Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of cancer and the third most common cause of
cancer-associated mortality, worldwide (1,2). The
highest number of patients with HCC was identified in Asian
countries (3). Radical resection
surgery is the most effective means of treatment for HCC (4). However, due to the rapid spread of
cancer or severe liver dysfunction associated with HCC, ~80% of
patients are unable to receive radical resection surgery and the
mortality rate is high (5). Previous
studies revealed that the five-year survival rate for advanced HCC
is only 5% (6–8); therefore, there is an urgent requirement
to research more efficient and safer treatments for HCC.
Tumor-targeting drug delivery systems may selectively transport
cytotoxic agents to the tumor site by exploiting subtle
morphological and physiological differences between healthy and
malignant cells (9). Therefore,
tumor-targeting drug delivery systems have become one of the most
attractive anticancer strategies (10).
Nanopharmaceutics is a branch of nanotechnology that
has developed rapidly, mainly involving nanocarrier drugs and
nanoscale active ingredient research, which may provide targeted
treatment for some diseases (11).
Nanocarrier drugs include nano-liposomes, nano-polymeric micelles
and nanoparticles, which achieve sustained and targeted release of
drugs and effectively improve the water-solubility and safety of
drugs (12).
Chitosan is one of the most plentiful biomaterials
prepared from N-deacetylation of chitin, and has attracted
significant interest in the biotechnology field due to its low
toxicity, high biocompatibility and biodegradability (13–15).
Additionally, chitosan-based nanoparticles have been exploited as
delivery system for liver disease targeting (16,17).
However, chitosan has poor solubility in neutral media, leading to
low loading capacities for nonionic hydrophobic drugs (18). To overcome these disadvantages,
chitosan may be amphiphilically modified by the attachment of both
hydrophobic and hydrophilic segments to the backbone (19). Previous studies demonstrated that
O-carboxymethyl chitosan (CC) is a water-soluble chitosan
derivative with favorable biocompatibility, similar to chitosan
(20). Lactose has been demonstrated
to have a ligand targeting function, including liver disease
targeting (21). Alginic acid, a high
molecular weight polysaccharide obtained from seaweed, is composed
of mannuronic acid and guluronic acid (22); previous studies have demonstrated the
presence of mannose receptors in the cell membranes of liver
non-parenchymal cells (23). Due to
the mannose residues contained in alginic acid and the similarity
of its structure to the glycosaminoglycan in the extracellular
matrix, low molecular weight algal polysaccharides may have liver
targeting properties (24).
In the present study, amphiphilically modified
chitosan derivatives, lactose myristoyl carboxymethyl chitosan
(LMCC) and algal polysaccharide myristoyl carboxymethyl chitosan
(AMCC), were designed and the potential of polymeric micelles
composed of LMCC or AMCC as liver-targeting carriers of Adriamycin
(ADM) was evaluated. The drug-loading capacity, stability, in
vitro drug release and liver targeting were studied in detail.
In addition, the safety and cytotoxicity of blank nanoparticles and
the ADM-loaded nanoparticles was determined.
Materials and methods
Materials
Adriamycin hydrochloride (ADM·HCl) was purchased
from Zhejiang Hisun Pharmaceutical Co., Ltd. (Taizhou, China). The
HU7 human hepatocarcinoma cell line, H22 murine hepatocarcinoma
cell line and the HT22 murine hippocampal neuron cell lines were
obtained from the Shanghai Cell Bank of the Chinese Academy of
Sciences (Shanghai, China). Dulbecco's modified Eagle's medium
(DMEM), fetal bovine serum (FBS), 0.25% trypsin solution and PBS
were provided by Gibco (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). Radioimmunoprecipitation lysis assay (RIPA) lysate, BCA
protein assay kit, and hematoxylin and eosin (H&E) staining kit
were provided by Beyotime Institute of Biotechnology Co., Ltd.
(Hangzhou, China). Rhodamine B isothiocyanate (RBITC) was purchased
from Aladdin Reagent Co., Ltd. (Shanghai, China). All other
reagents were analytical grade and were used without further
purification. Double distilled water was used in the current
study.
Kunming (KM) mice (n=90, 18–22 g, 4 weeks) were
supplied by the Laboratory Animal Centre of Xuzhou Medical
University (Xuzhou, China), and raised under specific pathogen-free
conditions with a 12 h light/dark cycle, constant temperature
(25°C) and humidity (60%), and free access to standard food and
water. New Zealand rabbits (n=3, 2.5 kg, 12 weeks, female) were
supplied by the Laboratory Animal Centre of Xuzhou Medical
University (Xuzhou, China), and raised under conventional
conditions with a 12 h light/dark cycle, constant temperature
(25°C) and humidity (60%), and free access to standard food and
water. All animal experiments were performed according to the
Guiding Principles for the Care and Use of Experiment Animals in
Xuzhou Medical University.
Preparation and characterization of
ADM-loaded nanoparticles
Preparation of ADM
ADM was prepared as previously described (25). Briefly, 10 mg ADM·HCl was dissolved in
5 ml dimethylformamide. Appropriate triethylamine (TEA, 5 µl) was
added into the solution and the mixture was stirred by a magnetic
stirrer overnight in the dark at room temperature. The reaction
solution was loaded into a dialysis bag with a molecular weight
cut-off (MWCO) of 3,500 Da (Spectrum Laboratories, Inc., Rancho
Dominguez, CA, USA) and dialyzed against water (3 times, 2 L per
time) for 24 h in the dark. The suspension in the dialysis bag was
filtrated by filter paper and the filter cake was washed three
times with water. Subsequent to drying under vacuum at 40°C for 24
h, ADM was obtained as a deep red powder.
Preparation of ADM-loaded LMCC and AMCC
nanoparticles
LMCC and AMCC were prepared by the Jiangsu Key
Laboratory of New Drug Research and Clinical Pharmacy (data not
presented). A total of 10 mg ADM and 200 mg LMCC or AMCC were
dissolved in dimethyl sulfoxide. The solution was dialyzed (MWCO:
3500 Da) against water (3 times, 2 l per time) for 24 h in the
dark. Following this, the suspension was filtered through a 0.45 µm
Millipore filter to remove insoluble ADM and ADM-loaded LMCC or
ADM-loaded AMCC nanoparticles solution was obtained.
Characterization of ADM-loaded LMCC or AMCC
nanoparticles
The morphology of LMCC or AMCC nanoparticles was
observed by transmission electron microscopy (TEM) via an H-600A
transmission electron microscope (Hitachi Ltd., Tokyo, Japan). The
nanoparticle size and zeta potential were further determined using
a Nicomp™380 ZLS (Particle Sizing Systems, Inc., Port
Richey, FL, USA), by measuring the dynamic light scattering in a 1
mg/ml suspension of ADM-LMCC or ADM-AMCC.
Determination of drug loading and entrapment
efficiency
The drug loading capacity (LC) and entrapment
efficiency (EE) were determined by quantifying the amount of ADM
encapsulated into the LMCC or AMCC nanoparticles using fluorescence
analysis. All fluorescence quantitative analyses in this article
were performed using a fluorescence spectrophotometer (F4600;
Hitachi, Ltd., Tokyo, Japan) at excitation and emission bandwidths
of 5 nm. Briefly, 10 mg freeze-dried ADM-LMCC or ADM-AMCC
nanoparticles were sonicated in 10 ml 6% hydrochloric acid alcohol
solution [a mixture of 36% hydrochloric acid and ethanol (200
proof, anhydrous, ≥99.5%; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) in a volume ratio of 6:94] for 5 min, and centrifuged at
13,400 × g at room temperature for 10 min. The fluorescence of ADM
in the supernatant was determined by the fluorescence
spectrophotometer (F4600; Hitachi, Ltd.) at excitation and emission
wavelengths of 501 and 590 nm, respectively. The LC and EE were
calculated according to the standard curve of ADM fluorescent
intensity to ADM concentration.
ADM release from nanoparticles in vitro
ADM release from LMCC or AMCC nanoparticles in
vitro was detected according to a previously described dialysis
method (26). PBS (pH 5.5 and 7.4,
0.15 M) containing 0.1% (w/v) SDS was used as a release medium, and
the release profiles of the nanoparticles were investigated.
Briefly, 1 ml of ADM-loaded LMCC or ADM-loaded AMCC nanoparticles
were added into a dialysis bag (MWCO, 3,500 Da). Subsequently, the
end-sealed dialysis bag containing the nanoparticles was placed in
50 ml fresh medium (PBS buffer with 0.1% SDS) at 37°C and 100 r/min
in a shaker (THZ-100; Yiheng Technical Co., Ltd., Shanghai, China).
At predetermined intervals, including 0.5, 1, 2, 3, 4, 6, 8, 12 and
24 h, 2 ml external release medium was extracted and an equal
volume of fresh release medium was added. The amount of released
ADM was determined by fluorescence measurement (λex=501
nm, λem=590 nm). The release experiments were conducted
in triplicate.
Cell uptake of ADM-LMCC
nanoparticles
Preparation of RBIT-labeled LMCC
A total of 0.5 g LMCC and 0.5 g RBITC
(Sigma-Aldrich; Merck KGaA) were dissolved in 10 ml PBS (pH 7.4).
Following stirring for 24 h in the dark, the reaction solution was
transferred into the dialysis bag (MWCO 3500) and dialyzed against
water (6 times, 2 l per time) for 48 h in the dark. The solution in
the dialysis bag was freeze dried by a lyophilizer (FD-1-50;
Biocool, Ltd., Beijing, China) for 24 h and RBITC-labeled LMCC
(RBITC-LMCC) was obtained as a red powder (27).
Cellular uptake
HU7 cells or HT22 cells were seeded in 24-well
plates at a density of 2.0×105 cells/well in 1 ml
complete DMEM and cultured at 37°C with 5% CO2 for 24 h.
To study the effect of nanoparticles' concentration on uptake, the
cells were treated with various concentrations (150, 300 and 600
µg/ml) of RBITC-LMCC nanoparticles at 37°C and 5% CO2.
At 1, 2 and 4 h, the cells were washed three times with PBS.
Fluorescent microimages of the cells were obtained with a
fluorescent microscope (Leica DMI4000B; Leica Microsytems GmbH,
Wetzlar, Germany). Following observation, the cells were lysed with
RIPA lysate following the manufacturer's protocol and centrifuged
at 12,000 × g at 4°C for 10 min. The fluorescence intensities of
RBITC (λex=547 nm, λem=582 nm) in
supernatants were determined by the fluorescence spectrophotometer
(F4600; Hitachi, Ltd.). Fluorescence intensity was normalized with
respect to total protein content. The protein content of the
supernatant was determined using BCA protein assay kit according to
the method specified by the manufacturer.
Antitumor efficacy in vivo
The in vivo antitumor efficacy of ADM-LMCC
and ADM-AMCC was evaluated with a subcutaneous H22 xenograft tumor
model in mouse as previously described (28). KM mice (n=20, 18–22 g, female) were
injected subcutaneously in the axilla of right anterior limb with
0.2 ml cell suspension, containing 1×106 H22 cells in
PBS. After 3 days (day 0), the mice were weighed and randomly
divided into four groups (n=5): (1)
negative control group (saline group); (2) positive control group (ADM, 5 mg/kg);
(3) ADM-LMCC (ADM, 5 mg/kg);
(4) ADM-AMCC (ADM, 5 mg/kg). Drug
administrations were performed three times via tail vein injection
every other day (day 0, 2 and 4). The tumor sizes were measured in
all mice at day 0, 2, 4 and 6. The tumor volume was calculated by
the following formula: (S2 × L)/2, where S was the short
diameter and L was the long diameter. At day 8, all the mice were
anesthetized with 5% chloral hydrate solution (380 mg/kg body
weight) by intraperitoneal injection and sacrificed by cervical
vertebra dislocation followed by separation and measurement of the
tumor block. The antitumor efficacies of each formulation were
evaluated by the tumor inhibition rate, which was calculated by the
following formula: (1 - tumor weight of the treatment group/tumor
weight of the negative control group) × 100%. In addition, the
separated tumor block was fixed in 4% (w/v) paraformaldehyde at 4°C
for 4 h, washed with PBS and embedded in paraffin. Tissue samples
were then cut into 8 µm thick sections, which were placed onto
gelatin-coated slides and stained with H&E staining kits
following the manufacturer's protocol for histological
examination.
Biodistribution
To assess the tissue distribution of ADM-loaded
nanoparticles, female KM mice bearing H22 hepatoma (~200
mm3) were weighed and randomly divided into three groups
(n=12): i) ADM·HCl group (ADM, 10 mg/kg); ii) ADM-LMCC group (ADM,
10 mg/kg); iii) ADM-AMCC group (ADM, 10 mg/kg). The ADM·HCl
solution and ADM-loaded nanoparticles were intravenously
administrated via tail vein at doses of 10 mg/kg. At 0.5, 1, 4 and
8 h post-injection (n=3 at each time point), the mice were
anaesthetized and blood was collected by retro-orbital bleeding.
Then, the mice were sacrificed and the tumors and major organs
(heart, liver, spleen, lungs, and kidneys) were dissected from the
mice. Tissue samples were blotted with paper towel, rinsed in
saline, blotted to remove excess fluid, weighed and stored at
−20°C.
ADM plasma concentration was determined by measuring
ADM fluorescence. Briefly, 0.2 ml plasma and 0.8 ml of 6%
hydrochloric acid alcohol were mixed by vortexing for 5 min,
following which they were separated by centrifugation at 13,400 × g
at room temperature for 10 min. Subsequently, 100 µl of the
supernatant was examined to determine the ADM content using the
F4600 fluorescence spectrophotometer at excitation and emission
wavelengths of 501 and 590 nm, respectively.
ADM tissue concentration was also determined by the
fluorescence method described above. A tissue sample (0.1 g) was
homogenized with 1 ml of 6% hydrochloric acid alcohol. The
homogenate was mixed by vortexing for 5 min. Following
centrifugation at 13,400 × g for 10 min at room temperature, the
fluorescent intensity of ADM in the supernatant was determined by
the fluorescence spectrophotometer (λex=501 nm,
λem=590 nm). The concentration of ADM in samples were
calculated according to the standard curve of ADM fluorescent
intensity to ADM concentration (29).
Safety of nanoparticles and ADM-loaded
nanoparticles
Hemolysis test
Rabbit blood was used to test the hemolysis effect
of nanoparticles. Blood (10 ml) was obtained via the ear vein from
the arteria cruralis of healthy New Zealand rabbits (2.5 kg, 12
weeks, female). The red blood cells (RBC) were collected by
centrifugation at 402 × g at room temperature for 10 min. The RBC
pellets at the bottom of the centrifugation tube were washed 3
times with 0.15 M PBS (pH 7.4). Following repeated washing and
centrifugation, an adequate quantity of 0.15 M PBS (pH 7.4) was
added into the RBC pellets to give a 2.5% RBC suspension, this was
then stored at 4°C. Subsequently, 2.5% RBC suspensions were
incubated with the equal volume of nanoparticles solutions with
different concentrations (2, 4, 6, 8 and 10 mg/ml) for 1 h in a
37°C water bath. The suspension was then centrifuged at 1,610 × g
at room temperature for 10 min to remove intact RBC. The
supernatant was collected and analyzed for the presence of released
hemoglobin using a spectrophotometer (UV-2450; Shimadzu, Kyoto,
Japan) at 541 nm (n=3). To obtain 0 and 100% hemolysis, saline and
distilled water were added in equal volumes to the RBC suspension,
respectively. The degree of hemolysis was calculated with the
following equation: Hemolysis ration (HR%) = [optical density
(OD)sample - ODnegative control)]
× 100%/(ODpositive control - ODnegative
control). The turbidity of the samples was compensated for by
using a sample solution without RBC as a blank control.
Acute toxicity
To determine the acute toxicity of the
nanoparticles, the toxic effects of ADM-LMCC and ADM-AMCC on major
organs was examined. The nanoparticle suspension (100 mg/ml) was
obtained by re-dissolving the freeze-dried powder of ADM-LMCC and
ADM-AMCC in saline. KM mice (male, n=10; female, n=10; 18–22 g)
were housed under normal conditions with free access to food and
water. Mice were randomly divided into two groups (n=10), and 0.5
ml ADM-LMCC or ADM-AMCC nanoparticles suspension were intravenously
administrated via the tail vein. Mice were observed for two weeks
in all groups, and the number of mice that survived was recorded.
At day 14, all the mice were sacrificed by cervical vertebra
dislocation followed by separation and observation of major organs
such as the heart, liver, spleen, lung, kidney and brain.
Statistical analysis
Data are expressed as the mean ± standard deviation.
A one-way analysis of variance followed by Dunnett's post-test was
used for statistical analysis. Data analysis was performed using a
SPSS 19.0 software package (IBM SPSS, Armonk, NY, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
Synthesis and characterization of
ADM-loaded LMCC and AMCC
In the present study, ADM was obtained from ADM·HCl.
ADM was incorporated into LMCC and AMCC using the dialysis method.
Fig. 1 presents the morphology of the
prepared nanoparticles as determined by TEM. The results revealed
that the morphology of the prepared nanoparticles was spherical,
with a lightly rough surface. The mean size of the LMCC particles
was ~20 nm, which was slightly smaller than that of AMCC at ~30
nm.
The size distribution and zeta potential of the
nanoparticles with or without ADM were investigated by dynamic
light scattering. As presented in Table
I, the LMCC and AMCC nanoparticles had a mean particle size of
~67.5 and ~80.4 nm respectively, which were generally larger than
that obtained by TEM. Following drug loading, the mean particle
size of ADM-loaded LMCC and AMCC nanoparticles recorded increased
to ~86.8 and ~102.5 nm, respectively. It was revealed that the LMCC
and AMCC nanoparticles have a negative surface charge, with a zeta
potential of ~-17.5 and ~-18.7 mV (Table
I) due to the carboxyl (-COOH) dissociation on the CC.
Following drug loading, the zeta potential of the LMCC and AMCC
nanoparticles exhibited no significant changes (P=0.570 and 0.679,
respectively).
 | Table I.Size distribution and zeta potential
of the nanoparticles (n=3). |
Table I.
Size distribution and zeta potential
of the nanoparticles (n=3).
| Nanoparticles | Mean diameter
(nm) | Zeta potential
(mV) |
|---|
| LMCC | 67.5±11.1 | −17.5±3.7 |
| AMCC | 80.4±13.1 | −18.7±5.4 |
| ADM loaded
LMCC | 86.8±10.3 | −15.8±2.6 |
| ADM loaded
AMCC | 102.5±18.9 | −17.2±3.8 |
Drug-loading and entrapment efficiency
of prepared nanoparticles
The drug-loading capacities of specific
nanoparticles are listed in Table
II. The LC of LMCC and AMCC was ~3.8% for each. In addition,
the EE of ADM for LMCC and AMCC was ~80 and ~70%, respectively. The
results suggest that ADM may be effectively loaded into LMCC or
AMCC nanoparticles using the dialysis method.
| Table II.Drug-loading and entrapment
efficiency of LMCC and AMCC (n=3). |
Table II.
Drug-loading and entrapment
efficiency of LMCC and AMCC (n=3).
| Sample | LC (wt %) | EE (wt %) |
|---|
| LMCC | 3.8±0.3 | 81.6±1.2 |
| AMCC | 3.4±0.2 | 70.3±0.6 |
Results of ADM release from
nanoparticles in vitro
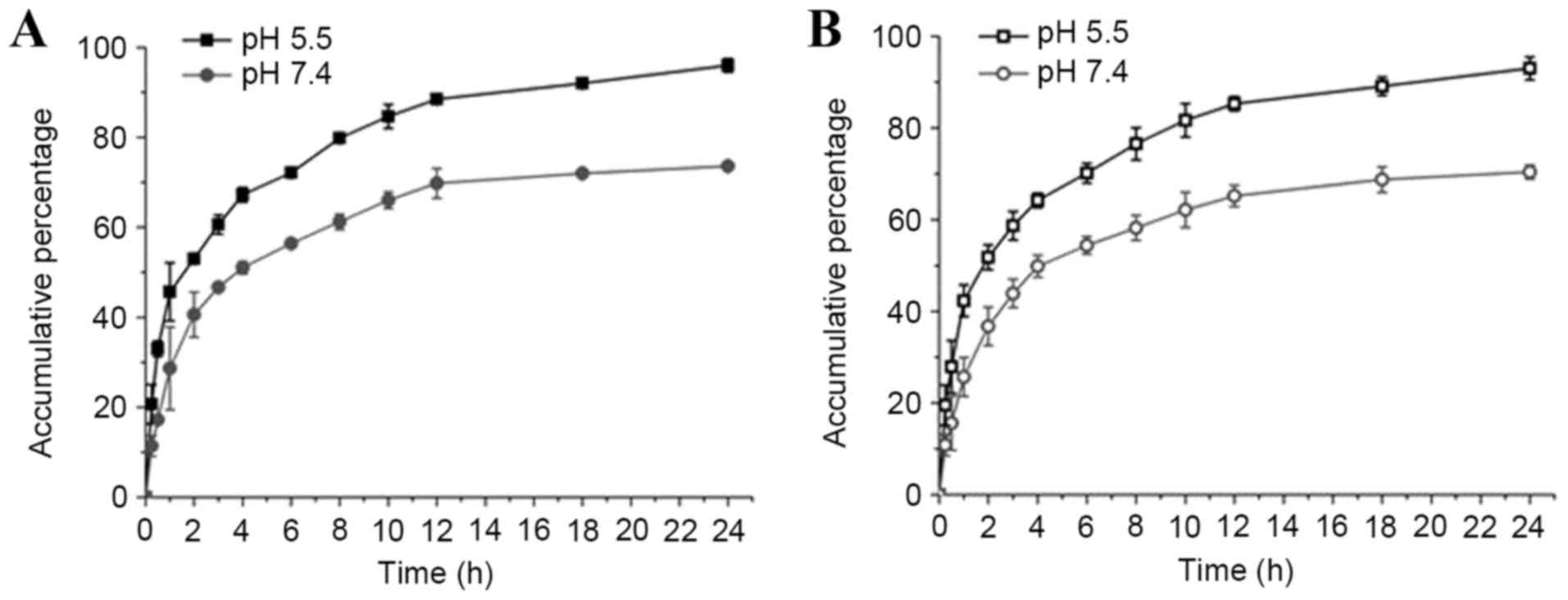
The drug release behaviors of nanomedicines from
LMCC or AMCC nanoparticles were investigated in PBS at pH 5.5 and
7.4, mimicking the physiological pH levels present in the tumor
extracellular microenvironment and normal tissues, respectively.
The ADM release rate increased as the pH decreased from pH 7.4–5.5
in LMCC and AMCC nanoparticles (Fig.
2). Additionally, ADM-loaded AMCC nanoparticles had lower drug
release rates compared with ADM-loaded LMCC nanoparticles, which
may be associated with the particle size.
Cell uptake of ADM-LMCC
nanoparticles
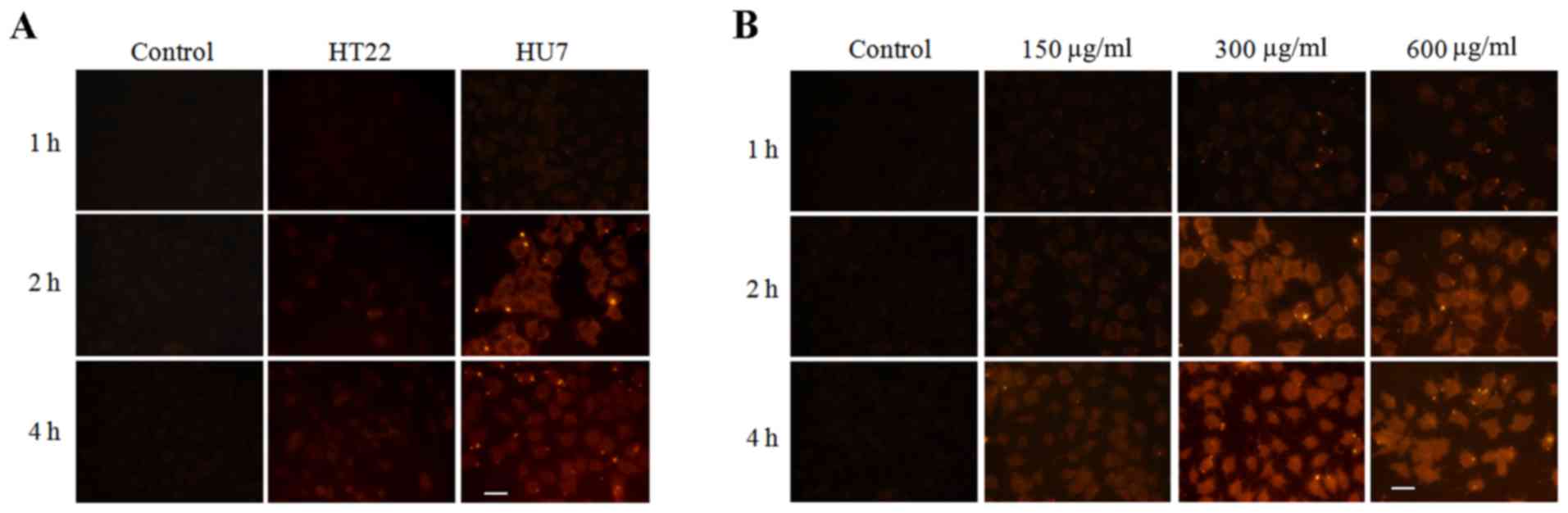
To evaluate the liver targeting of the prepared
nanoparticles, the cellular uptake of LMCC in HU7 and HT22 cells
was determined by fluorescent microscopy. LMCC was marked with red
fluorescence by RBITC. Fig. 3A
demonstrated the fluorescent micro-images of HU7 and HT22 cells
co-incubated with RBITC-LMCC nanoparticles or free RBITC solution
for 1, 2 and 4 h. As predicted, the fluorescence intensity
determined in HU7 cells co-incubated with RBITC-LMCC nanoparticles
was significantly higher, compared with the fluorescence in HT22
cells at 2 and 4 h (P<0.01), which indicated that the prepared
nanoparticles had a specific interaction with HU7 cells. Fig. 3B indicated that enhancing the
concentration of nanoparticles resulted in significantly increased
uptake of RBITC-LMCC in HU7 cells from 150 to 300 µg/ml
(P<0.01). However, the fluorescence intensity did not exhibit a
marked improvement when the concentration of nanoparticles was
additionally increased to 600 µg/ml (P>0.50), which resulted in
the uptake of RBITC-LMCC by HU7 cells being saturated >300
µg/ml. In addition, the highest intensity of the fluorescence in
HU7 cells occurring at 4 h, indicating that the uptake of LMCC
nanoparticles was time-dependent.
Antitumor efficacy in vivo
The evaluations of the hepatoma-targeting ability
and antitumor efficacy of ADM-loaded nanoparticles in vivo
were performed using an H22 xenograft mouse model. During the
complete experimental period (6 days), the weight of the mice body
was determined and no significant body weight loss was observed
following the administration of ADM or ADM-loaded nanoparticles
compared with the initial body weights of the tumor-bearing mice,
indicating that the nanoparticles were well tolerated at the dosage
level used.
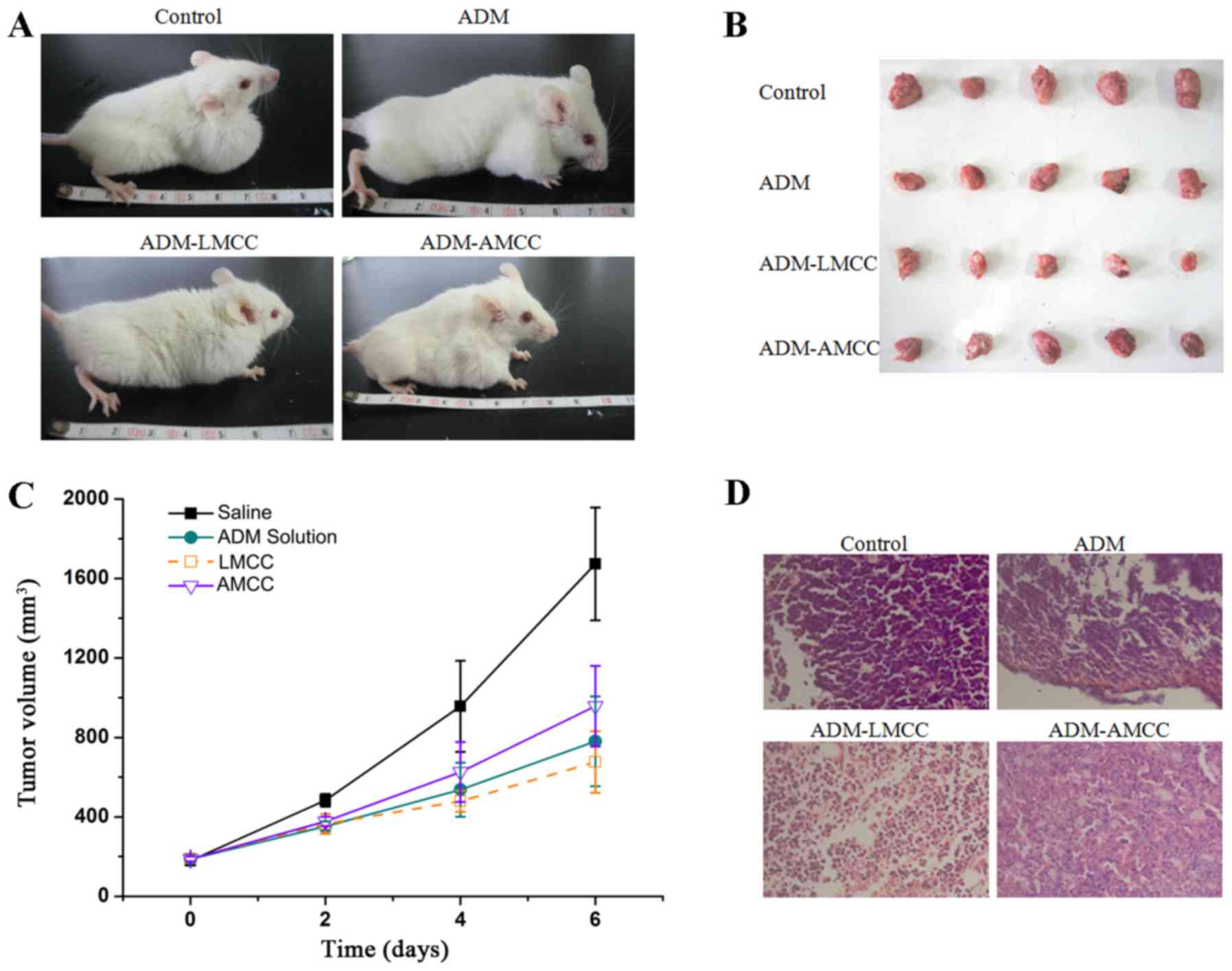
The tumor volume was monitored over a treatment
period of 6 days and the weight of tumors was determined at day 6.
As presented in Fig. 4, the ADM
(P<0.001), ADM-LMCC nanoparticles (P<0.001), and ADM-AMCC
(P<0.001) significantly reduced the tumor volume compared with
the saline group, in particular at day 6. Notably, the tumor volume
of the ADM-LMCC group was the smallest (679.1±128.2
mm3). The histopathological changes of the tumor tissues
are presented in Fig. 4. ADM-LMCC
nanoparticles greatly improved the damage to tumor tissues in H22
xenograft mice; as compared with mice treated with ADM alone.
Additionally, the antitumor efficacy of ADM-loaded
nanoparticles was evaluated by calculating the tumor inhibition
rate of the mice following tumor implantation, compared with the
saline group or ADM group. As presented in Table III, the ADM-LMCC group had the
highest tumor inhibition rate at 62.7%, followed by the ADM and
ADM-AMCC groups at 51.2 and 42.5%, respectively.
| Table III.Antitumor efficacy of ADM or
ADM-loaded nanoparticle in H22 xenograft mice. |
Table III.
Antitumor efficacy of ADM or
ADM-loaded nanoparticle in H22 xenograft mice.
|
|
| Body weight
(g) |
|
|
|---|
|
|
|
|
|
|
|---|
| Drug | Dose (mg/kg) | Prior to
administration | Following
administration | Tumor weight
(g) | Inhibition (%) |
|---|
| Saline | – | 24.3±1.9 | 29.1±2.3 | 1.29±0.34 | – |
| ADM.HCl | 5 | 24.0±0.9 | 27.4±1.7 |
0.63±0.10a | 51.2±7.7 |
| LMCC | 5 | 23.9±1.3 | 26.1±2.1 |
0.48±0.07b | 62.7±5.4 |
| AMCC | 5 | 23.3±1.2 | 26.4±2.2 |
0.74±0.19b | 42.5±14.7 |
Biodistribution
The tissue distribution profiles of ADM-loaded
nanoparticles and ADM following intravenous administration were
compared in the mice. The concentration of ADM in each tissue was
determined using the fluorescence method. As presented in Fig. 5, ADM was widely and rapidly
distributed into the majority of tissues following intravenous
administration of the nanoparticles, and the highest concentration
of ADM was identified in liver tissue, followed by lung, kidney,
spleen, heart, brain and plasma tissues in decreasing order at 1 h
subsequent to administration. Following 8 h, the drug concentration
was decreased in the following order: Liver > lung > kidney
> spleen > brain > plasma > heart; ADM concentrations
in the heart, brain and plasma tissues were low. However, following
administration of ADM hydrochloride at 1 h, the maximum
concentration of the drug was observed in the liver, followed by
the kidney and lung. The exposure of the organs to ADM at 8 h
decreased in the following order: Kidney > liver > spleen
> lung > heart > plasma > brain.
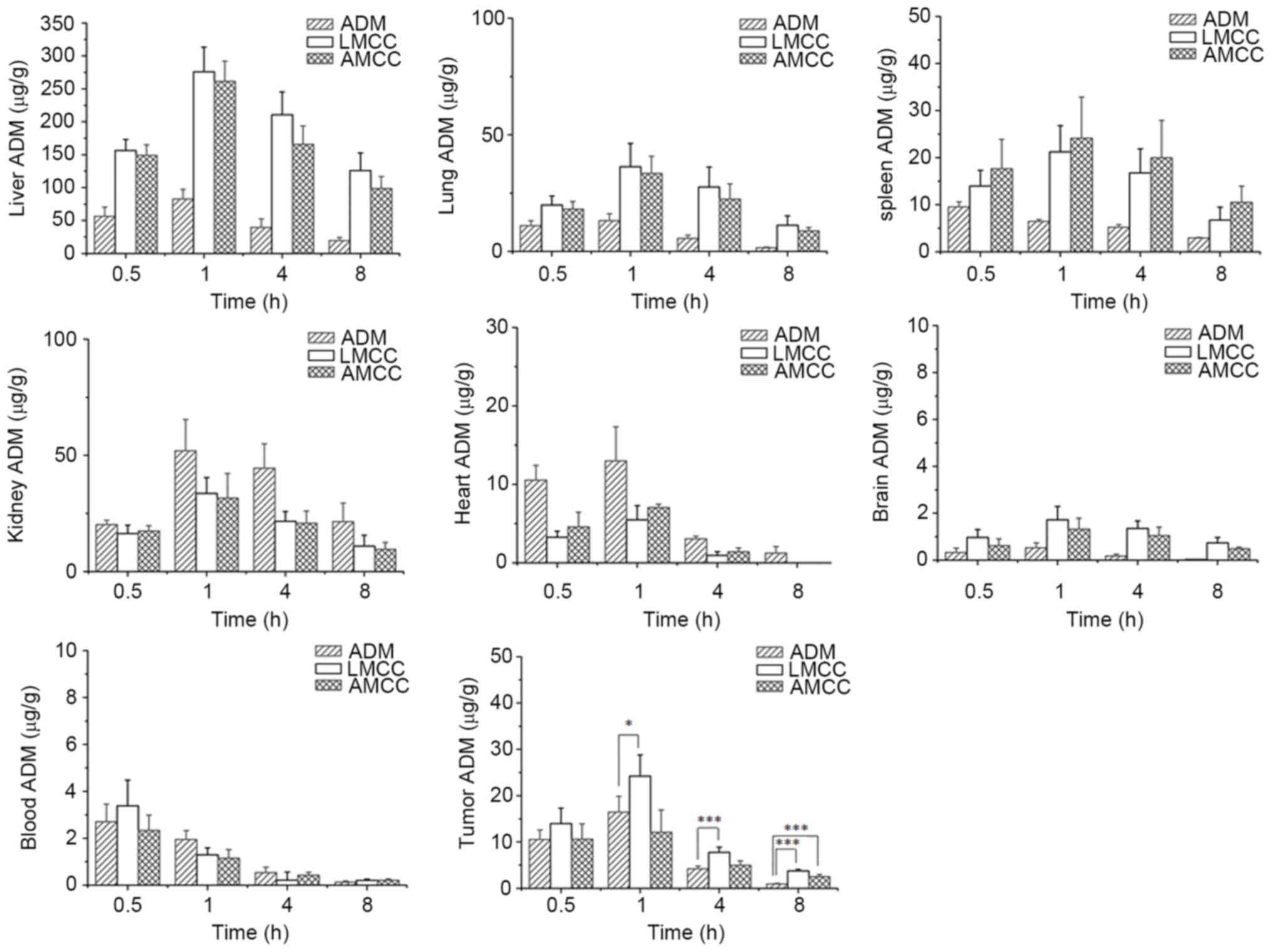 | Figure 5.Tissue distribution and plasma
concentration of ADM among various organs (liver, lung, spleen,
kidney, heart, brain, tumor and plasma) at various sampling times
following intravenous administration at a dose of 10 mg/kg in mice.
Each column represents the mean ± standard deviation (n=12).
*P<0.05, **P<0.01, ***P<0.001. ADM, Adriamycin; LMCC,
lactose myristoyl carboxymethyl chitosan; AMCC, algal
polysaccharide myristoyl carboxymethyl chitosan. |
Safety of nanoparticles and ADM-loaded
nanoparticles
Hemolysis test
The amphiphilic compounds may solubilize lipids or
be inserted into phospholipid membranes to destabilize them, which
may lead to hemolysis of RBC. Thus, the hemolysis test is required
for amphiphilic LMCC and AMCC nanoparticles (30). The HR of LMCC and AMCC was revealed to
be <5%, including at concentrations ≤10 mg/ml, which indicates
that the hemolysis of LMCC and AMCC was negligible (Table IV).
| Table IV.Hemolysis rate of LMCC and AMCC. |
Table IV.
Hemolysis rate of LMCC and AMCC.
|
| Hemolysis rate
(%) |
|---|
|
|
|
|---|
| Concentration
(mg/ml) | LMCC | AMCC |
|---|
| 2 | 0.52 | 0.74 |
| 4 | 1.37 | 1.63 |
| 6 | 1.79 | 2.03 |
| 8 | 2.95 | 2.13 |
| 10 | 4.51 | 3.97 |
Acute toxicity
The acute toxicity evaluation in vivo was
performed on the mice following the administration of
nanoparticles. Following an injection of 2,000 mg/kg nanoparticles,
no mortalities occurred in the treatment group; however, the
decrease in general distress in test mice was observed, such as
inactivity, shortness of breath, and non-eating, which returned to
normal 2 h subsequent to administration. Mortality was not observed
until the end of the experiment. The autopsy of the treated mice
did not reveal any macroscopic changes in the major organs,
including the heart, liver, spleen, lung and kidney.
Discussion
The present study represents the first report on the
LMCC or AMCC nanoparticles as a targeted delivery system for ADM to
treat HCC. The drug-loading capacity of LMCC nanoparticles was
slightly higher compared with that of AMCC. This may be associated
with the alternate morphologies of LMCC and AMCC. The TEM images
indicated that the surface of LMCC was rough, whereas that of AMCC
was uniform. The rough surface allowed for easier drug loading, due
to the presence of more binding sites.
The ADM release rate from LMCC and AMCC
nanoparticles was higher at pH 5.5 compared with pH 7.4, which
indicated that the drug release behaviors of the nanoparticles were
influenced by pH. This was consistent with the ADM-loaded poly
(OEGylated L-glutamate)-block-poly (L-glutamic acid) and (poly
(galactosylated L-glutamate)-block-poly (L-glutamic acid) micelles
with pH-dependent release behavior (31). The tumor extracellular
microenvironment is often acidic (32), thus, the pH-dependent release behavior
may contribute to tumor targeting. In addition, the TEM images
indicated that the mean particle size of LMCC and AMCC were 30–50
nm. The zeta potential of these two nanoparticles was approximately
−17.5 and −18.7 mV, respectively. The particle size, negative
surface charge and pH-dependent release behavior contributed to the
liver targeting of ADM.
The cellular uptake of ADM-loaded LMCC nanoparticles
in HU7 cells was higher compared with HT22 cells in vitro,
which indicated that ADM-loaded nanoparticles may effectively
target HCC cells. In vivo, ADM-loaded LMCC was the most
effective combination at suppressing tumor growth, compared with
free ADM and ADM-AMCC nanomedicine in the H22 xenograft mouse
model. These results suggest that the lactose modified LMCC
nanoparticles may significantly improve the antitumor effect and
liver targeting of ADM. Although ADM was widely distributed into
the majority of tissues following intravenous administration of the
nanoparticles, the highest ADM levels were revealed to be in the
liver, lung, kidney and spleen. The localization of ADM to the
liver was consistent with uptake by the HU7 cells, which further
demonstrated that LMCC nanoparticles facilitated the effective
liver targeting of ADM.
With respect to safety evaluation, nanoparticle
hemolysis was negligible due to the hemolysis rate, which was
<5% at 10 mg/ml. Additionally, there was no significant body
weight loss, mouse mortality or organ damage following the
administration of nanoparticles, compared with the initial body
weights of the tumor-bearing mice during the entire experimental
period. The safety evaluation results indicated that nanoparticles
were well tolerated at the dosage level investigated.
In conclusion, amphiphilic LMCC and AMCC
nanoparticles were developed and characterized as a liver targeting
delivery system for ADM. In vitro and in vivo studies
demonstrated that LMCC nanoparticles have superior antitumor
capability, and higher affinity for HU7 cells and liver tissues.
Furthermore, no acute toxicity or hemolysis was revealed. The
results indicate that LMCC nanoparticles may be useful in the
development of a liver targeting drug delivery system.
Acknowledgements
The present study was supported by the Science and
Technology Project of Xuzhou City (grant no. XZZD1351), National
Natural Science Foundation of China (grant no. 81102381).
References
|
1
|
Kimhofer T, Fye H, Taylor-Robinson S,
Thursz M and Holmes E: Proteomic and metabonomic biomarkers for
hepatocellular carcinoma: A comprehensive review. Br J Cancer.
112:1141–1156. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Clark T, Maximin S, Meier J, Pokharel S
and Bhargava P: Hepatocellular Carcinoma: Review of epidemiology,
screening, imaging diagnosis, response assessment, and treatment.
Curr Probl Diagn Radiol. 44:479–486. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goh GB, Chang PE and Tan CK: Changing
epidemiology of hepatocellular carcinoma in Asia. Best Pract Res
Clin Gastroenterol. 29:919–928. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ikeda K: Current therapy for
hepatocellular carcinoma. Nihon Rinsho. 68:1129–1136. 2010.(In
Japanese). PubMed/NCBI
|
|
5
|
Chu KK and Cheung TT: Update in management
of hepatocellular carcinoma in Eastern population. World J Hepatol.
7:1562–1571. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang Y, Wang C, Lu Y, Bai W, An L, Qu J,
Gao X, Chen Y, Zhou L, Wu Y, et al: Outcomes of ultrasound-guided
percutaneous argon-helium cryoablation of hepatocellular carcinoma.
J Hepatobiliary Pancreat Sci. 19:674–684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Furuta M, Kozaki K, Tanimoto K, Tanaka S,
Arii S, Shimamura T, Niida A, Miyano S and Inazawa J: The
tumor-suppressive miR-497-195 cluster targets multiple cell-cycle
regulators in hepatocellular carcinoma. PLoS One. 8:e601552013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mizuguchi Y, Takizawa T, Yoshida H and
Uchida E: Dysregulated miRNA in progression of hepatocellular
carcinoma: A systematic review. Hepatol Res. 46:391–406. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guaragna A, Chiaviello A, Paolella C,
D'Alonzo D and Palumbo G and Palumbo G: Synthesis and evaluation of
folate-based chlorambucil delivery systems for tumor-targeted
chemotherapy. Bioconjug Chem. 23:84–96. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vinh NQ, Naka S, Cabral H, Murayama H,
Kaida S, Kataoka K, Morikawa S and Tani T: MRI-detectable polymeric
micelles incorporating platinum anticancer drugs enhance survival
in an advanced hepatocellular carcinoma model. Int J Nanomedicine.
10:4137–4147. 2015.PubMed/NCBI
|
|
11
|
Pérez-Herrero E and Fernández-Medarde A:
Advanced targeted therapies in cancer: Drug nanocarriers, the
future of chemotherapy. Eur J Pharm Biopharm. 93:52–79. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Puntawee S, Theerasilp M, Reabroi S,
Saeeng R, Piyachaturawat P, Chairoungdua A and Nasongkla N:
Solubility enhancement and in vitro evaluation of PEG-b-PLA
micelles as nanocarrier of semi-synthetic andrographolide analogue
for cholangiocarcinoma chemotherapy. Pharm Dev Technol. 21:437–444.
2016.PubMed/NCBI
|
|
13
|
Prabaharan M: Chitosan-based nanoparticles
for tumor-targeted drug delivery. Int J Biol Macromol.
72:1313–1322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang JJ, Zeng ZW, Xiao RZ, Xie T, Zhou GL,
Zhan XR and Wang SL: Recent advances of chitosan nanoparticles as
drug carriers. Int J Nanomedicine. 6:765–774. 2011.PubMed/NCBI
|
|
15
|
Tiyaboonchai W: Chitosan nanoparticles: A
promising system for drug delivery. Naresuan Uni J: Sci Technol.
11:51–66. 2003.
|
|
16
|
Tian Q, Wang XH, Wang W, Zhang CN, Wang P
and Yuan Z: Self-assembly and liver targeting of sulfated chitosan
nanoparticles functionalized with glycyrrhetinic acid.
Nanomedicine. 8:870–879. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guan M, Zhou Y, Zhu QL, Liu Y, Bei YY,
Zhang XN and Zhang Q: N-trimethyl chitosan
nanoparticle-encapsulated lactosyl-norcantharidin for liver cancer
therapy with high targeting efficacy. Nanomedicine. 8:1172–1181.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Richardson KE, Xue Z, Huang Y, Seo Y and
Lapitsky Y: Physicochemical and antibacterial properties of
surfactant mixtures with quaternized chitosan microgels. Carbohydr
Polym. 93:709–717. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huo M, Zhang Y, Zhou J, Zou A, Yu D, Wu Y,
Li J and Li H: Synthesis and characterization of low-toxic
amphiphilic chitosan derivatives and their application as micelle
carrier for antitumor drug. Int J Pharm. 394:162–173. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen SC, Wu YC, Mi FL, Lin YH, Yu LC and
Sung HW: A novel pH-sensitive hydrogel composed of
N,O-carboxymethyl chitosan and alginate cross-linked by genipin for
protein drug delivery. J Control Release. 96:285–300. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma P, Liu S, Huang Y, Chen X, Zhang L and
Jing X: Lactose mediated liver-targeting effect observed by ex vivo
imaging technology. Biomaterials. 31:2646–2654. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sellimi S, Younes I, Ayed HB, Maalej H,
Montero V, Rinaudo M, Dahia M, Mechichi T, Hajji M and Nasri M:
Structural, physicochemical and antioxidant properties of sodium
alginate isolated from a Tunisian brown seaweed. Int J Biol
Macromol. 72:1358–1367. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Otter M, Kuiper J, Bos R, Rijken D and van
Berkel TJ: Characterization of the interaction both in vitro and in
vivo of tissue-type plasminogen activator (t-PA) with rat liver
cells. Biochem J. 284:545–550. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Puxbaum V, Nimmerfall E, Bäuerl C, Taub N,
Blaas PM, Wieser J, Mikula M, Mikulits W, Ng KM, Yeoh GC and Mach
L: M6P/IGF2R modulates the invasiveness of liver cells via its
capacity to bind mannose 6-phosphate residues. J Hepatol.
57:337–343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bertin PA, Smith D and Nguyen ST:
High-density doxorubicin-conjugated polymeric nanoparticles via
ring-opening metathesis polymerization. Chem Commun (Camb).
14:3793–3795. 2005. View
Article : Google Scholar
|
|
26
|
Fontana MC, Beckenkamp A, Buffon A and
Beck RC: Controlled release of raloxifene by nanoencapsulation:
Effect on in vitro antiproliferative activity of human breast
cancer cells. Int J Nanomedicine. 9:2979–2991. 2014.PubMed/NCBI
|
|
27
|
Yao YC, Zhan XY, Zhang J, Zou XH, Wang ZH,
Xiong YC, Chen J and Chen GQ: A specific drug targeting system
based on polyhydroxyalkanoate granule binding protein PhaP fused
with targeted cell ligands. Biomaterials. 29:4823–4830. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Peng W, Hu C, Shu Z, Han T, Qin L and
Zheng C: Antitumor activity of tatariside F isolated from roots of
Fagopyrum tataricum (L.) Gaertn against H22 hepatocellular
carcinoma via up-regulation of p53. Phytomedicine. 22:730–736.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Van Lancker MA, Bellemans LA and De
Leenheer AP: Quantitative determination of low concentrations of
adriamycin in plasma and cell cultures, using a volatile extraction
buffer. J Chromatogr. 374:415–420. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Du P, Viswanathan UM, Xu Z, Ebrahimnejad
H, Hanf B, Burkholz T, Schneider M, Bernhardt I, Kirsch G and Jacob
C: Synthesis of amphiphilic seleninic acid derivatives with
considerable activity against cellular membranes and certain
pathogenic microbes. J Hazard Mater. 269:74–82. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ding J, Xiao C, Li Y, Cheng Y, Wang N, He
C, Zhuang X, Zhu X and Chen X: Efficacious hepatoma-targeted
nanomedicine self-assembled from galactopeptide and doxorubicin
driven by two-stage physical interactions. J Control Release.
169:193–203. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kato Y, Ozawa S, Miyamoto C, Maehata Y,
Suzuki A, Maeda T and Baba Y: Acidic extracellular microenvironment
and cancer. Cancer Cell Int. 13:892013. View Article : Google Scholar : PubMed/NCBI
|