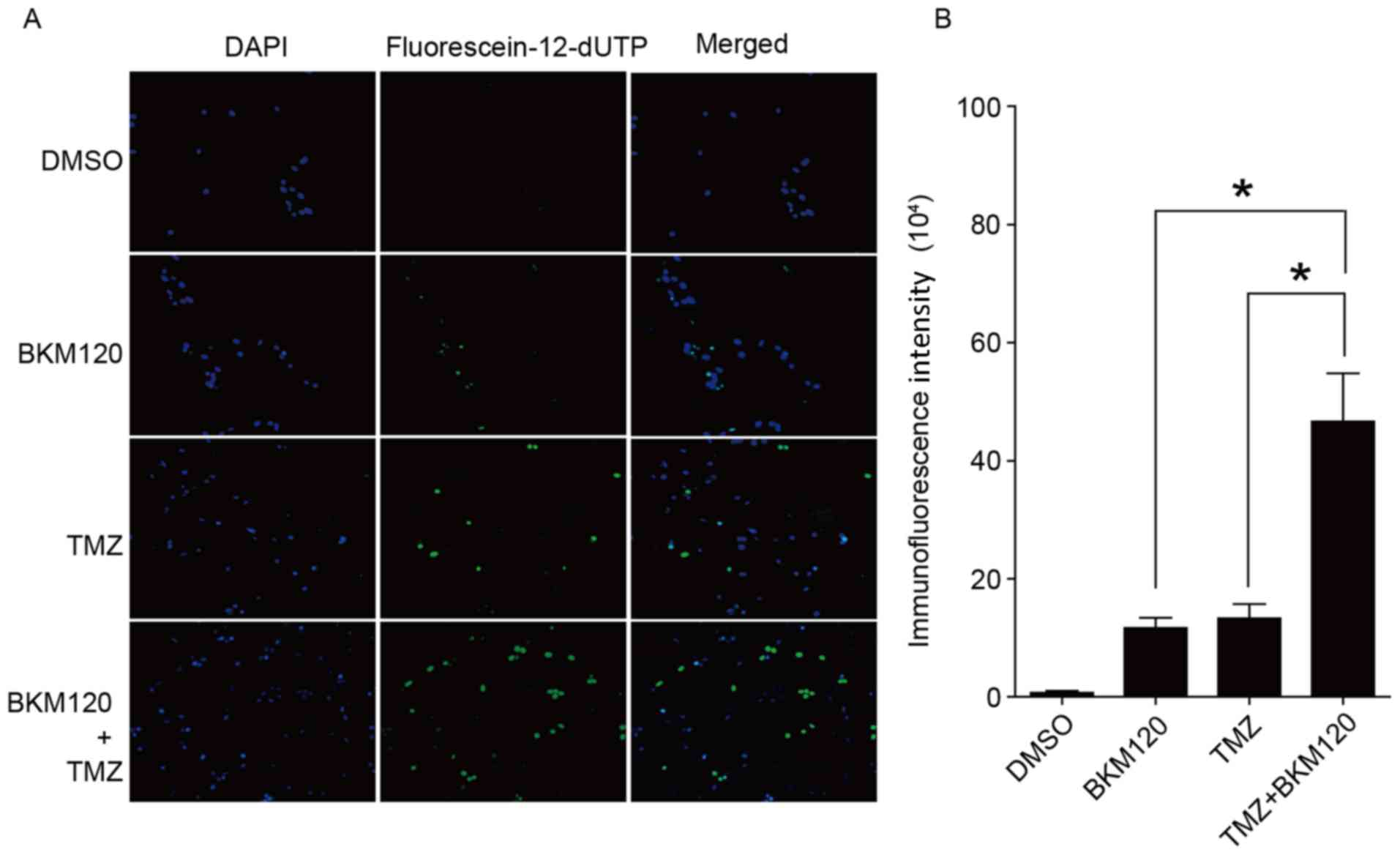
ANOVA; Fig. 3B). In addition, TUNEL
staining was performed to quantify the fraction of cells that were
apoptotic. The analysis indicated that the immunofluorescence
intensity of the combination treatment group was significantly
(P<0.05, ANOVA) higher compared with the TMZ group or the BKM120
group (Fig. 4A and B). The present
study also determined the level of expression of
apoptosis-associated proteins (cleaved caspase-3 and Bax) by
western blotting. The results confirmed that the combination
strategy induced increased expression of cleaved caspase-3 and Bax
expression compared with TMZ or BKM120 monotherapy (Fig. 3C). These results suggest that BKM120
increased TMZ-induced apoptosis in C6 glioma cells.
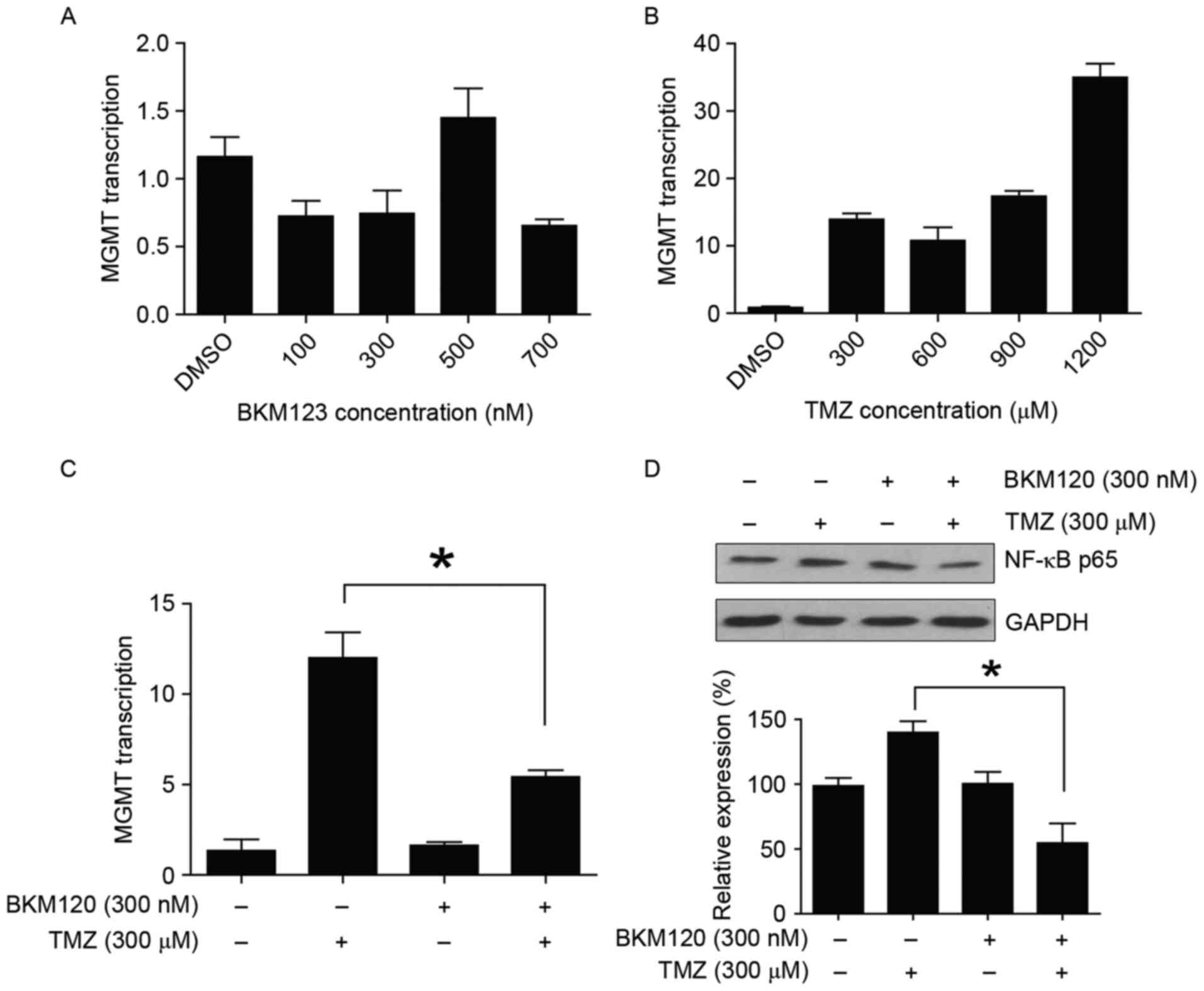
BKM120 suppresses TMZ-induced MGMT
transcription
RT-qPCR analysis demonstrated that exposure to
increasing doses of TMZ for 24 h significantly increased the level
of MGMT transcription. C6 glioma cells exposed to BKM120 alone
revealed no significant effects on the level of MGMT transcription
(Fig. 5A and B). However, when the
cells were treated with a combination of 300 nM BKM120 and 300 nM
TMZ, the level of MGMT transcription was significantly
downregulated compared with treatment with TMZ monotherapy (P=0.03,
one-way ANOVA; Fig. 5C). Taken
together, these experimental results demonstrated that BKM120
suppressed TMZ-induced MGMT transcription in C6 glioma cells.
NF-κB p65 contributes to
the therapeutic potential of combination strategy
The mechanism of the observed reversal in
TMZ-resistance was further investigated by detecting the levels of
NF-κB p65 expression. NF-κB p65 is a downstream of PI3K and an
important mediator of MGMT transcription. Using western blotting,
it was observed that the expression of NF-κB p65 was increased in
the TMZ monotherapy group compared with the combination treatment
group. It was also indicated that the expression of NF-κB p65 was
significantly decreased in the TMZ and BKM120 combination treatment
group from 139.8±15.1 to 54.6±26.1% compared with the TMZ
monotherapy group (P=0.008, one-way ANOVA), which was consistent
with the changes observed in the level of MGMT transcription
(Fig. 5D). This result suggested that
NF-κB p65 contributed to the therapeutic potential of the
combination strategy.
Discussion
Glioblastoma is the most rapidly growing and
aggressive type of intracranial neoplasm in adults (8). Surgical resection, radiotherapy and TMZ
chemotherapy constitute standard therapy for patients with
glioblastoma (9). TMZ is able to
cross the brain-blood barrier and achieve therapeutic concentration
in brain tissues. However, glioblastoma is often resistant to TMZ
due to MGMT expression in glioma cells (10). MGMT is highly expressed in solid types
of cancer, and its expression removes the TMZ-induced cytotoxic
O6-alkylguanine DNA adducts. MGMT promoter methylation status is an
important prognosis indicator for patients with glioblastoma
(11). Therefore, investigating
combination treatments of TMZ with other drugs is required in order
to inhibit unexpected DNA repair. A number of MGMT inhibitors,
including O6-benzylguanine, streptozotocin and PaTrin-2, have been
studied in combination treatment with TMZ to develop a novel
chemotherapy strategy to sensitize cancer cells to TMZ (12). However, although these drug
combinations are able to effectively inhibit MGMT expression in
cancer cells, serious drug reactions and systematic toxicity
indicated a disconsolate future (13). There is an urgent requirement to
develop well-tolerated drugs with the capacity to reverse
TMZ-resistance. Therefore, drugs targeting proteins with the
capacity to mediate MGMT expression levels are actively
studied.
A previous study demonstrated that over-activation
of the PI3K/AKT signaling pathway in glioblastoma was associated
with chemotherapy resistance (14).
p-Akt is the primary mediator of PI3K-initiated signaling, and a
number of its downstream substrates contribute to the resistance of
chemotherapy, including Bcl2-associated agonist of cell death, p53,
NF-κB and glycogen synthase kinase-3β (15). NF-κB is a component, which initiates
the inflammatory transcription pathway in various types of solid
cancer. NF-κB p65 is the key subunit in this family of
transcription factors (16).
Typically, NF-κB is bound to IκBα and exists in the cytosol
(17). Due to over-activation of the
PI3K/AKT signaling pathway, NF-κB p65 translocates into the nucleus
and subsequently initiates transcription of numerous genes,
including MGMT (18). Excessive
activation of NF-κB has been previously reported in glioblastoma,
and NF-κB activity has been observed to be higher in tumor tissues
compared with normal brain tissues (19). These observations support the
hypothesis that combination treatment with TMZ and inhibitors of
Akt signaling pathway may achieve promising clinical benefit.
The present study focused on the possible mechanism
of BKM120 (a potent selective PI3K inhibitor) in sensitizing C6
glioma cells to TMZ. The findings revealed that BKM120 induced
significant downregulation of the PI3K/AKT signaling pathway in C6
glioma. However, the BKM120 monotherapy required a high
concentration in order to inhibit viability of C6 cells.
Combination treatment with BKM120 and TMZ resulted in a strong
synergistic cytotoxicity effect on C6 glioma cells. A low
concentration of BKM120 was able to sensitize glioma cells to TMZ.
The IC50 of TMZ significantly decreased from
1,137.67±271.53 to 346.67±134.53 µM, and the CDIs were close to 0.7
in various TMZ concentrations, which indicated that the TMZ and
BKM120 combination treatment had a strong synergistic effect on C6
glioma cells.
Apoptotic defects are present in numerous types of
solid cancer, including glioblastoma (20). A previous study demonstrated that
apoptosis serves a critical role in TMZ-induced cell death
(21). The present study performed
Hoechst 33342 and TUNEL staining to investigate morphological
changes and rates of apoptosis in various treatment groups. There
was a higher level of apoptosis in cells treated with BKM120 and
TMZ compared with cells treated with TMZ alone. In addition,
principal apoptosis proteins, including caspase enzymes and Bax
family, were determined. The present study evaluated the level of
cleaved caspase-3 expression, which is the most frequently
activated protein in the apoptosis process, as well as the
expression of Bax in glioma cells treated with TMZ and/or BKM120.
Results from western blotting revealed that TMZ and/or BKM120
treatment increased the expression of cleaved caspase-3 and Bax,
and this change was more prominent in the combination treatment
group.
The present study investigated MGMT and NF-κB p65
expression in different treatment groups, with the aim of
investigating the association between the PI3K/Akt/NF-κB signaling
pathway and MGMT transcription. Marked increases in MGMT
transcription were identified in the TMZ monotherapy group, which
was in agreement with a previous study (18). Secondly, no significant change was
revealed in the BKM120 monotherapy group compared with the vehicle
control group. Notably, MGMT transcription levels were suppressed
in cells treated with combination treatment compared with cells
treated with TMZ monotherapy. This finding suggested that the
BKM120-induced inhibition of MGMT transcription may only be
stimulated when activation of MGMT transcription is excessive and
aberrant, for example when induced by TMZ. The expression of NF-κB
p65 was increased following treatment with TMZ monotherapy. When
treated with BKM120 alone, there were no significant changes in the
level of NF-κB p65 expression. However, the combination treatment
induced a significant decrease in NF-κB p65 expression compared
with TMZ treatment alone. This finding was similar to the changes
observed in the level of MGMT transcription, which suggests that
the PI3K/AKT/NF-κB/MGMT signaling pathway may be involved in the
synergistic effect of combination treatment.
Due to the limitations of the experimental
conditions, the present study did not use RNA interference to
demonstrate the direct role of NF-κB p65 in the BKM120-induced MGMT
reduction. In addition, whether other cancer cell death mechanisms,
including autophagy, were involved in the progress of combination
treatment requires additional investigation. In the present study,
the synergistic cytotoxicity of TMZ and BKM120 on C6 glioma cells
was confirmed. Compared with TMZ monotherapy, a combination of TMZ
and BKM120 decreased the survival of C6 glioma cells. Additionally,
cleaved caspase-3 and Bax expression was increased in cells treated
with the combination treatment compared with TMZ monotherapy. There
were also significantly higher levels of apoptosis. Furthermore,
the present study suggested that BKM120 may be able to inhibit
TMZ-induced MGMT transcription, which may be associated with
downregulation of NF-κB p65. In conclusion, combination therapy of
BKM120 and TMZ may be a promising strategy for the treatment of
glioblastoma. Furthermore, the present study suggested a mechanism
of action for the BKM120 and TMZ combination treatment, for the
application of PI3K/Akt inhibitors to reverse TMZ-resistance in
clinical treatment.
References
|
1
|
Tate MC and Aghi MK: Biology of
angiogenesis and invasion in glioma. Neurotherapeutics. 6:447–457.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grauer OM, Wesseling P and Adema GJ:
Immunotherapy of diffuse gliomas: Biological background, current
status and future developments. Brain Pathol. 19:674–693. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Erickson LC, Bradley MO, Ducore JM, Ewig
RA and Kohn KW: DNA crosslinking and cytotoxicity in normal and
transformed human cells treated with antitumor nitrosoureas. Proc
Natl Acad Sci USA. 77:pp. 467–471. 1980; View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pegg AE, Dolan ME and Moschel RC:
Structure, function, and inhibition of O6-alkylguanine-DNA
alkyltransferase. Prog Nucleic Acid Res Mol Biol. 51:167–223. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen L, Han L, Shi Z, Zhang K, Liu Y,
Zheng Y, Jiang T, Pu P, Jiang C and Kang C: LY294002 enhances
cytotoxicity of temozolomide in glioma by down-regulation of the
PI3K/Akt pathway. Mol Med Rep. 5:575–579. 2012.PubMed/NCBI
|
|
6
|
Rodon J, Braña I, Siu LL, De Jonge MJ,
Homji N, Mills D, Di Tomaso E, Sarr C, Trandafir L, Massacesi C, et
al: Phase I dose-escalation and -expansion study of buparlisib
(BKM120), an oral pan-Class I PI3K inhibitor, in patients with
advanced solid tumors. Invest New Drugs. 32:670–681. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Butowski NA, Sneed PK and Chang SM:
Diagnosis and treatment of recurrent high-grade astrocytoma. J Clin
Oncol. 24:1273–1280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sarkaria JN, Kitange GJ, James CD, Plummer
R, Calvert H, Weller M and Wick W: Mechanisms of chemoresistance to
alkylating agents in malignant glioma. Clin Cancer Res.
14:2900–2908. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brandes AA, Franceschi E, Tosoni A, Blatt
V, Pession A, Tallini G, Bertorelle R, Bartolini S, Calbucci F,
Andreoli A, et al: MGMT promoter methylation status can predict the
incidence and outcome of pseudoprogression after concomitant
radiochemotherapy in newly diagnosed glioblastoma patients. J Clin
Oncol. 26:2192–2197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Turriziani M, Caporaso P, Bonmassar L,
Buccisano F, Amadori S, Venditti A, Cantonetti M, D'Atri S and
Bonmassar E: O6-(4-bromothenyl) guanine (PaTrin-2), a novel
inhibitor of O6-alkylguanine DNA alkyl-transferase, increases the
inhibitory activity of temozolomide against human acute leukaemia
cells in vitro. Pharmacol Res. 53:317–323. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Quinn JA, Jiang SX, Reardon DA, Desjardins
A, Vredenburgh JJ, Rich JN, Gururangan S, Friedman AH, Bigner DD,
Sampson JH, et al: Phase II trial of temozolomide plus
o6-benzylguanine in adults with recurrent, temozolomide-resistant
malignant glioma. J Clin Oncol. 27:1262–1267. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chakravarti A, Zhai G, Suzuki Y, Sarkesh
S, Black PM, Muzikansky A and Loeffler JS: The prognostic
significance of phosphatidylinositol 3-kinase pathway activation in
human gliomas. J Clin Oncol. 22:1926–1933. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Misra UK, Deedwania R and Pizzo SV:
Activation and cross-talk between Akt, NF-kappaB, and unfolded
protein response signaling in 1-LN prostate cancer cells consequent
to ligation of cell surface-associated GRP78. J Biol Chem.
281:13694–13707. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kumar A, Takada Y, Boriek AM and Aggarwal
BB: Nuclear factor-kappaB: Its role in health and disease. J Mol
Med (Berl). 82:434–448. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Senftleben U, Cao Y, Xiao G, Greten FR,
Krähn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC and Karin M:
Activation by IKKalpha of a second, evolutionary conserved,
NF-kappa B signaling pathway. Science. 293:1495–1499. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lavon I, Fuchs D, Zrihan D, Efroni G,
Zelikovitch B, Fellig Y and Siegal T: Novel mechanism whereby
nuclear factor kappaB mediates DNA damage repair through regulation
of O(6)-methylguanine-DNA-methyltransferase. Cancer Res.
67:8952–8959. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang H, Lin H, Zhang X and Li J:
Resveratrol reverses temozolomide resistance by downregulation of
MGMT in T98G glioblastoma cells by the NF-κB-dependent pathway.
Oncol Rep. 27:2050–2056. 2012.PubMed/NCBI
|
|
20
|
Igney FH and Krammer PH: Death and
anti-death: Tumour resistance to apoptosis. Nat Rev Cancer.
2:277–288. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roos WP, Batista LF, Naumann SC, Wick W,
Weller M, Menck CF and Kaina B: Apoptosis in malignant glioma cells
triggered by the temozolomide-induced DNA lesion O6-methylguanine.
Oncogene. 26:186–197. 2007. View Article : Google Scholar : PubMed/NCBI
|