Introduction
Melanoma is a particularly aggressive skin cancer
and its incidence is increasing faster than any other cancer
worldwide (1). At present, the
10-year survival rate of patients diagnosed with advanced (stage
IV) metastatic melanoma is <10% (2), which highlights the importance of early
detection and treatment. The Food and Drug Administration approved
the chemotherapeutic agent dacarbazine (DTIC) for the treatment of
metastatic melanoma in 1975, and it remains the only licensed
chemotherapeutic agent in use today (1). DTIC is a methylating agent which causes
DNA damage, cell cycle arrest and apoptosis. Despite this, only 2%
of all patients with metastatic melanoma receiving this treatment
demonstrate a significant response and only 11.2% demonstrate a
partial response (3). Resistance to
DTIC has been associated with the upregulation of pro-survival
signals and anti-apoptotic molecules in cancer cells. Despite its
moderate effects, DTIC continues to be the standard treatment for
metastatic melanoma as no other chemotherapeutic treatment has been
demonstrated to have a significantly increased chance of survival
when compared with DTIC (4,5). Provided the limited efficacy of the
current metastatic melanoma chemotherapies in addition to the
increasing incidence of melanoma cases, there appears to be a need
for the development of more effective treatment strategies.
Statins are a group of drugs commonly used for the
reduction of cholesterol levels (6).
They work by inhibiting 3-hydroxy-3-methyl-glutaryl-coenzyme A
reductase, a critical enzyme in the mevalonate pathway, which is
responsible for cholesterol synthesis (6,7). In
addition, statins have been demonstrated to serve a function in
immune regulation and cancer prevention (8). Evidence from in vitro and in
vivo studies has revealed that statins have a wide range of
anticancer activities in various types of cancer (6,9,10). In multiple myeloma cells, simvastatin
has been demonstrated to induce S phase cell cycle arrest through
the downregulation of cell division cycle 25a, cyclin A and cyclin
dependent kinase expression and the activation of checkpoint kinase
1 (9). This cell cycle arrest was
accompanied by intrinsic apoptosis as demonstrated by diminished
B-cell lymphoma 2 (Bcl-2) protein levels, increased cytosolic
cytochrome c and active caspase 9 and caspase 3 levels. In human
glioblastoma cells, previous studies have revealed that
erivastatin, pitavastatin and fluvastatin are potent
anti-proliferative agents (10,11). In
addition, one clinical trial revealed that fluvastatin reduced
tumour proliferation and increased apoptotic activity in
high-grade, stage 0/1 breast cancer (12). Pitavastatin has been demonstrated to
exert a cytotoxic effect on U87 glioblastoma tumour growth in
vivo (10). On a molecular level,
pitavastatin treatment has been demonstrated to upregulate the cell
cycle regulator p21 and to inhibit nuclear factor-κB (NF-κB) in
different tumour cells, which resulted in cell cycle arrest and
apoptosis (13,14). Finally, in glioma cells, autophagic
cell death was demonstrated to be a potential mechanism of
pitavastatin-induced cytotoxicity by also resulting in the
inhibition of NF-κB (15). However,
the exact molecular mechanisms underpinning the anticancer activity
of pitavastatin remain mostly unknown.
Given the shortage of treatments for metastatic
melanoma, the present study therefore aimed to explore the effects
of combined pitavastatin and DTIC treatment in human melanoma
cells. The present study demonstrated that this combined treatment
results in the synergistic inhibition of cell survival and further
demonstrated that this occurs through the induction of intrinsic
apoptosis and autophagy cell death pathways.
Materials and methods
Cell culture and treatments
The human melanoma cell lines A375 and WM115,
sourced from the Department of Human Biology, University of Cape
Town (Cape Town, South Africa), were maintained in Dulbecco's
modified Eagle's medium, Biological Industries Israel Beit-Haemek
(Kibbutz Beit-Heamek, Israel) supplemented with 10% foetal bovine
serum, Biological Industries Israel Beit-Haemek in a humidified 5%
CO2 balanced air incubator at 37°C. Pitavastatin (Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) and DTIC (Sigma-Aldrich,
Merck KGaA, Darmstadt, Germany) were dissolved in dimethyl
sulfoxide (DMSO) to give stock concentrations of 5 mM, which were
stored for no more than 5 days. Control cells were treated with
equivalent concentrations of DMSO (vehicle). Autophagy inhibitor
3-methyl adenine (10 mM) (3MA; Sigma-Aldrich; Merck KGaA) was added
at 37°C for 1 h prior to pitavastatin/DTIC treatment.
Cytotoxicity assays
Cells were seeded in 96-well plates at a density of
4×103-5×103 cells per well and allowed to
settle for 48 h at 37°C. Cells were treated with a range of
pitavastatin (0–5.0 µM) and/or DTIC (0.0–100 µM) concentrations or
vehicle for 48 h at 37°C. Cytotoxicity was assessed using an MTT
assay kit as per the manufacturer's protocol (Roche Diagnostics
GmbH, Mannheim, Germany) (16).
Briefly, 10 µl MTT solution was added to each well and cells were
incubated at 37°C for 4 h, followed by addition of 100 µl
solubilisation buffer [10% SDS; Sigma-Aldrich; Merck KGaA) in 0.01
M hydrochloric acid (HCl) (Sigma-Aldrich, Israel)] and incubation
for 16 h at 37°C. Absorbance at 585 nm was determined for each well
and the mean cell viability was calculated as a percentage of the
mean vehicle control.
Cell cycle analysis
Cells were plated at a density of
3×105-4×105 cells per 6-cm dish and allowed
to settle for 24 h at 37°C. Log-phase cultures were exposed to
drugs or vehicle for 48 h at 37°C. Cells were then trypsinised,
washed with PBS and fixed in 95% ethanol at 4°C overnight, followed
by RNase A (50 µg/ml; Sigma-Aldrich; Merck KGaA) treatment for 15
min at 37°C and and immediately stained for 30 min at room
temperature with propidium iodide (PI; Sigma-Aldrich; Merck KGaA).
Cellular DNA content was determined using flow cytometry with
individual samples subjected to a FACSCalibur flow cytometer with a
488 nm coherent laser (BD Biosciences, San Jose, CA, USA).
Cellquest Pro version 5.2.1 software (BD Biosciences) was used for
data acquisition and analyses were performed using Modfit version
2.0 software (BD Biosciences).
Apoptosis detection
Log-phase melanoma cultures were treated with 1.0 µM
pitavastatin, 40.0 µM DTIC, combined pitavastatin (1.0 Μm)-DTIC
(40.0 µM) or vehicle for 48 h at 37°C. Adherent and floating cells
were collected and double-labelled with Annexin V-Fluorescein
isothiocyanate (FITC) and PI using Annexin V-FITC Apoptosis
Detection Kit (Sigma-Aldrich; Merck KGaA) as per the manufacturer's
protocol. Annexin V-FITC was used to quantitatively determine the
percentage of apoptotic cells while PI was used to stain all dead
cells. Cells were analysed by flow cytometry with a 488 nm coherent
laser equipped with FACStation running version 3.3 Cell Quest
software (BD Biosciences).
Cytochrome c release
Melanoma cells treated as aforementioned with
vehicle or pitavastatin-DTIC for 48 h at 37°C were trypsinized,
re-suspended in HB-7S buffer [1 mmol/l EGTA Na-free, 5 mmol/l
Tris-HCl (pH 7.4), 1 mmol/l DTT, and 11% sucrose] (Sigma-Aldrich;
Merck KGaA) and subcellular fractions were collected as described
previously (17).
Western blotting
Cells were harvested and the protein was prepared as
described previously (16). Primary
antibodies used were as follows: Anti-PARP1/2 (cat no. sc-7150),
anti-p53 (cat no. sc-126), anti-p21 (cat no. sc-756),
anti-Bcl2-associated X, apoptosis regulator (Bax; cat no. sc-7480),
anti-cyclin D (cat no. sc-753), anti-cytochrome c (cat no.
sc-65396), anti-COXIV (cat no. sc-69359) and anti-cyclin E (cat no.
sc-247; Santa Cruz Biotechnology, Inc.), anti-actin (cat. no.,
A4700; Sigma-Aldrich; Merck KGaA),
anti-LC3-phosphatidylethanolamine conjugate (cat no. 2775),
anti-BCL-2 (cat no. 2876), anti-Caspase-3 (cat no. 9661) and
anti-Caspase-8 (cat no. 9746; Cell Signaling Technology, Inc.,
Danvers, MA, USA). Following primary antibody incubation, membranes
were incubated with appropriate horseradish peroxidase-conjugated
secondary antibodies (1:5,000; Bio-Rad Laboratories, Inc.,) and
antibody-reactive proteins were visualized using the
electrochemiluminescence reaction detection system as previously
described (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
(18).
Autophagy assays
Autophagy was confirmed by the presence of
fluorescent puncta in cells transfected with a green fluorescence
protein (GFP)-light chain 3 LC3) expression vector (cat. no.,
24920; Addgene, Inc., Cambridge, MA, USA), as previously described
(16).
Statistical analysis
Results are presented as the mean ± standard error
of the mean (SEM) of the three independent experiments. Statistical
analysis of data was performed using the two-sample t-test in
Microsoft Excel 2013 (Microsoft Corporation, Redmond, WA, USA) or a
one-way ANOVA with Tukey's post hoc test in Graph Pad Prism v.5.
(GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Pitavastatin and DTIC synergistically
inhibit melanoma cell survival
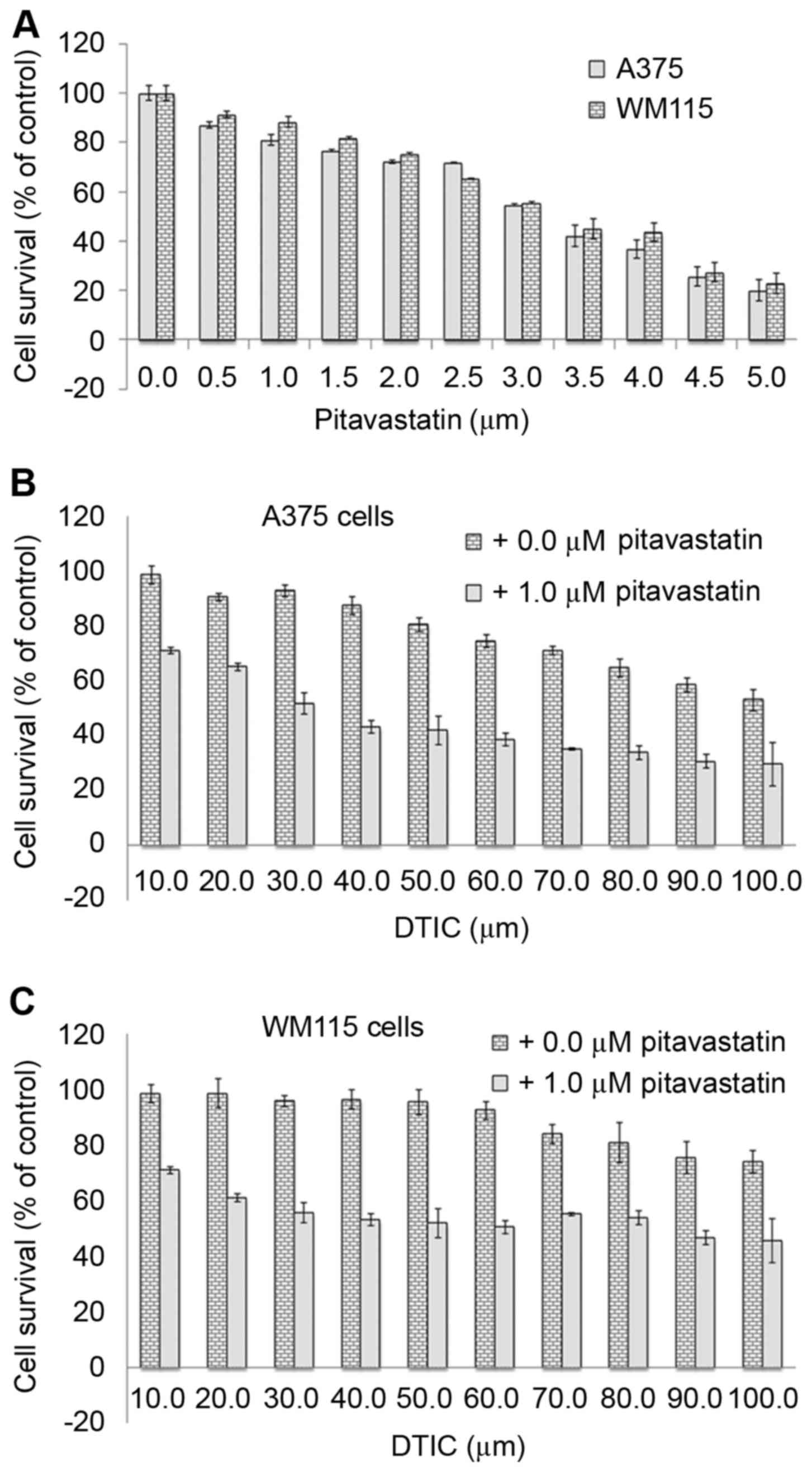
To investigate the anti-cancer effect of
pitavastatin on melanoma, human A375 and WM115 melanoma cells were
treated with increasing concentrations (0–5 µM) of pitavastatin for
48 h. An MTT assay was used to determine cell viability and a
dose-dependent decrease in cell survival was observed in A375 and
WM115 cells (Fig. 1A). The results
revealed that 4 µM pitavastatin treatment resulted in the death of
>50% of cells, suggesting that it exerts potent cytotoxic
effects against melanoma cells specifically at high concentrations.
DTIC has previously been demonstrated to inhibit A375 cell survival
but only at a high concentrations of 25–100 µM (19). Therefore, the present study aimed to
determine whether the combined treatment of pitavastatin and DTIC
may have a greater anti-cancer effect on A375 and WM115 cells. To
this end, cells were treated with 1 µM pitavastatin for 1 h
followed by treatment with increasing concentrations (10–100 µM) of
DTIC for 48 h. Fig. 1B and C
demonstrate that the combined treatment resulted in enhanced
anti-cytotoxic activity compared with DTIC treatment alone. Whilst
40 µM DTIC resulted in the death of <15% of melanoma cells, when
cells were pre-treated with 1 µM pitavastatin, it resulted in the
death of ~50% of melanoma cells at the same DTIC concentration. The
results of the present study demonstrate that combined pitavastatin
and DTIC treatment results in a synergistic cytotoxic effect in
melanoma cells.
Combined pitavastatin and DTIC
treatment induces G1 cell cycle arrest in melanoma cells
The present study aimed to investigate the mechanism
by which combined pitavastatin-DTIC treatment synergistically
inhibits melanoma cell survival. To this end, A375 and WM115 cells
were treated with vehicle, pitavastatin (1.0 µM), DTIC (40.0 µM) or
pitavastatin-DTIC (1.0 and 40.0 µM, respectively) and the effect on
the cell cycle profile was determined by flow cytometry. Fig. 2A demonstrates that combined
pitavastatin-DTIC treatment induced a significantly greater G1 cell
cycle arrest than either treatment alone. To further explore this,
A375 and WM115 cells were treated with pitavastatin and DTIC and
markers of cell cycle arrest were analysed by the use of western
blotting. Fig. 2B demonstrates the
p53 response elicited by combined pitavastatin-DTIC treatment. In
the two cell lines, treatment with only pitavastatin resulted in an
increase in p21 levels, however, when WM115 cells were treated with
pitavastatin-DTIC, p21 levels were even further increased. For A375
and WM115 cells pitavastatin-DTIC treatment corresponded with a
decrease in cyclin D1 and cyclin E1, which are required for the
transition from G1 to S phase. These results provide compelling
evidence that combined pitavastatin-DTIC treatment results in G1
cell cycle arrest in melanoma cells.
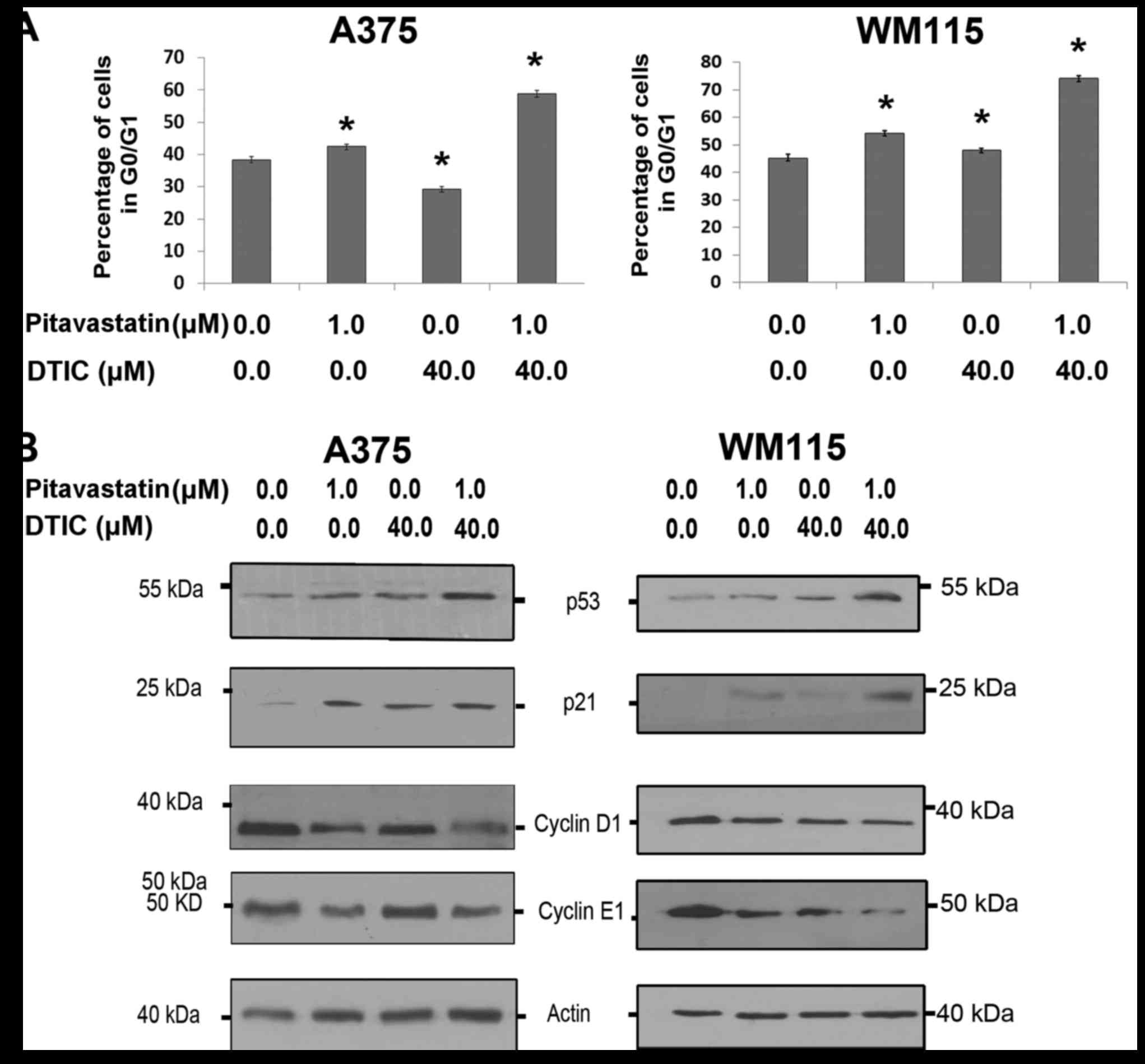 | Figure 2.Combined pitavastatin and DTIC
treatment induces G1 cell cycle arrest in melanoma cells. (A) Cell
cycle analysis of A375 and WM115 cells treated with vehicle,
pitavastatin (1.0 µM), DTIC (40.0 µM) or pitavastatin-DTIC (1.0 and
40.0 µM, respectively). The proportion of cells at G0/G1 phase was
expressed as a percentage of the total number of cells analysed and
presented in the graphs as the mean ± standard error of the mean of
three independent experiments (*P<0.01 as compared with vehicle,
one-way ANOVA with Tukey's post hoc test). (B) A375 and WM115 cells
were treated with 1.0 µM pitavastatin, 40.0 µM DTIC,
pitavastatin-DTIC (1.0 and 40.0 µM, respectively) or vehicle.
Protein extracts were analysed by SDS-PAGE (8–15%) and western
blotting using antibodies against p53, p21, Cyclin E1 and Cyclin
D1. Actin was detected as a loading control. DTIC, dacarbazine. |
Combined pitavastatin-DTIC treatment
activates intrinsic apoptosis
The presence of sub-G1 peaks on DNA content
histograms is generally accepted to represent apoptotic cells
(20). As cell cycle analysis
demonstrated that combined pitavastatin-DTIC treatment resulted in
sub-G1 peaks (Fig. 2A), the present
study aimed to determine whether pitavastatin-DTIC treatment
induced cell death by apoptosis. To this end, A375 and WM115 cells
were treated with pitavastatin-DTIC and western blotting was
performed to assess PARP cleavage and active caspase 3 levels,
which are molecular markers of apoptosis. Fig. 3A demonstrates that levels of the
active cleaved PARP and caspase 3 proteins notably increased in
response to combined pitavastatin-DTIC treatment, suggesting that
apoptosis is indeed activated.
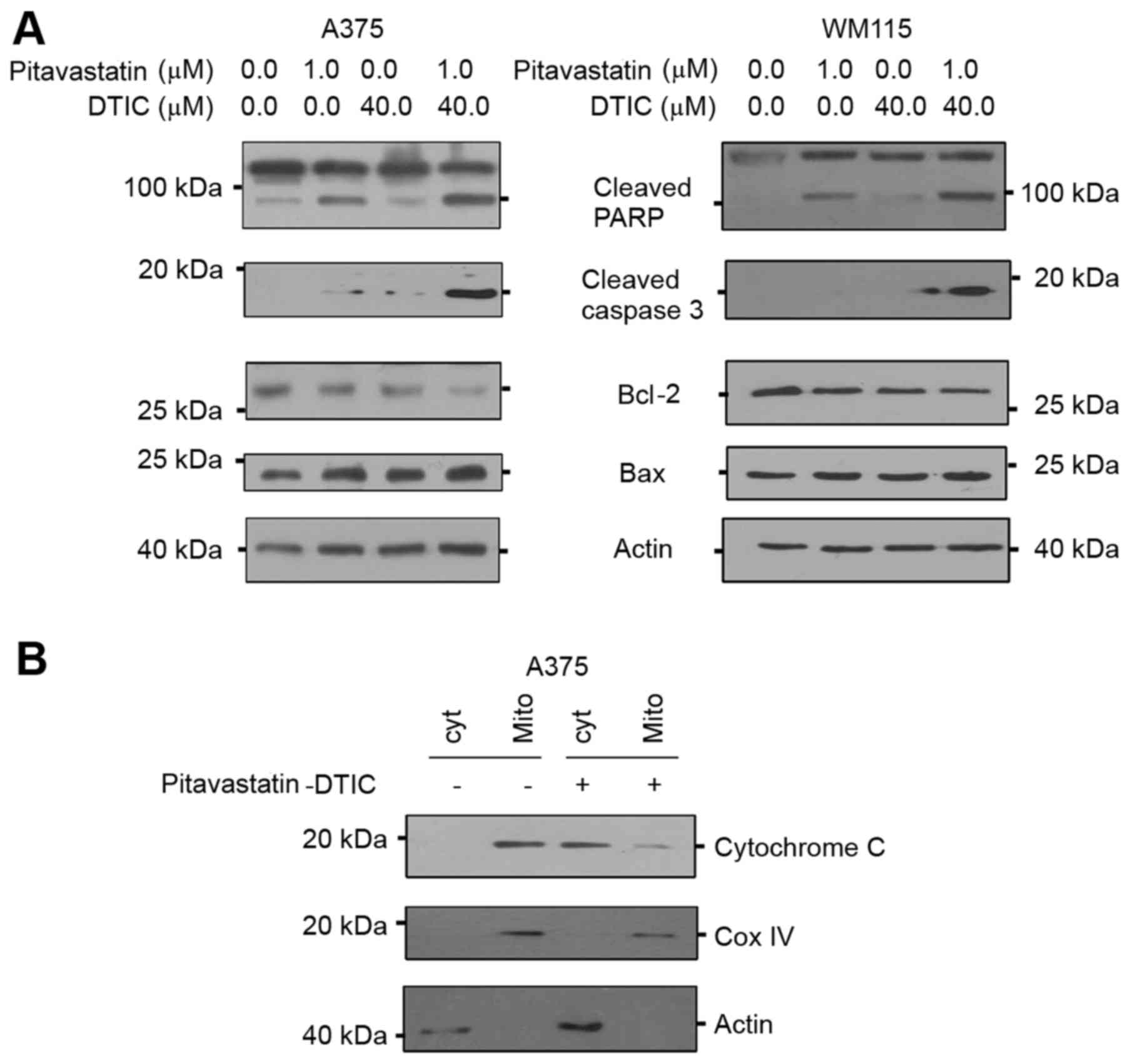 | Figure 3.Combined treatment of
pitavastatin-DTIC activates intrinsic apoptosis. (A) A375 and WM115
cells were treated with either vehicle, 1.0 µM pitavastatin, 40.0
µM DTIC or pitavastatin-DTIC (1.0 and 40.0 µM, respectively).
Protein extracts were analysed by SDS-PAGE (8–15%) and western
blotting using antibodies to cleaved PARP, active caspase 3, Bax
and Bcl-2. Actin was detected as a loading control. (B) Protein
extracts from mitochondrial and cytoplasmic fractions were prepared
from A375 cells treated with vehicle or Pitavastatin-DTIC (1.0 and
40.0 µM, respectively), resolved by SDS-PAGE (8–15%) and probed
with the anti-cytochrome c, Cox IV and actin antibodies. DTIC,
dacarbazine; CoxIV, cyclooxygenase IV; Bax, Bcl-2 associated X,
apoptosis regulator; Bcl-2, B-cell lymphoma 2; PARP, poly
(ADP-ribose) polymerase. |
Apoptosis may be activated through two main
pathways, namely the extrinsic and intrinsic pathways (21). Whilst extrinsic apoptosis is mediated
by death receptors and characterized by caspase 8 activation
(22), intrinsic apoptosis is
mitochondria mediated and usually triggered by intracellular
signals including hypoxia and DNA damage (23). During intrinsic apoptosis, the
overexpression of pro-apoptotic Bcl-2 proteins disrupts the
mitochondrial membrane and eventually leads to the release of
cytochrome c (24). In order to
determine whether combined Pitavastatin-DTIC treatment activated
either one of the apoptotic pathways, levels of intrinsic and
extrinsic apoptotic molecular markers were measured by western
blotting. The results of the present study demonstrate that
combined pitavastatin-DTIC treatment did not activate the
pro-apoptotic factor caspase 8, suggesting that the extrinsic
apoptotic pathway is not activated (data not shown). On the other
hand, combined pitavastatin-DTIC treatment induced expression of
the intrinsic pro-apoptotic factor Bax and decreased expression of
the anti-apoptotic protein Bcl-2. Furthermore, Fig. 3B demonstrates that pitavastatin-DTIC
treatment led to a notable increase in cytoplasmic cytochrome c
protein levels and a notable decrease in mitochondrial cytochrome c
protein level in A375 cells. Cytochrome oxidase IV was used as a
mitochondrial marker and actin was detected as a marker for
cytoplasmic fraction. Collectively, the results of the present
study demonstrate that combined pitavastatin-DTIC treatment induces
cell death through intrinsic apoptosis in melanoma cells.
Combined pitavastatin-DTIC treatment
induces autophagy
Increasing evidence has revealed that autophagy is
activated in response to statins (25) and large vacuoles indicative of
autophagy were observed in A375 and WM115 cells treated with
pitavastatin alone or and pitavastatin-DTIC combined (data not
shown). In order to explore this, protein extracts from cells
treated with pitavastatin, DTIC, pitavastatin-DTIC or vehicle were
assessed for LC3II, a molecular marker of autophagy, by western
blotting. Fig. 4A demonstrated that
pitavastatin alone or in combination with DTIC induces high levels
of LC3II. In order to further confirm that combined
pitavastatin-DTIC treatment induces autophagy, A375 and WM115 cells
were transiently transfected with a GFP-LC3 expression vector and
autophagosome formation was monitored by confocal microscopy. As a
result, the combined treatment of pitavastatin-DTIC led to a
significant accumulation of GFP-LC3 puncta (Fig. 4B).
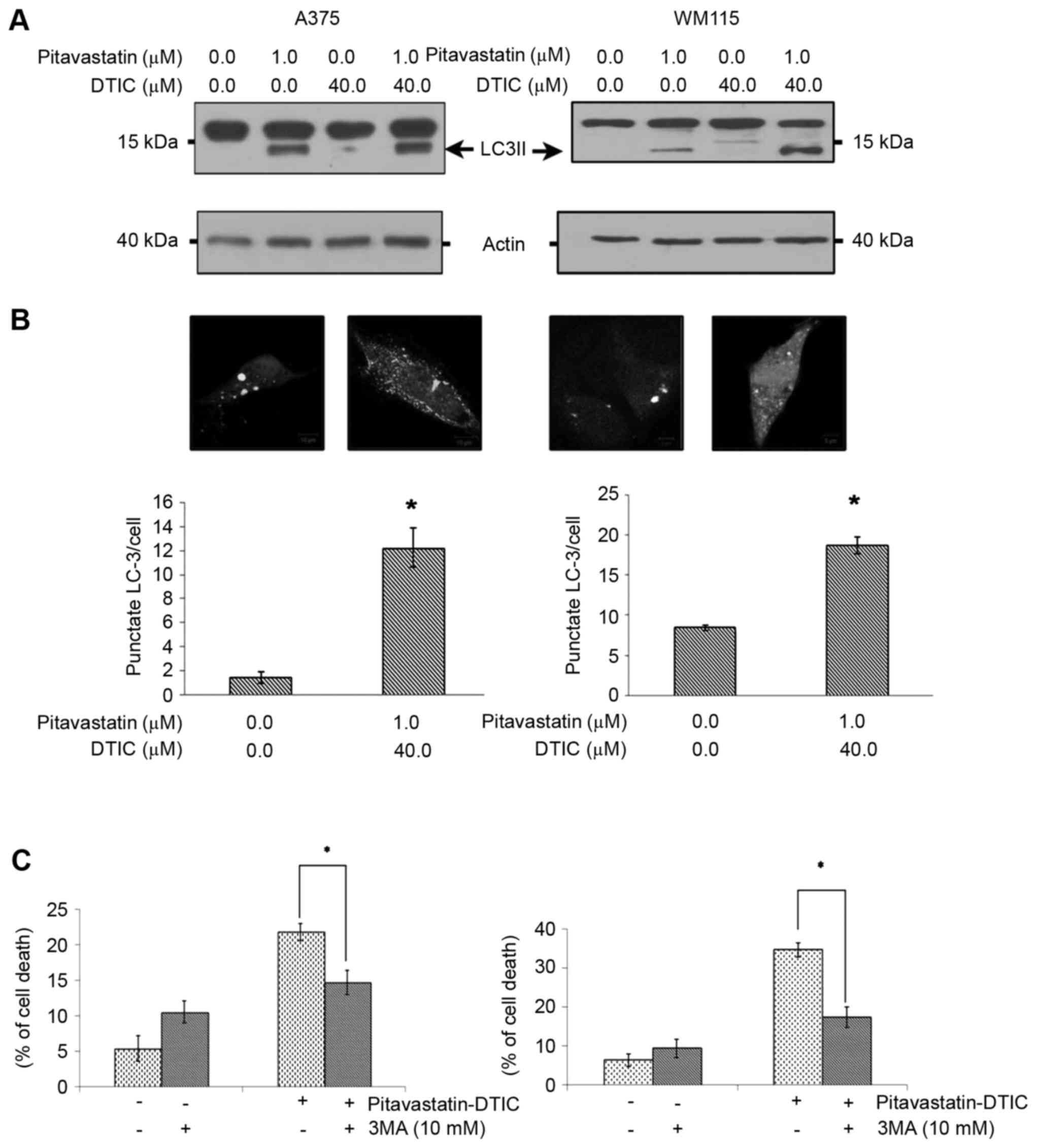 | Figure 4.Combined pitavastatin-DTIC treatment
induces autophagy. (A) A375 and WM115 cells were treated with
either vehicle, 1.0 µM pitavastatin, 40.0 µM DTIC or
Pitavastatin-DTIC (1.0 and 40.0 µM, respectively). Protein extracts
were analysed using SDS-PAGE (8–15%) and western blotting using
antibodies against LC3II and actin was used as a loading control.
(B) A375 and WM115 cells were transiently transfected with a
GFP-LC3 plasmid and treated with vehicle or pitavastatin-DTIC (1.0
and 40.0 µM, respectively). Autophagy was quantified by counting
the number of GFP-LC3 puncta in twenty fields of view at
magnification, ×400, and dividing by the total number of
transfected cells within these fields. The number of GFP-LC3
puncta/cell are presented in the graphs as the mean ± standard
error of the mean of three independent experiments (*P<0.001 as
compared with vehicle, Student's t-test). (C) Annexin V/propidium
iodide double staining assay presenting the percentage of cell
death induced by pitavastatin-DTIC when autophagy was inhibited by
10 mM 3-methyladenine and cells treated with pitavastatin-DTIC.
(*P<0.05 with comparisons indicated by lines, Student's t-test).
DTIC, dacarbazine; GFP, green fluorescence protein; LC3II,
LC3-phosphatidylethanolamine conjugate; LC3, light chain 3. |
Whilst previous studies have suggested that
chemotherapy may induce cell death via apoptosis and autophagy
(26), other studies have
demonstrated that this may induce autophagy, which attenuates
apoptosis leading to drug resistance (27). Therefore, the present study examined,
using Annexin V assays, whether the pitavastatin-DTIC-induced
autophagy is a cell death or a cell survival mechanism. Fig. 4C demonstrates that pharmacological
inhibition of autophagy, through the use of 3MA, significantly
reduced the cytotoxicity and the total cell death induced by
combined pitavastatin-DTIC treatment. Taken together these
observations suggest that pitavastatin-DTIC induced autophagy
favours cell death and contributes to cytotoxicity.
Discussion
Malignant melanoma incidence is rapidly increasing
and has almost doubled in the previous decade (2). Whilst DTIC chemotherapy is the standard
treatment, <20% of patients respond at all to the treatment and
<5% continue to respond to long-term treatment (28,29).
Previous studies have demonstrated that, in the case of melanoma,
resistance to DTIC is associated with low levels of apoptosis and
increasing levels of anti-apoptotic proteins (19,29,30).
Statins, which are anti-lipid agents, have been
demonstrated to exert anti-proliferative and anti-cancer effect
against a range of types of tumour (9,15,31). The present study therefore explored
the in vitro efficacy of a combined treatment of DTIC with
pitavastatin in human melanoma. The results of the present study
provide several lines of evidence to suggest that pitavastatin may
synergistically enhance the anti-cancer effects of DTIC. The
half-maximal inhibitory concentration (IC50) of DTIC in
several melanoma cell lines, including A375, A875, SB2, MeWo,
B16-F1 and SK-MEL-5, has been demonstrated to be relatively high at
>100 µM (5,32). Similar results were also obtained from
the present study, where the IC50 of DTIC in the A375
and WM115 cell lines was >100 µM. On the other hand,
pitavastatin has also been demonstrated to be cytotoxic on glioma,
breast, myeloma and colon cancer with IC50 values of
<10 µM (9,10,15).
Whilst previous studies have not described the cytotoxic effect of
pitavastatin on melanoma cells, the present study demonstrates that
pitavastatin is cytotoxic in melanoma cells and treatment with 4 µM
pitavastatin results in ~50% cell death. The present study further
demonstrated that melanoma cells pre-treated with pitavastatin (1
µM) are sensitised to DTIC (40 µM) resulting in ~50% cell
death.
Furthermore, the present study revealed that the
mechanism of action by which pitavastatin-DTIC combined treatment
inhibits melanoma growth involves cell cycle arrest, apoptosis and
autophagy. Previous studies have indicated that high doses of DTIC
(2 mM) result in the death of cancer cells through apoptosis
(33). In addition, in uveal melanoma
cell lines, DTIC treatment (5 µg/ml) primarily led to cell cycle
arrest in the G1 phase (34). Another
previous study demonstrated that DTIC (1 mM) induces S-phase cell
cycle arrest in A375 cells and results in a slight increase in the
sub-G1 peak (35). The same previous
study demonstrated that melanoma cells exposed to parthenolide, a
sesquiterpene lactone derived from the leaves of feverfew
(Tanacetum parthenium), combined with 2 mM DTIC accumulated in the
G2/M phase. Notably, pitavastatin was also demonstrated to inhibit
the proliferation of human U937 monocytic tumour cells in a
dose-dependent manner and to induce S-phase cell cycle arrest
(14). According to the same previous
study, pitavastatin upregulated p21 protein levels but did not
affect the expression levels of cyclin D1. The results of the
present study reveal that whilst cells treated with DTIC (40 µM)
demonstrated slight alterations to the cell cycle profile, combined
pitavastatin-DTIC treated cells accumulated in the G1 phase as
evidenced by flow cytometry and cell cycle regulator analyses. The
results of the present study suggest that the induction of cell
cycle arrest by DTIC may be concentration dependent and that
pitavastatin enhances the ability of DTIC to halt cell cycle
progression. Furthermore, the present study demonstrated that the
cytotoxic effect of pitavastatin-DTIC includes the induction of
apoptosis. Notably, whilst DTIC treatment (40 µM) did not induce
apoptosis, combined pitavastatin-DTIC treatment resulted in
significant levels of apoptosis as evidenced by the increased level
of apoptosis markers, cleaved PARP and active caspase 3, as well as
annexin V staining. The results of the present study are in
agreement with a number of studies which demonstrate that high
concentrations of DTIC are necessary to induce apoptosis in
melanoma cells (19,32,36).
Whilst there has been insufficient research to determine which type
of apoptosis is induced by DTIC, sensitization to the intrinsic
apoptotic pathway has been revealed to augment DTIC-induced
melanoma tumour cell death (37). In
accordance with these observations, the results of the present
study suggest that pitavastatin treatment may enhance DTIC
cytotoxicity through augmentation of the intrinsic apoptosis
pathway.
Furthermore, the results of the present study
demonstrated that melanoma cells treated with combined
pitavastatin-DTIC expressed a high level of L3II, a marker of
autophagy. Chemical inhibition of autophagy resulted in enhancement
of cell viability, suggesting that pitavastatin-DTIC-induced
autophagy occurs as a cell death mechanism. In support of this,
previous studies have suggested that DTIC and pitavastatin may
serve a function in the induction of autophagy and specifically as
a mode of cell death (15,19,38).
In summary, results from the present study suggest
that the combined treatment of pitavastatin-DTIC provides a
synergistic anti-cancer effect through apoptosis and autophagy, and
this should be further examined for melanoma treatment.
Acknowledgements
The present study was supported by the Qatar Charity
under the Ibhath project for research grants, which is funded by
the Cooperation Council for the Arab States of the Gulf through the
Islamic Development Bank. The authors would like to thank Dr Aretha
Cooper for her critical reading and comments on this
manuscript.
References
|
1
|
Bhatia S, Tykodi SS and Thompson JA:
Treatment of metastatic melanoma: An overview. Oncology (Williston
Park). 23:488–496. 2009.PubMed/NCBI
|
|
2
|
Li W and Melton DW: Cisplatin regulates
the MAPK kinase pathway to induce increased expression of DNA
repair gene ERCC1 and increase melanoma chemoresistance. Oncogene.
31:2412–2422. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lui P, Cashin R, Machado M, Hemels M,
Corey-Lisle PK and Einarson TR: Treatments for metastatic melanoma:
Synthesis of evidence from randomized trials. Cancer Treat Rev.
33:665–680. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bedikian AY, Millward M, Pehamberger H,
Conry R, Gore M, Trefzer U, Pavlick AC, DeConti R, Hersh EM, Hersey
P, et al: Bcl-2 antisense (oblimersen sodium) plus dacarbazine in
patients with advanced melanoma: The Oblimersen Melanoma Study
Group. J Clin Oncol. 24:4738–4745. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lev DC, Onn A, Melinkova VO, Miller C,
Stone V, Ruiz M, McGary EC, Ananthaswamy HN, Price JE and Bar-Eli
M: Exposure of melanoma cells to dacarbazine results in enhanced
tumor growth and metastasis in vivo. J Clin Oncol. 22:2092–2100.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gopalan A, Yu W, Sanders BG and Kline K:
Simvastatin inhibition of mevalonate pathway induces apoptosis in
human breast cancer cells via activation of JNK/CHOP/DR5 signaling
pathway. Cancer Lett. 329:9–16. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Warita K, Warita T, Beckwitt CH, Schurdak
ME, Vazquez A, Wells A and Oltvai ZN: Statin-induced mevalonate
pathway inhibition attenuates the growth of mesenchymal-like cancer
cells that lack functional E-cadherin mediated cell cohesion. Sci
Rep. 4:75932014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Simon A: Cholesterol metabolism and
immunity. N Engl J Med. 371:1933–1935. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tu YS, Kang XL, Zhou JG, Lv XF, Tang YB
and Guan YY: Involvement of Chk1-Cdc25A-cyclin A/CDk2 pathway in
simvastatin induced S-phase cell cycle arrest and apoptosis in
multiple myeloma cells. Eur J Pharmacol. 670:356–364. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee YP, Wang CW, Liao WC, Yang CR, Yeh CT,
Tsai CH, Yang CC and Tzeng YM: In vitro and in vivo anticancer
effects of mevalonate pathway modulation on human cancer cells.
Anticancer Res. 32:2735–2745. 2012.PubMed/NCBI
|
|
11
|
McFarland AJ, Anoopkumar-Dukie S, Arora
DS, Grant GD, McDermott CM, Perkins AV and Davey AK: Molecular
mechanisms underlying the effects of statins in the central nervous
system. Int J Mol Sci. 15:20607–20637. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garwood ER, Kumar AS, Baehner FL, Moore
DH, Au A, Hylton N, Flowers CI, Garber J, Lesnikoski BA, Hwang ES,
et al: Fluvastatin reduces proliferation and increases apoptosis in
women with high grade breast cancer. Breast Cancer Res Treat.
119:137–144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang J, Tokoro T, Higa S and Kitajima I:
Anti-inflammatory effect of pitavastatin on NF-kappaB activated by
TNF-alpha in hepatocellular carcinoma cells. Biol Pharm Bull.
29:634–639. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fujino M, Miura S, Matsuo Y, Tanigawa H,
Kawamura A and Saku K: Pitavastatin-induced downregulation of CCR2
and CCR5 in monocytes is associated with the arrest of cell-cycle
in S phase. Atherosclerosis. 187:301–308. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tsuboi Y, Kurimoto M, Nagai S, Hayakawa Y,
Kamiyama H, Hayashi N, Kitajima I and Endo S: Induction of
autophagic cell death and radiosensitization by the pharmacological
inhibition of nuclear factor-kappa B activation in human glioma
cell lines. J Neuosurg. 110:594–604. 2009. View Article : Google Scholar
|
|
16
|
Aliwaini S, Swarts AJ, Blanckenberg A,
Mapolie S and Prince S: A novel binuclear palladacycle complex
inhibits melanoma growth in vitro and in vivo through apoptosis and
autophagy. Biochem Pharmacol. 86:1650–1663. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bagnoli M, Balladore E, Luison E, Alberti
P, Raspagliesi F, Marcomini B, Canevari S and Mezzanzanica D:
Sensitization of p53-mutated epithelial ovarian cancer to
CD95-mediated apoptosis is synergistically induced by cisplatin
pretreatment. Mol Cancer Ther. 6:762–772. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006.Bedia C, Casas J, Andrieu-Abadie N, Fabriàs G
and Levade T: Acid ceramidase expression modulates the sensitivity
of A375 melanoma cells to dacarbazine. J Biol Chem 286:
28200–28209, 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bedia C, Casas J, Andrieu-Abadie N,
Fabriàs G and Levade T: Acid ceramidase expression modulates the
sensitivity of A375 melanoma cells to dacarbazine. J Biol Chem.
286:28200–28209. 2011.Kim YJ, Brox T, Feiden W and Weickert J:
Fully automated segmentation and morphometrical analysis of muscle
fiber images. Cytometry A 71: 8–15, 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim YJ, Brox T, Feiden W and Weickert J:
Fully automated segmentation and morphometrical analysis of muscle
fiber images. Cytometry A. 71:8–15. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ghatage DD, Gosavi SR, Ganvir SM and
Hazarey VK: Apoptosis: Molecular mechanism. J Orofac Sci.
4:103–107. 2012. View Article : Google Scholar
|
|
23
|
Surova O and Zhivotovsky B: Various modes
of cell death induced by DNA damage. Oncogene. 32:3789–3797. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kang MH and Reynolds CP: Bcl-2 inhibitors:
Targeting mitochondrial apoptotic pathways in cancer therapy. Clin
Cancer Res. 15:1126–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: Therapeutic implications. Mol
Cancer Ther. 10:1533–1534. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tomic T, Botton T, Cerezo M, Robert G,
Luciano F, Puissant A, Gounon P, Allegra M, Bertolotto C, Bereder
JM, et al: Metformin inhibits melanoma development through
autophagy and apoptosis mechanisms. Cell Death Dis. 2:e1992011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zeng Y, Yang X, Wang J, Fan J, Kong Q and
Yu X: Aristolochic acid I induced autophagy extenuates cell
apoptosis via ERK 1/2 pathway in renal tubular epithelial cells.
PLoS One. 7:e303122012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Patel PM, Suciu S, Mortier L, Kruit WH,
Robert C, Schadendorf D, Trefzer U, Punt CJ, Dummer R, Davidson N,
et al: Extended schedule, escalated dose temozolomide versus
dacarbazine in stage IV melanoma: Final results of a randomised
phase III study (EORTC 18032). Eur J Cancer. 47:1476–1483. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Risberg K, Fodstad O and Andersson Y:
Anti-melanoma activity of the 9.2.27PE immunotoxin in dacarbazine
resistant cells. J Immunother. 33:272–278. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fuller TL and Canada RG: Enhancement of
cisplatin cytotoxicity by terbium in cisplatin-resistant MDA/CH
human breast cancer cells. Cancer Chemother Pharmacol. 44:249–252.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang PM and Chen CC: Life or death?
Autophagy in anticancer therapies with statins and histone
deacetylase inhibitors. Autophagy. 7:107–108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jin JL, Gong J, Yin TJ, Lu YJ, Xia JJ, Xie
YY, Di Y, He L, Guo JL, Sun J, et al: PTD4-apoptin protein and
dacarbazine show a synergistic antitumor effect on B16-F1 melanoma
in vitro and in vivo. Eur J Pharmacol. 654:17–25. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sanada M, Hidaka M, Takagi Y, Takano TY,
Nakatsu Y, Tsuzuki T and Sekiguchi M: Modes of actions of two types
of anti-neoplastic drugs, dacarbazine and ACNU, to induce
apoptosis. Carcinogenesis. 28:2657–2663. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cun B, Song X, Jia R, Zhao X, Wang H, Ge S
and Fan X: Combination of oncolytic adenovirus and dacarbazine
enhances antitumor ability against uveal melanoma cells via cell
cycle block. Cancer Biol Ther. 13:77–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Koprowska K, Hartman ML, Sztiller-Sikorska
M and Czyz ME: Parthenolide enhances dacarbazine activity against
melanoma cells. Cancer Biol. 24:835–845. 2013.
|
|
36
|
Ishibashi M, Arai M, Tanaka S, Onda K and
Hirano T: Antiproliferative and apoptosis-inducing effects of
lipophilic vitamins on human melanoma A375 cells in vitro. Biol
Pharm Bull. 35:10–17. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Anvekar RA, Asciolla JJ, Lopez-Rivera E,
Floros KV, Izadmehr S, Elkholi R, Belbin G, Sikora AG and Chipuk
JE: Sensitization to the mitochondrial pathway of apoptosis
augments melanoma tumor cell responses to conventional
chemotherapeutic regimens. Cell Death Dis. 3:e4202012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Aliwaini S, Peres J, Kröger WL,
Blanckenberg A, de la Mare J, Edkins AL, Mapolie S and Prince S:
The palladacycle, AJ-5, exhibits anti-tumour and anti-cancer stem
cell activity in breast cancer cells. Cancer Lett. 357:206–218.
2015. View Article : Google Scholar : PubMed/NCBI
|