Introduction
Breast cancer is the most frequently diagnosed
cancer in females and is currently the second most common cause of
cancer-associated mortality behind lung cancer (1,2). The
American Cancer Society estimated that, in the USA, 40,290 of
231,840 patients with newly diagnosed invasive breast cancer would
succumb to breast cancer in 2015 (3).
It was also predicted that, in 2015, <12% of females in the USA
would develop invasive breast cancer over their lifetime (4). One type of breast cancer,
triple-negative breast cancer (TNBC), is characterized by the
absence of the following genes: Estrogen receptor α (ERα),
progesterone receptor and human epidermal growth factor receptor 2
(HER2) (5). These genes are common
targets for therapy, which makes treatment of TNBC a challenge, due
to the lack of expression of these target genes in TNBC patients
(5). Patients with TNBC are
considered to possess poorer clinical outcomes and exhibit an
increased rate of distant recurrence, compared with other types of
breast cancer.
Understanding types of cancer has enabled novel and
systemic treatments to be offered patients with breast cancer in
order to improve their prognosis. Cytokine-induced killer (CIK)
therapy is a new and widely used tumor-adoptive immunotherapeutic
method which has been combined with surgery, chemotherapy and
radiation therapy to enhance the antitumor effects (6–8).
Autologous CIK cells have been widely accepted as an effective
therapeutic method for solid and hematological malignances. The
advantages of this therapy are numerous: CIK therapy exhibits
antitumor effects, is easily produced in vitro, possesses
greater efficacy with fewer side effects compared with radiotherapy
or chemotherapy, and major histocompatibility complex-unrestricted
cytotoxicity (9). However, further
studies are required to investigate the signaling pathway and
underlying molecular mechanisms of CIK therapy.
The tumor suppressor and transcription factor p53 is
encoded by the tumor protein p53 (TP53) gene, and triggers cell
cycle arrest, cell proliferation, apoptosis and senescence in
response to a variety of cellular stress signals (10–12).
Stress signals activate the transcriptional activity of p53 and may
also induce stabilization of the protein through post-translational
modifications including phosphorylation (11–13).
Degradation of p53 by proteasomes is mediated by the ubiquitination
of murine double minute 2 (MDM2) and this process may be enhanced
by the interaction of MDM2 and phosphatase and tensin homolog
deleted on chromosome 10 (PTEN) (14–16). TP53
gene mutations always result in a loss of wild-type p53
oncosuppressive properties, therefore numerous mechanisms have been
studied, including p53 mutations and deletions, and MDM2
amplification. p53 forms a homotetramer prior to serving as a
transcription factor and so expression of TP53 mutant variants may
lead to novel oncogenic gain-of-function (GOF) activities in cancer
cells (17). Previous studies have
demonstrated that the incidence of p53 mutations differs between
various types of cancer, ranging from near universal (~96% for
serous ovarian cancer) to rare occurrence (<10% for thyroid
cancer) (18). The incidence of p53
mutations is between 18 and 25% for primary breast carcinomas
(19). It has also been demonstrated
that GOF mutant p53 may be able to upregulate CXC chemokines and
enhance cell migration (20–23).
MDM2, also known as human double minute (HDM) 2 in
humans, is a negative regulator of p53 and may promote p53
degradation by binding to the protein using its N-terminal
p53-binding domain (24). At the
C-terminus of MDM2, the really interesting new gene (RING) domain
is responsible for p53 degradation, serving as an E3 ligase and
aiding the formation of polyubiquitin chains (23). MDM2 is also able to inhibit the
transcriptional activation of p53 by binding to the protein at the
N-terminus and promoting p53 degradation (25). In a number of human cancers, MDM2 is
overexpressed, and is associated with poor prognosis in cancers
including sarcoma, acute lymphocytic leukemia and glioma (26). MDM2 may interfere with p53-mediated
apoptosis in tumors (27) and cause
carcinogenesis independently of the p53 signaling pathway (25). Previous studies have identified that
loss of MDM2 mimics the inhibition of the MDM2-p53 interaction and
increases the stabilization of mutant p53 and the incidence of
metastasis (28,29).
Although CIK therapy is administered to patients
with breast cancer, and accepted as a form of therapy, the
molecular mechanisms and pathways of CIK therapy remain unclear.
Previously, it was proposed to use RNA sequencing (RNA-Seq) to
detect the gene expression changes in 2 patients with breast cancer
(Zhou et al, unpublished data). Samples from a third patient
with breast cancer were analyzed using the quantitative polymerase
chain reaction (qPCR) to assess the reliability of RNA-Seq results.
Samples of blood were collected prior to and following CIK therapy
from the patients with breast cancer. In the present study, the
expression levels of 9 genes were determined [TP53, p85, MDM2,
MDM4, ribosomal protein L11 (RPL11), ribosomal protein S23 (RPS23),
sirtuin 1 (SIRT1), histone deacetylase 1 (HDAC1), tuberous
sclerosis complex 1 (TSC1) and mechanistic target of rapamycin
(mTOR)]. The expression levels of genes which may regulate the
expression or activity of p53 (8/9 genes aforementioned) were
determined using RNA-Seq. MDM2 only demonstrated a marked
alteration in expression levels subsequent to CIK therapy For the
genes that were able to regulate the expression or activities of
p53, the expression levels of 8 genes out of 9 (except p85) were
determined using RNA-Seq, with only MDM2 identified to be
significantly altered in expression levels following CIK therapy.
The present study demonstrated that in a number of breast cancers,
alterations in the expression of TP53 were associated with
alterations in the expression of MDM4. This pattern of association
was also observed between the RPL11 and RPS23 genes, and the TSC1
and mTOR genes.
Materials and methods
Patient samples
In the present study, mononuclear cells were
collected from three female patients with breast cancer (age range,
37–55 years) in March 2016 in Wuhan Integrated TCM and Western
Medicine Hospital (Wuhan, China), 2 weeks after the patients had
completed chemotherapy or radiation. The cells were separated from
the patient's peripheral blood and cultured for 14 days, following
which the CIK cell number, viability and phenotype were determined
to confirm safety for infusion. Patients were administered three
intravenous infusions of >5×109 CIK cells at 1 day
intervals. The peripheral blood from 3 patients with breast cancer
was collected prior to and following CIK therapy. The present study
was approved by the ethical review board of the Wuhan Integrated
TCM and Western Medicine Hospital (Wuhan, China), and written
informed consent was obtained from all participants.
cDNA preparation
Peripheral blood, collected from the patients, was
first separated using lymphocyte separation medium (GE Healthcare
Bio-Sciences, Pittsburgh, PA, USA) and lymphocytes were collected
for cDNA preparation. TRIzol (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA), trichloromethane, isopropanol and ethyl
alcohol, were used to extract RNA, according to the manufacturer's
protocol (Invitrogen; Thermo Fisher Scientific, Inc.). A total of
40 µl nuclease-free water was used to resuspend RNA and a NanoDrop
2000 instrument (Thermo Fisher Scientific, Inc.) was used to
determine the quantity and purity of RNA. The absorbance at 260 nm
was determined and the absorbance ratio at 260/280 was calculated.
Finally, a Moloney murine leukemia virus Reverse Transcriptase kit
(Promega Corporation, Madison, WI, USA) was used to generate the
final cDNA library from the total extracted RNA.
Two paired samples were selected for RNA-Seq, termed
L_1 (prior to CIK therapy) and L_2 (following CIK therapy) for one
patient and Z_1 (prior to CIK therapy) and Z_2 (following CIK
therapy) for the other patient. Samples from another patient were
used for qPCR analysis.
qPCR validation
qPCR was conducted to re-analyze selected genes
listed in Table I and to determine
the validity of sequencing data. PCR amplifications were performed
using 200 ng cDNA, 10 µl SYBR® qPCR Mix (CWBIO, Beijing,
China) and 0.4 µl (10 µM) primers (Table
I). For each gene tested, 3 replicates were performed and the
GAPDH gene was used as the internal reference gene. RNase-free
H2O (CWBIO) was added to the mixture to a final volume
of 20 µl. Following heating to 95°C for 10 min, 40 cycles of 5°C
for 15 sec, 55°C for 30 sec and 72°C for 30 sec were applied. The
2−ΔΔCq method (30) was
used for relative gene expression level analysis.
 | Table I.Primers used in the quantitative
polymerase chain reaction. |
Table I.
Primers used in the quantitative
polymerase chain reaction.
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|
| TP53 |
TCAGCATCTTATCCGAGTGGAA |
AGGGCACCACCACACTATGTC |
| TSC1 |
GAAAGCCGCCTATCGGAAA |
TCCGTTTTTGGGAGGTATCAAG |
| RPL11 |
CGCATCCGCAAACTCTGTCT |
TCCGGATGCCAAAGGATCT |
| RPS23 |
CGAGACCAGAAGTGGCATGA |
GCATGAGAAGCACCTCCAAAAG |
| MDM2 |
CAGGCAGGGGAGAGTGATA |
GTGATGGAAGGGGGGGATT |
| GAPDH |
CATGAGAAGTATGACAACAGCCT |
AGTCCTTCCACGATACCAAAGT |
Results
Data from RNA-Seq
From a previous study (Zhou et al,
unpublished data), the MDM2 gene was detected in the 2 paired
samples using RNA-Seq. Expression changes were demonstrated in the
2 patients prior to and following CIK therapy, with an increase of
2.1-fold for 1 patient and 3.05-fold for the other patient
(Table II).
| Table II.Detailed information of the MDM2 gene
for two patients using RNA sequencing. |
Table II.
Detailed information of the MDM2 gene
for two patients using RNA sequencing.
| MDM2 gene | Patient 1 | Patient 2 |
|---|
| Fold change |
2.3 |
3.0 |
| Log2
(fold-change) |
1.2 |
1.6 |
| P-value |
2.3×10−11 |
2.3×10−3 |
| FDR |
5.9×10−11 |
4.6×10−3 |
| Expression
change | Up | Up |
| Expression
following therapy | 40.9 | 44.0 |
| Expression prior to
therapy | 17.8 | 14.5 |
| Description | E3
ubiquitin-protein ligase MDM2 | E3
ubiquitin-protein ligase MDM2 |
Vijayakumaran et al (31) demonstrated that p53 expression,
particularly mutant p53 in cancer cells, may be regulated by
several proteins and microRNAs (miRs). The proteins that may
control p53 proteasomal degradation include MDM2, MDM4, RPL11,
RPS23, p85, cell division control protein 42 (CDC42), SIRT1, HDAC1,
TSC1 and the mTOR signaling pathway. In the present study, attempts
to identify additional proteins associated with p53, other than
MDM2, in CIK therapy were carried out. The data collected using
RNA-Seq (Table III) demonstrated
alterations in the expression levels of the TP53, MDM4, RPL11,
RPS23, SIRT1, HDAC1, TSC1 and mTOR genes identified in the two
paired samples. Analysis of results revealed that only the RPL11
and RPS23 pseudogenes were detected with significant alterations in
expression [false discovery rate (FDR) <0.05] in the two paired
samples. Additionally, marked changes in the expression level of
the mTOR gene was determined in one of the paired samples which was
possibly due to individual differences.
| Table III.Detailed information of gene
expression levels for two patients using RNA sequencing. |
Table III.
Detailed information of gene
expression levels for two patients using RNA sequencing.
| mRNA | L_1 | L_2 | Z_1 | Z_2 |
|---|
| CDC42 | 165 | 252 | 389 | 248 |
| HDAC1 | 134 | 294 | 344 | 288 |
| MDM4 | 51 | 120 | 141 | 81 |
| mTOR | 75 | 128 | 147 | 53 |
| RPL11 | 2,045 | 3,260 | 4,589 | 3,790 |
| RPS23 | 490 | 722 | 1,008 | 695 |
| SIRT1 | 8 | 17 | 29 | 18 |
| TP53 | 26 | 119 | 96 | 57 |
| TSC1 | 72 | 105 | 122 | 79 |
qPCR validation of gene
transcription
To confirm the validity of RNA-Seq data and previous
studies (Zhou et al, unpublished data) on p53 regulation,
the expression levels of genes, including TP53, MDM2, RPL11, RPS23
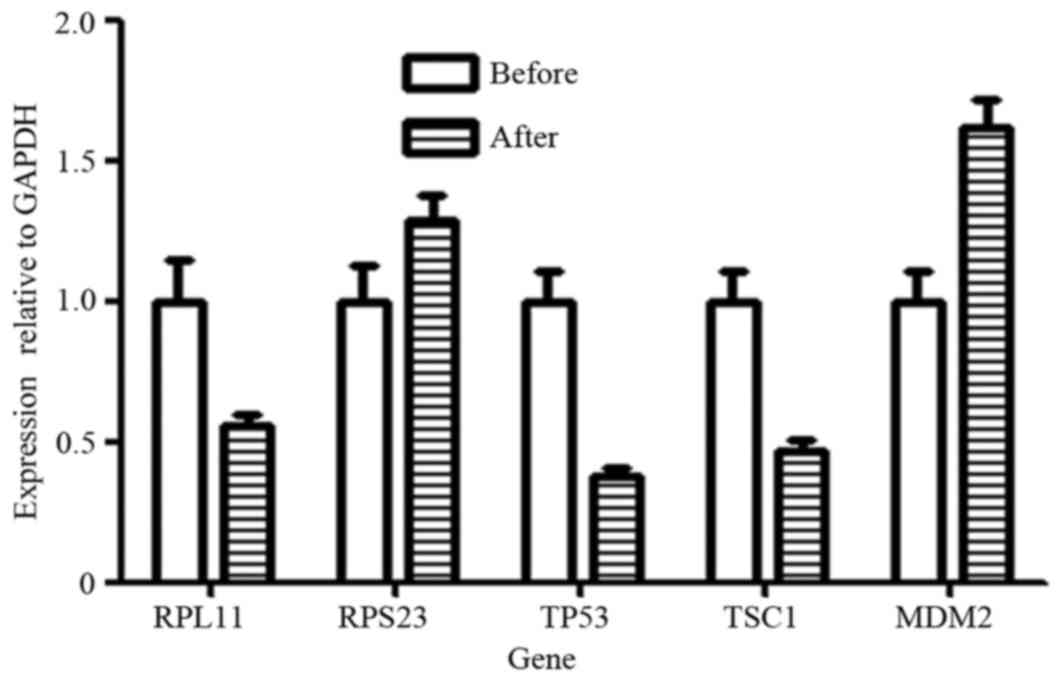
and TSC1, were all analyzed by qPCR. The results identified that
the RPL11, TP53 and TSC1 genes were all downregulated following
therapy for one patient, and the RPS23 and MDM2 genes were
upregulated following CIK therapy (Fig.
1).
Discussion
RNA-Seq and qPCR identified 3 genes, MDM2, TP53 and
TSC1, of the third patient with similar alterations to those of one
of the patients with samples termed L_2 and Z_2. The distinctions
were possibly due to individual variations.
In normal cells, transcription factor p53 is
regulated and maintained at low expression levels by MDM2. As an E3
ubiquitin ligase, MDM2 is able to bind to p53 specifically by its
N-terminus, form ubiquitin chains on p53 by the catalytic RING
domain at the MDM2 C-terminus and result in proteasomal degradation
of p53 (24). The MDM2-p53
interaction is complex, and is regulated at a number of levels by
numerous cellular proteins and epigenetic mechanisms. In ~50% of
human cancers, the gene TP53 is mutated, including deletion,
loss-of-function and gain-of function mutations; however, ~17% of
tumors exhibit MDM2 gene amplification. Although p53 induces the
expression of MDM2, overexpression of MDM2 may promote the
degradation and inhibit the cellular activity of p53. Therefore,
increased expression levels of MDM2 may lead to the inhibition of
MDM2-p53 interactions, which will lead to downregulation of p53
pathways by promoting the degradation of wild-type and mutated p53
proteins.
In the present study, the expression levels of MDM2
were determined by RNA-Seq in the two samples with markedly
upregulated alterations following CIK therapy. Similar alterations
in the third patient were confirmed by qPCR. The primary function
of MDM2 is to promote degradation of p53, and, therefore, the
results of the present study indicated that the signaling pathways
and underlying molecular mechanisms of CIK therapy for breast
cancer enhanced apoptosis of cells, particularly in cancer cells
with p53 mutations. Subsequently, the expression levels of the TP53
gene were analyzed and it was revealed that TP53 was expressed in
the two paired samples, with no marked alterations following CIK
therapy. However, results from one paired sample demonstrated that
the expression of TP53 was upregulated following therapy, whereas
the other sample revealed downregulation following treatment. As in
~50% of human cancers, the TP53 gene is mutated in a number of
ways, including simple mutation, deletion, loss-of-function and
gain-of function. It was hypothesized that if the patients
developed breast cancer due to p53 mutations, the pathways and
underlying molecular mechanisms of CIK therapy may be associated to
degrade mutant p53 proteins and to promote apoptosis of cells with
the TP53 mutants. This hypothesis, that downregulated expression of
p53 was caused by elimination of non-recoverable p53 mutants,
requires additional study.
The expression of wild-type p53 may lead to
apoptosis of cells by upregulating target genes including p21,
B-cell lymphoma 2 (Bcl-2)-like protein, p53 upregulated modulator
of apoptosis and phorbol 12-myristate 13-acetate-induced protein 1
(32). However, the pathways and
underlying molecular mechanisms of p53-mediated tumor suppression
remain unclear. In breast cancers with mutant p53, the signaling
pathways associated with p53 for apoptosis differ from those in
normal cells. In a previous study of acute myeloid leukemia, Bcl-2,
inhibited by p53-mediated signaling pathways, may induce apoptosis
in mitochondria (33). In addition,
the upregulated expression of MDM2 may induce apoptosis through
this aforementioned signaling pathway by enhancing the degradation
of p53. These hypotheses suggest additional signaling pathways for
CIK therapy.
MDM2 may negatively regulate the transcriptional
activity of p53 by monoubiquitin to trigger p53 from nuclear
exportation (34). It may be an
additional signaling pathway mediated by MDM2 and p53 for CIK
therapy with non-recoverable p53 mutations.
MDM4 is a primary negative regulator of p53, similar
to MDM2, and is known as MDMX and HDMX. MDM4 is a homologous MDM2
protein that may be induced by p53 and assist in the maintenance of
low p53 expression levels in normal cells (35). MDM4, similarly to MDM2, is
overexpressed in human cancers including sarcoma and breast cancer
with incidences of ~20 and 15%, respectively (17). The MDM2-MDM4 heterodimer is formed via
the binding of the C-terminus of the two proteins, although MDM4
does not possess E3 ligase activity at C-terminus, unlike MDM2.
MDM4 may enhance ubiquitination on p53 by forming MDM2-MDM4
heterodimers (36) and inhibit the
transcriptional activity of p53 by binding to it via its N-terminus
(37). In the present study, the
expression levels of MDM4 were altered in the two paired samples
with FDR values >0.05, which means that these alterations were
not marked. The alterations in MDM4 expression levels were distinct
between patients, similar to that observed in TP53, with expression
upregulated for one patient and downregulated for another. It was
identified that the patient with upregulated expression levels of
TP53 additionally exhibited upregulated expression of MDM4 and the
other patient exhibited downregulated expression of these two
genes. The results demonstrate that, for a number of patients with
breast cancer, alterations in the expression levels of TP53 were
associated with that of MDM4. Therefore, it was hypothesized that
the trend of expression levels observed in patients with cancer
following CIK therapy were due to the distinct expression patterns
in the MDM4 gene, as MDM4 may enhance p53 degradation by forming
MDM2-MDM4 heterodimers (35). The
results may validate the previous hypothesis that MDM2 serves a
function in CIK therapy, through p53-mediated signaling pathways
including leading to apoptosis.
The expression levels of p53, MDM2 and MDM4 may all
be regulated by distinct miRs. For example, miR-192, −194, −215,
−143, −145 and −605 may enhance the expression levels of p53
through the suppression of MDM2 expression, and subsequently the
expression levels of these miRs may be upregulated by p53 (38). Additional studies are required to
investigate the regulation mechanisms of p53, MDM2 and MDM4
following CIK therapy for patients with cancer.
Expression levels of p53 may be additionally
regulated by other proteins, including RPL11, RPS23, p85, CDC42,
SIRT1, HDAC1, TSC1 and the mTOR signaling pathway (31). According to a previous study, RPL11
may enhance the expression levels of p53 through inhibiting MDM2,
whereas RPS23 may enhance the activity of RPL11 by inducing
ribosomal stress (31). In the
present study, the expression of the RPL11 and RPS23 genes were
determined using RNA-Seq in two paired samples and identified
increased expression levels, particularly for the RPL11 gene
(Table III). The expression levels
of these two aforementioned genes did not markedly alter, with an
FDR value >0.05. The expression level of RPL11 was upregulated
in the two paired samples, whereas the expression level of RPS23
was upregulated for one patient and downregulated for another. The
results of the present study revealed that the expression level of
RPL11 was markedly altered for one patient when the expression
level of RPS23 was upregulated. However, when the expression level
of RPS23 was downregulated, the expression levels of RPL11 did not
alter as much following therapy. As a ribosomal protein, RPL11 may
bind to MDM2 and block its inhibition of p53 (39), which may lead to a novel hypothesis
that the expression levels of RPL11 were associated with those of
RPS23. Additional studies are required to determine the functions
of these aforementioned proteins in p53- and MDM2-mediated pathways
for patients with cancer following CIK therapy.
TSC1 may enhance the activities of p53 through
inhibition of the mTOR signaling pathway (31). In the present study, the expression
levels of two genes, TSC1 and mTOR, were upregulated in one paired
sample and downregulated in another patient following CIK therapy.
These results suggest that the expression levels of mTOR may be
associated with those of TSC1. Elucidation of the function and
underlying molecular mechanisms of TSC1 and mTOR in p53-mediated
pathways in patients with cancer following CIK therapy requires
additional study.
p85, CDC42, SIRT1 and HDAC1 may inhibit the activity
of p53 directly. According to the RNA-Seq data, the trends in
expression levels of CDC42 and HDAC1 were similar to those of TSC1
and mTOR. The expression levels of SIRT1 were decreased, with
upregulation in the two patients. However, no signals were
determined with respect to the expression of p85, suggesting that
p85 may not be expressed in these two patients.
In the present study, expression levels of 9 genes
were analyzed, including TP53, MDM2, MDM4, RPL11, RPS23, SIRT1,
HDAC1, TSC1 and mTOR. For the genes that may regulate the
expression or activity of p53, the expression levels of 8/9 genes
were determined by RNA-Seq, with only MDM2 identified as markedly
altered following CIK therapy. In addition, the alterations in the
expression levels of TP53, RPLI1 and TSC1 were associated with that
of MDM4, RPS23 and mTOR, respectively. The aforementioned proteins
may regulate the expression and activity of p53 at different levels
in signaling pathways. Elucidation of the function and underlying
molecular mechanisms of these proteins in p53-mediated signaling
pathways in patients with cancer following CIK therapy requires
additional study. Furthermore, the expression levels and activity
of these proteins are regulated by additional proteins and miRNAs.
Additional studies are required to investigate the upstream
signaling pathways mediated by the aforementioned 8 genes through
the expression changes of target genes for patients with cancer
following CIK therapy.
Acknowledgements
The authors thank the Department of Research, Wuhan
Hamilton Biotechnology Co., Ltd. (Wuhan, China), for their
assistance, including the provision of laboratory apparatus and
assistance in data analyses.
References
|
1
|
American Cancer Society: Cancer facts
& figures. 2015, https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2015/cancer-facts-and-figures-2015.pdf
|
|
2
|
Edwards BK, Noone AM, Mariotto AB, Simard
EP, Boscoe FP, Henley SJ, Jemal A, Cho H, Anderson RN, Kohler BA,
et al: Annual Report to the Nation on the status of cancer,
1975–2010, featuring prevalence of comorbidity and impact on
survival among persons with lung, colorectal, breast, or prostate
cancer. Cancer. 120:1290–1314. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015: CA Cancer J Clin. 65:5–29. 2015.
|
|
4
|
Lo-Fo-Wong DN, Sitnikova K, Sprangers MA
and de Haes HC: Predictors of health care use of women with breast
cancer: A systematic review. Breast J. 21:508–513. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rakha EA, El-Sayed ME, Green AR, Lee AH,
Robertson JF and Ellis IO: Prognostic markers in triple-negative
breast cancer. Cancer. 109:25–32. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li H, Wang C, Yu J, Cao S, Wei F, Zhang W,
Han Y and Ren XB: Dendritic cell-activated cytokine-induced killer
cells enhance the anti-tumor effect of chemotherapy on non-small
cell lung cancer in patients after surgery. Cytotherapy.
11:1076–1083. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hui KM: CIK cells-current status, clinical
perspectives and future prospects-the good news. Expert Opin Biol
Ther. 12:659–661. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma Y, Zhang Z, Tang L, Xu YC, Xie ZM, Gu
XF and Wang HX: Cytokine-induced killer cells in the treatment of
patients with solid carcinomas: A systematic review and pooled
analysis. Cytotherapy. 14:483–493. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang X, Yu W, Li H, Yu J, Zhang X, Ren X
and Cao S: Can the dual-functional capability of CIK cells be used
to improve antitumor effects? Cell Immunol. 287:18–22. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartkova J, Horejsí Z, Koed K, Krämer A,
Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et
al: DNA damage response as a candidate anti-cancer barrier in early
human tumorigenesis. Nature. 434:864–870. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harms K, Nozell S and Chen X: The common
and distinct target genes of the p53 family transcription factors.
Cell Mol Life Sci. 61:822–842. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao Y, Chaiswing L, Velez JM,
Batinic-Haberle I, Colburn NH, Oberley TD and St Clair DK: p53
translocation to mitochondria precedes its nuclear translocation
and targets mitochondrial oxidative defense protein-manganese
superoxide dismutase. Cancer Res. 65:3745–3750. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chipuk JE, Kuwana T, Bouchier-Hayes L,
Droin NM, Newmeyer DD, Schuler M and Green DR: Direct activation of
Bax by p53 mediates mitochondrial membrane permeabilization and
apoptosis. Science. 303:1010–1014. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mayo LD and Donner DB: A
phosphatidylinositol 3-kinase/Akt pathway promotes translocation of
Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA.
98:pp. 11598–11603. 2001, View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gottlieb TM, Leal JF, Seger R, Taya Y and
Oren M: Cross-talk between Akt, p53 and Mdm2: Possible implications
for the regulation of apoptosis. Oncogene. 21:1299–1303. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oren M, Damalas A, Gottlieb T, Michael D,
Taplick J, Leal JF, Maya R, Moas M, Seger R, Taya Y and Ben-Ze'ev
A: Regulation of p53: Intricate loops and delicate balances.
Biochem Pharmacol. 64:865–871. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shtraizent N, Matsui H, Polotskaia A and
Bargonetti J: Hot spot mutation in TP53 (R248Q) causes oncogenic
gain-of-function phenotypes in a breast cancer cell line derived
from an African American patient. Int J Environ Res Public Health.
13:ijerph130100222015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Burgess A, Chia KM, Haupt S, Thomas D,
Haupt Y and Lim E: Clinical Overview of MDM2/X-Targeted Therapies.
Front Oncol. 6:72016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alsner J, Yilmaz M, Guldberg P, Hansen LL
and Overgaard J: Heterogeneity in the clinical phenotype of TP53
mutations in breast cancer patients. Clin Cancer Res. 6:3923–3931.
2000.PubMed/NCBI
|
|
20
|
Freed-Pastor WA and Prives C: Mutant p53:
One name, many proteins. Genes Dev. 26:1268–1286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Walerych D, Napoli M, Collavin L and Del
Sal G: The rebel angel: Mutant p53 as the driving oncogene in
breast cancer. Carcinogenesis. 33:2007–2017. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yeudall WA, Vaughan CA, Miyazaki H,
Ramamoorthy M, Choi MY, Chapman CG, Wang H, Black E, Bulysheva AA,
Deb SP, et al: Gain-of-function mutant p53 upregulates CXC
chemokines and enhances cell migration. Carcinogenesis. 33:442–451.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Haupt Y, Maya R, Kazaz A and Oren M: Mdm2
promotes the rapid degradation of p53. Nature. 387:296–299. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bond GL, Hu W and Levine AJ: MDM2 is a
central node in the p53 pathway: 12 years and counting. Curr Cancer
Drug Targets. 5:3–8. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Onel K and Cordon-Cardo C: MDM2 and
prognosis. Mol Cancer Res. 2:1–8. 2004.PubMed/NCBI
|
|
27
|
de Rozieres S, Maya R, Oren M and Lozano
G: The loss of mdm2 induces p53-mediated apoptosis. Oncogene.
19:1691–1697. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Steinman HA, Burstein E, Lengner C,
Gosselin J, Pihan G, Duckett CS and Jones SN: An alternative splice
form of Mdm2 induces p53-independent cell growth and tumorigenesis.
J Biol Chem. 279:4877–4886. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Terzian T, Suh YA, Iwakuma T, Post SM,
Neumann M, Lang GA, Van Pelt CS and Lozano G: The inherent
instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss.
Genes Dev. 22:1337–1344. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vijayakumaran R, Tan KH, Miranda PJ, Haupt
S and Haupt Y: Regulation of Mutant p53 Protein Expression. Front
Oncol. 5:2842015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vousden KH and Prives C: Blinded by the
light: The growing complexity of p53. Cell. 137:413–431. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kojima K, Konopleva M, Samudio IJ, Schober
WD, Bornmann WG and Andreeff M: Concomitant inhibition of MDM2 and
Bcl-2 protein function synergistically induce mitochondrial
apoptosis in AML. Cell Cycle. 5:2778–2786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li M, Brooks CL, Wu-Baer F, Chen D, Baer R
and Gu W: Mono-versus polyubiquitination: Differential control of
p53 fate by Mdm2. Science. 302:1972–1975. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shadfan M, Lopez-Pajares V and Yuan ZM:
MDM2 and MDMX: Alone and together in regulation of p53. Transl
Cancer Res. 1:88–89. 2012.PubMed/NCBI
|
|
36
|
Leslie PL, Ke H and Zhang Y: The MDM2 RING
domain and central acidic domain play distinct roles in MDM2
protein homodimerization and MDM2-MDMX protein heterodimerization.
J Biol Chem. 290:12941–12950. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brooks CL and Gu W: p53 ubiquitination:
Mdm2 and beyond. Mol Cell. 21:307–315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hoffman Y, Pilpel Y and Oren M: microRNAs
and Alu elements in the p53-Mdm2-Mdm4 regulatory network. J Mol
Cell Biol. 6:192–197. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kamio T, Gu BW, Olson TS, Zhang Y, Mason
PJ and Bessler M: Mice with a Mutation in the Mdm2 gene that
interferes with MDM2/Ribosomal protein binding develop a defect in
Erythropoiesis. PLoS One. 11:e01522632016. View Article : Google Scholar : PubMed/NCBI
|