Introduction
Estradiol (E2) has an essential role in
the development and progression of estrogen receptor (ER)-positive
breast cancer (1,2). Therefore, the use of aromatase
inhibitors (AIs), including letrozole, anastrozole and exemestane,
as adjuvants is regarded as a standard approach in postmenopausal
women with ER-positive breast cancer (3–5). However,
certain patients with breast cancer develop resistance to AIs
following long-term treatment (6).
Previous studies have revealed crosstalk between the activation of
the insulin-like growth factor-1 (IGF-I) signaling pathway and ERα
in long-term AI-treated breast cancer cells (7,8). One
mechanism of AI resistance is aberrant signaling through the
phosphatidylinositol 3-kinase (PI3K)/RAC serine/threonine-protein
kinase (Akt)/mechanistic target of rapamycin (mTOR) signaling
pathway (8,9) (Fig.
1A).
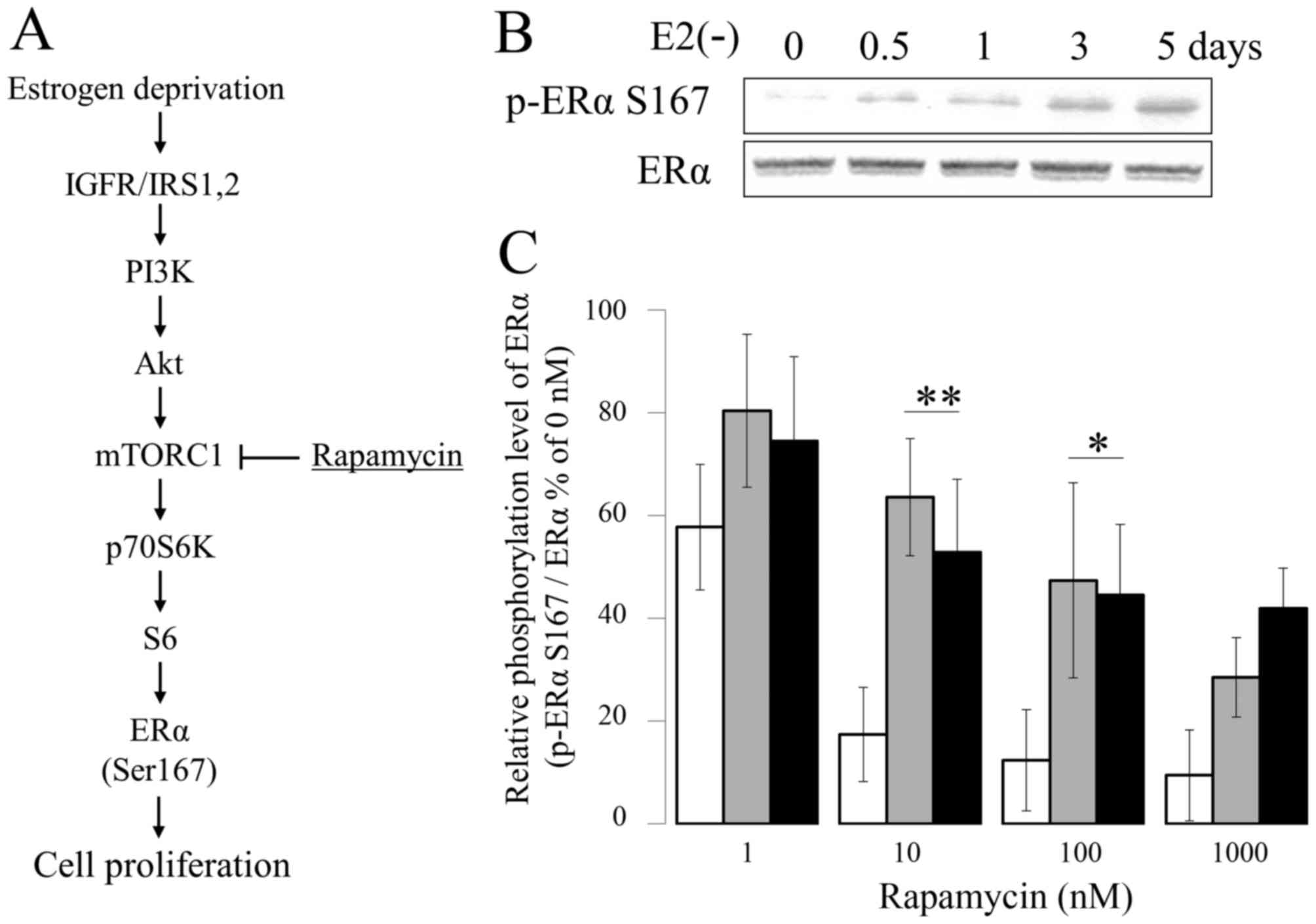 | Figure 1.Suppressive effect of Eve on ERα
Ser167 phosphorylation. (A) Schematic diagram of activated PI3K/Akt
pathway in E2-deprivation breast cancer. (B)
Phosphorylation of ERα Ser167 in MCF-7 cells cultured in
E2-deprived medium for 0–5 days. Calcium dependency of
the degradation of microsomal aromatase in the presence of cytosol.
MCF-7 (white bar), E2-deprived (5 days) MCF-7 (gray
bar), and LTED (black bar) MCF-7 cells were incubated with the
indicated concentration of rapamycin for 1 h (n=4). *P<0.01.
Eve, everolimus; ERα, estrogen receptor-α; E2,
estradiol; LTED, long-term E2-deprived; PI3K,
phosphoinositide 3-kinase; Akt, RAC serine/threonine-protein
kinase; mTORC1, mechanistic target of rapamycin complex 1; IGFR,
insulin-like growth factor receptor; IRS-1, insulin receptor
substrate 1. |
Accordingly, the interruption of PI3K/Akt/mTOR
signaling has been demonstrated in preclinical
E2-deprivation resistance models, in which an mTOR
inhibitor in combination with exemestane led to abrogation of
proliferation, induction of apoptosis and enhanced tumor regression
(10). A substrate of mTOR complex 1,
S6 kinase 1 (S6K), phosphorylates activation function domain 1 of
ERα, which is responsible for ligand-independent receptor
activation (7,8,11).
IGF-1-dependent activation of ERα was proposed as the reason for AI
resistance, and the role of S6 K was elucidated in previous studies
(7,12). Abnormal activation of ERα is dependent
on the phosphorylation of Ser104, Ser106, Ser118 and Ser167,
located in the amino terminal A/B domain of ERα (13,14). The
phosphorylation level of proteins is determined by the activity and
balance of protein kinases, and phosphatases. Using the phosphatase
inhibitor okadaic acid (OA) (15,16), a
previous study demonstrated that serine/threonine-protein
phosphatase 2A (PP2A) has an important role in the regulation of
ERα Ser167 phosphorylation and in the proliferation of MCF-7 cells
(17).
PP2A is a key tumor suppressor that regulates
signaling pathways relevant to a number of types of human cancer
(18,19). PP2A is a ubiquitously expressed member
of a phosphoserine- and phosphothreonine-specific protein
phosphatase family involved in the regulation of cell
proliferation, cell differentiation, RNA transcription, DNA repair
and apoptosis (20–22). As inhibition of its activity and loss
of certain functional subunits are characteristics of neoplastic
transformation, PP2A is widely designated as a tumor suppressor
(23). Forskolin (FSK) lacks
adenylate cyclase-activating function but retains the ability to
activate PP2A, which is necessary for growth inhibition and
induction of apoptosis induction in leukemic cells (23).
In the present study, E2 depletion
decreased PP2A expression and reduced the susceptibility of MCF-7
cells to mTOR inhibitors. Furthermore, activation of PP2A by FSK
enhanced the effect of everolimus (Eve) and strongly inhibited
long-term E2-deprived (LTED) cell proliferation.
Materials and methods
Cell culture
Human ER-positive breast cancer MCF-7 cells
(American Type Culture Collection., Manassas, VA, USA) were
maintained in RPMI 1640 medium (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum
(FBS; Nichirei Biosciences, Inc., Tokyo, Japan) and 1%
penicillin/streptomycin at 37°C in a 5% CO2-humidified
atmosphere incubator. Cells treated with 17β-estradiol
(E2) (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany),
Phos STOP (Sigma-Aldrich; Merck KGaA), OA, calyculin A (CalA),
rapamycin and Eve (Wako Pure Chemical Industries, Ltd., Osaka,
Japan) in Dimethyl sulfoxide (DMSO; Wako Pure Chemical Industries,
Ltd.) were cultured in phenol-red-free RPMI 1640 medium (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10%
dextran-coated charcoal (DCC)-treated FBS (Nichirei Biosciences,
Inc.) and 1% penicillin/streptomycin. MCF-7 cells cultured in
phenol-red-free RPMI 1640 with 10% dextran-coated charcoal
(DCC)-treated FBS and 10 nM E2 and then for 5 days
without E2 (MCF-7 5d) and 6 months without E2
(LTED) were used in the experiment. LTED cells modeling AIs
resistance were derived from a parental cell line by long-term
culture in the presence of RPMI 1640 medium containing 10%
DCC-treated FBS, as described previously (12,24,25). MCF-7
cells were cultured with E2 (10 nM), OA (100 nM), Cal A (1 nM),
FK506 (10 nM), or DMSO (0.1%, vehicle) in phenol red-free RPMI 1640
medium supplemented with 10% dextran-coated charcoal fetal bovine
serum for 5 days at 37°C. The cell viability of cultured cells was
determined using Cell Counting kit-8 (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) according to the
manufacturer's protocol.
Western blot analysis
Whole-cell lysates were collected using lysis buffer
[containing 62.5 mM Tris HCl pH 6.8, 5% 2-mercaptoethanol, 2%
sodium dodecyl sulfate, 5% sucrose and 0.01% Bromophenol Blue (Wako
Pure Chemical Industries, Ltd.)]. The protein content was
subsequently determined using a RC DC™ Protein Assay
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) with bovine serum
albumin (Sigma-Aldrich; Merck KGaA) as the standard. For western
blot analysis, solubilized proteins (5 µg of protein/lane) were
separated by 10% SDS-PAGE and transferred to a polyvinylidene
difluoride membrane (GE Healthcare, Chicago, IL, USA). Membranes
were pre-incubated with ImmunoBlock (DS Pharma Biomedical Co., Ltd.
Osaka, Japan) as a blocking regent at room temperature for 30 min
and then incubated at 4°C overnight with antibodies at 1:1,000
dilution directed against Akt (cat. no. 9272S) and phosphorylated
Akt Ser473 (cat. no. 4060S); Cell Signaling Technology, Inc.,
Danvers, MA, USA, ERα (cat. no. sc-543), phosphorylated ERα Ser167
(cat. no. sc-101676), and ERα Ser118 (cat. no. sc-101675), or a
b-actin antibody (cat. no. sc-47778; Santa Cruz Biotechnology,
Inc., Santa Cruz, California, USA). The membrane was subsequently
washed with TBS-Tween 20 (TBS-T) buffer (20 mmol/l Tris-HCl (pH
7.5), 150 mmol/l NaCl, 0.5% Tween-20) and incubated with a
horseradish peroxidase-labeled secondary anti-rabbit (cat. no.
170-6515; Bio-Rad Laboratories, Inc., Hercules, CA, USA) or
anti-mouse (cat. no. 330; MBL, Nagoya, Japan) IgG antibody for 1 h
at room temperature. All antibodies were diluted in Can Get Signal
Immunoreaction Enhancer solution (cat. no. NKB-101; Toyobo Life
Science, Osaka, Japan). Once the membrane was washed with TBS-T
buffer, immunoreactive bands were visualized using Immobilon
Western Chemiluminescent HRP substrate (EMD Millipore, Billerica,
MA, USA). The intensity of the chemiluminescence of specific bands
was digitized using Cool Saver software version 1.2 (ATTO
Corporation, Tokyo, Japan) and quantified.
Statistical analysis
All experimental data comparing more than two groups
were analyzed by one-way analysis of variance followed by Fisher's
protected least significant difference test. The software used for
statistical analyses was SPSS v24 (IBM SPSS, Armonk, NY, USA). When
differences were significant, subsequent analyses with post hoc
t-tests with Bonferroni correction were performed. Other
statistical comparisons were conducted by a two-tailed unpaired
t-test. Data are presented as the mean ± standard deviation.
P<0.05 was considered to indicate a statistically significant
difference.
Results
17β-estradiol depletion reduces the
sensitivity to mTOR inhibitor treatment
MCF-7 cells have previously been used as a model for
the study of the E2 response in vitro (26,27). In
vitro studies using E2 deprivation or chronic
exposure to anti-E2 have led to the isolation of hormone
therapy-resistant variants of MCF-7 cells (12,24,25). LTED
cells serve as a model of AIs-resistant breast cancer, and have
been generated by several laboratories (25). When MCF-7 cells were cultured in a
phenol-red-free RPMI 1640 with 10% dextran-coated charcoal
(DCC)-treated FBS medium, ERα Ser167 phosphorylation decreased in a
time-dependent manner (Fig. 1B).
Next, MCF-7, MCF-7 5d and LTED cells were evaluated for sensitivity
to mTOR inhibition. MCF-7, MCF7 5d and LTED were treated with
various amounts of the mTOR inhibitor rapamycin (concentrations of
1, 10, 100 or 1,000 nM) for 1 h at 37°C, and the number of cells
was measured with a Cell Counting kit 8. Following treatment of the
cells with 1 nM rapamycin for 1 h, phosphorylation of ERα Ser167
was determined by western blotting. The phosphorylation levels of
ERα Ser167 were ~58 and ~20% higher in cells treated with in 1, and
10 nM rapamycin, respectively, compared with that in
vehicle-treated MCF-7 control cells (Fig.
1C). By contrast, following culturing in the presence of 1,000
nM rapamycin for 1 h, the intracellular phosphorylation level of
ERα Ser167 in LTED cells decreased to ~50% of that observed in
vehicle-treated control cells (Fig.
1C).
PP2A inhibition leads to resistance to
E2 depletion via ERα Ser167 phosphorylation
Protein phosphorylation status is determined by the
balance between phosphorylation and dephosphorylation. Previous
studies have revealed that the mechanism of endocrine resistance
involves aberrant signaling through the PI3K/Akt/mTOR signaling
pathway (7,12). However, the identity of the
phosphatase involved in ERα phosphorylation remains unclear.
Western blot analysis was conducted using several protein
phosphatase inhibitors, which have been well characterized in
phosphorylation studies (17). At 1 h
after the addition of each inhibitor [Phos STOP (PS); protein
phosphatase inhibitor cocktail, OA and Cal A; PP2A inhibitor,
FK506; protein phosphatase type 2B inhibitor], phosphorylation of
ERα Ser167 was increased in the culture solution following PS, OA,
FK506 and Cal A treatment (Fig. 2A).
In addition, OA and Cal A treatment increased the number of cells
in the E2-free medium (Fig.
2B).
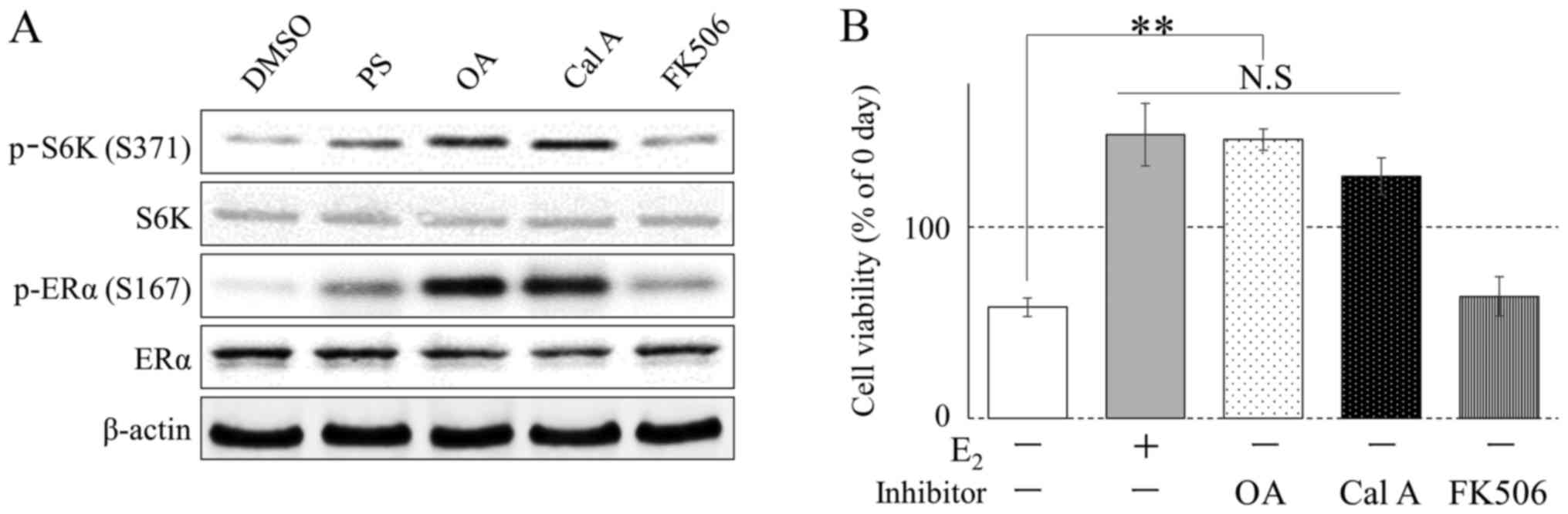 | Figure 2.Alteration of the phosphorylation
status of ERα and S6K in MCF-7 cells by phosphatase inhibitors. (A)
MCF-7 cells were incubated with PS, OA (100 nM), Cal A (1 nM),
FK506 (10 nM), or DMSO (0.1%, vehicle) in E2 (10 nM)
medium. Phosphorylation was determined by western blotting. (B)
MCF-7 cells were cultured with E2 (10 nM), OA (100 nM),
Cal A (1 nM), FK506 (10 nM), or DMSO (0.1%, vehicle) in phenol
red-free RPMI 1640 medium supplemented with 10% dextran-coated
charcoal fetal bovine serum for 5 days (n=4). Cell viability was
analyzed using Cell Counting kit 8. *P<0.01, **P<0.001. ERα,
estrogen receptor-α; DMSO, dimethyl sulfoxide; S6K, S6 kinase; OA,
okadaic acid; Cal A, calyculin A. |
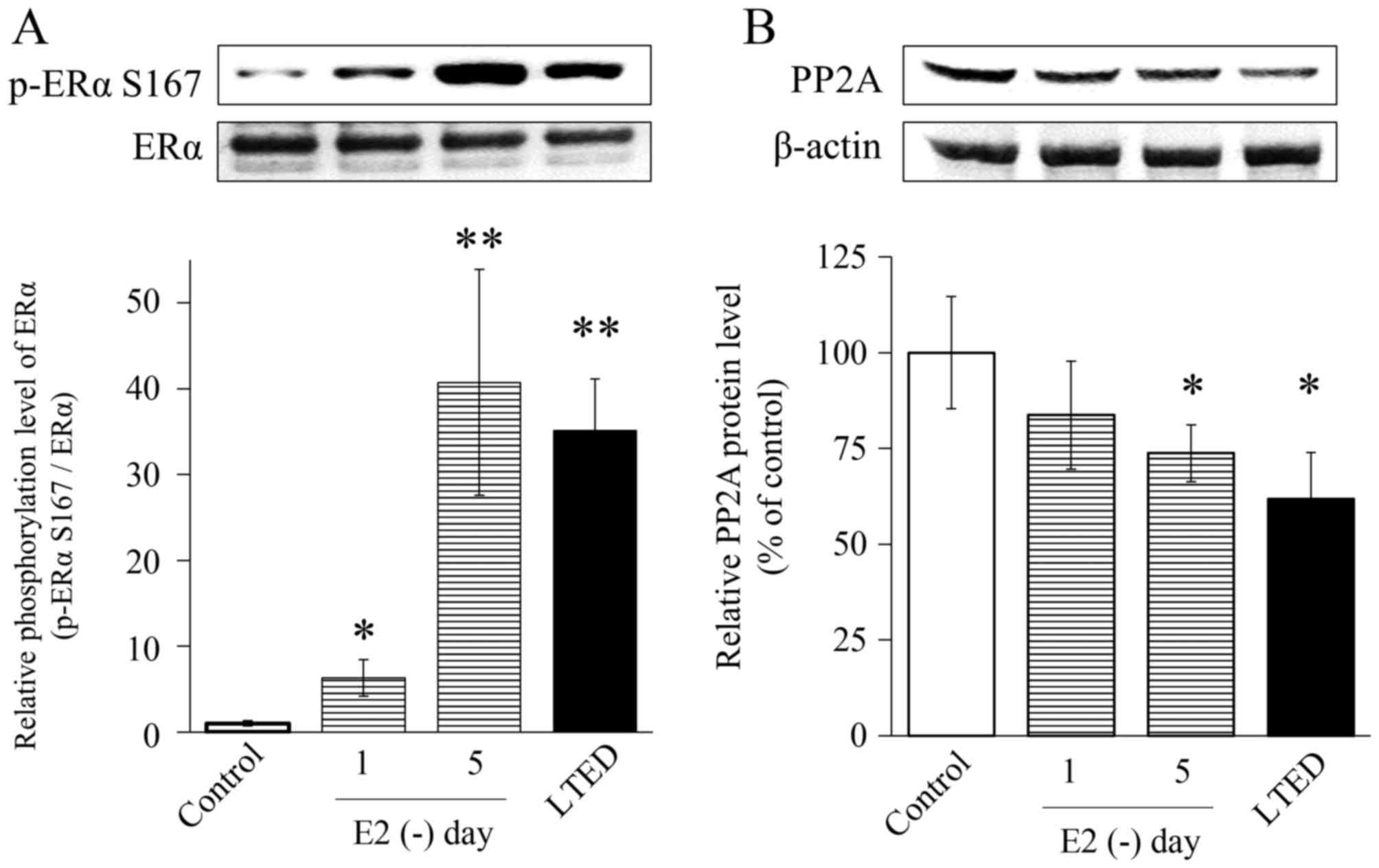
E2 deprivation reduces PP2A
levels in MCF-7 cells
PP2A is involved in endocrine therapy resistance
(28). Therefore, MCF-7 cells
cultured without steroids were examined after 1 or 5 days, which
activated mTOR. Levels of phosphorylated ERα Ser167 were analyzed
by western blotting. Phosphorylation of ERα Ser167 in LTED cells
was induced by long-term E2 deprivation in MCF-7
parental cells. ERα Ser167 phosphorylation in MCF-7 cells cultured
under E2 depletion for 1 day with LTED was increased
6-fold, whereas that in MCF-7 cells cultured under E2
depletion for 5 days with LTED increased by 35-fold or more,
relative to untreated cells (Fig.
3A). By contrast, after 1 day without E2 in the
medium, PP2A protein levels decreased to 60% of the baseline value
(Fig. 3B).
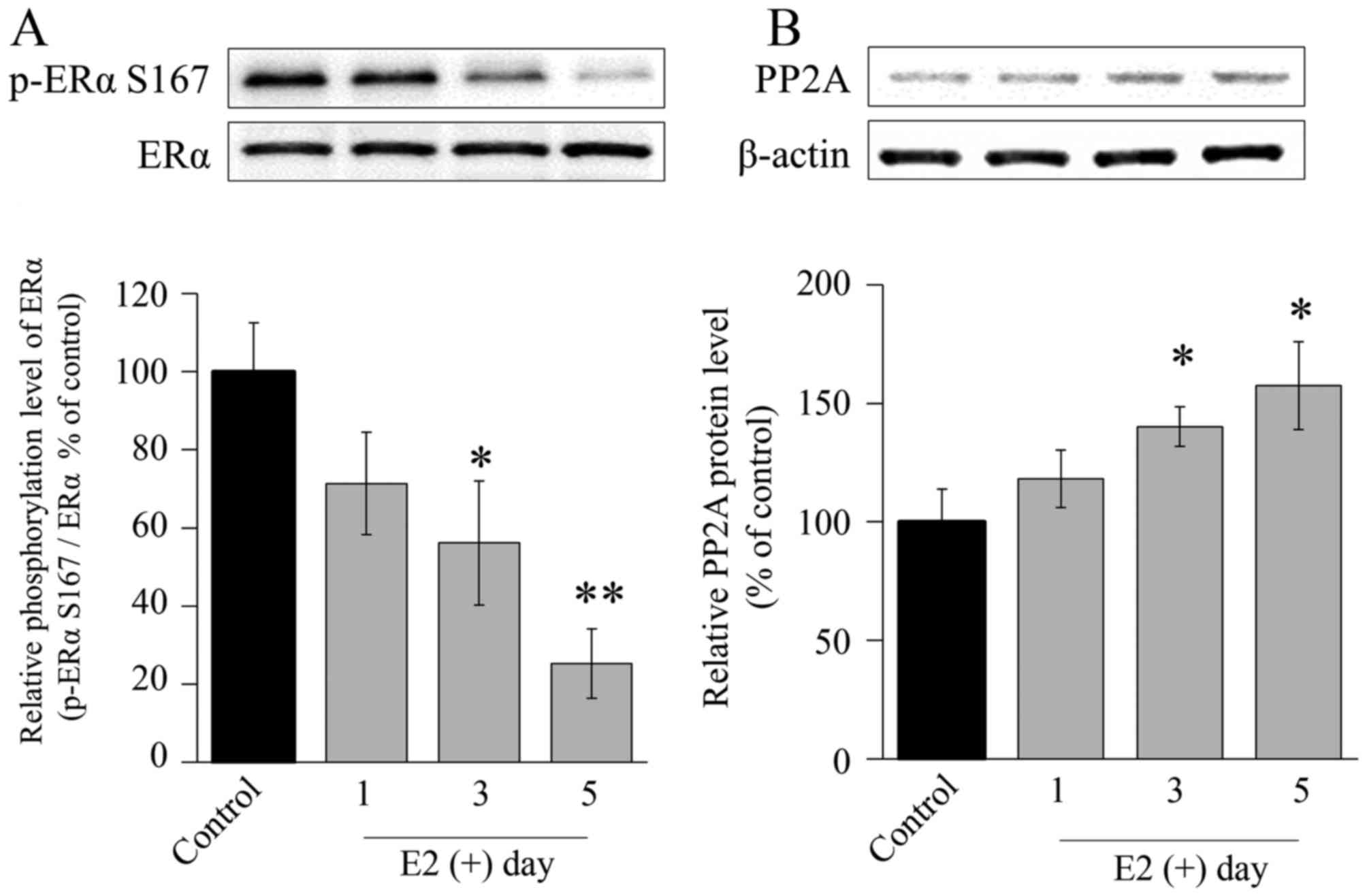
PP2A is upregulated by E2
under LTED conditions
Phosphorylation of ERα Ser167 in LTED cells was
reduced by E2 exposure, consistent with the results
observed in the parental MCF-7 cells. ERα Ser167 phosphorylation in
LTED cells was significantly decreased by exposure to E2
in a time-dependent manner (Fig. 4A).
However, 3 days after addition of E2 to the medium, PP2A
protein levels were significantly increased (Fig. 4B).
PP2A activation enhances the effect of
Eve
Following E2 treatment, PP2A expression
was increased in the medium. These results indicated that PP2A
expression was modulated by E2 and has a major role in
resistance to E2 depletion. Therefore, we hypothesized
that PP2A activation increases the effect of Eve. FSK is an
activator of PP2A. E2 induced cell growth in
ERα-positive LTED cells. To investigate the role of PP2A in this
process, the potential role of PP2A activation by FSK in cell death
was determined. LTED cells in E2-depleted medium were
treated with Eve (10 nM) and/or the PP2A activator FSK (5 mg/ml)
for 5 days. FSK and Eve significantly inhibited cell growth. In
addition, the combination of Eve and FSK significantly reduced cell
growth (Fig. 5A). E2 did
not suppress the growth of LTED cells despite increased PP2A
expression (Fig. 5A).
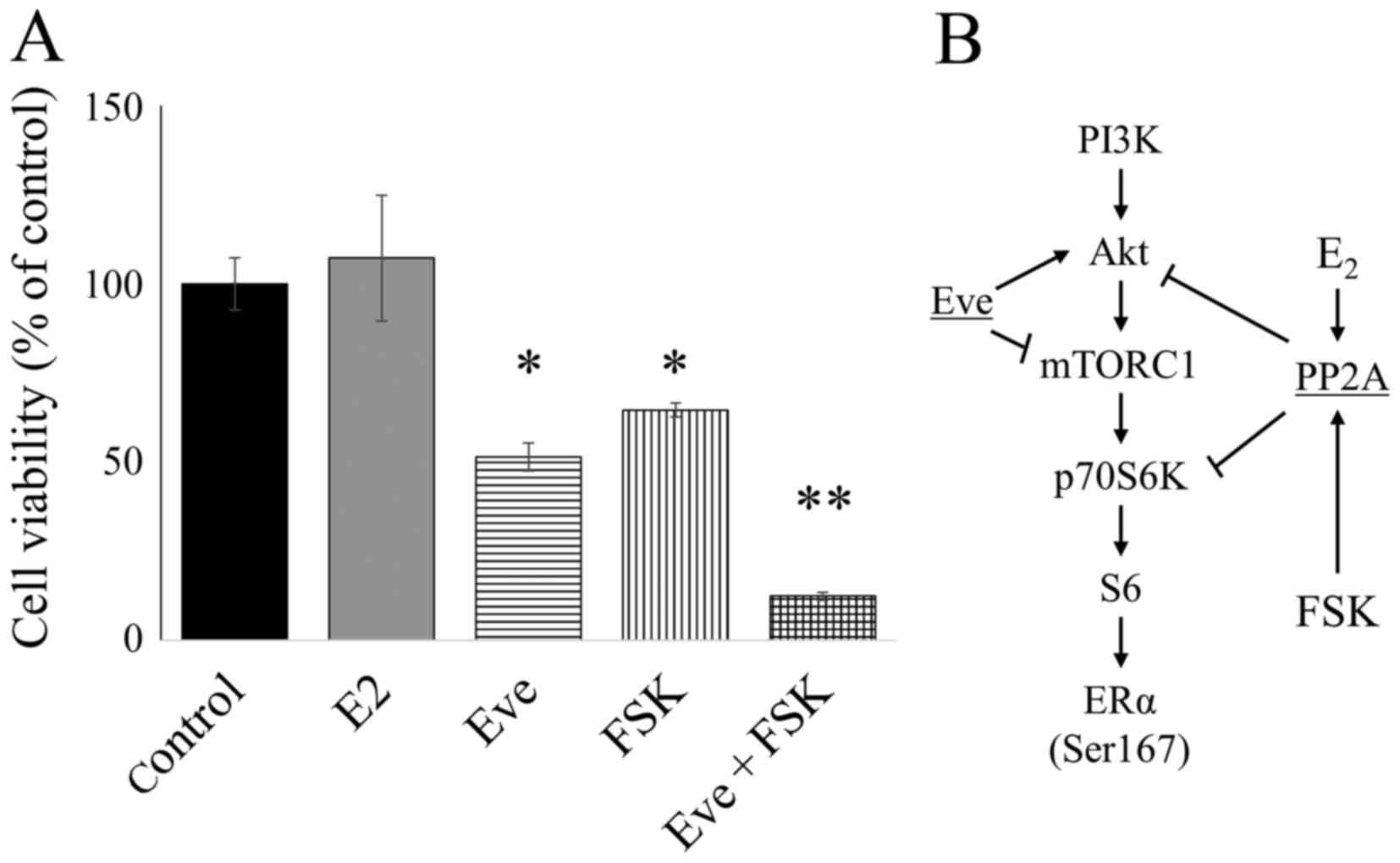 | Figure 5.Combination of Eve and FSK reduced
the viability of LTED cells. (A) Cell Counting kit-8 assay showing
the effect of E2 (10 nM), Eve (10 nM), and FSK (5
µg/ml), alone or in combination, in LTED cells; *P<0.01,
**P<0.001. (B) Schematic diagram of the signaling pathway in
E2-deprived breast cancer cells, showing the effects of
Eve, FSK and E2. Eve, everolimus; FSK, forskolin;
E2, estradiol; LTED, long-term E2-deprived;
PI3K, phosphoinositide 3-kinase; Akt, RAC serine/threonine-protein
kinase; mTORC1, mechanistic target of rapamycin complex 1. |
Discussion
Resistance to AIs is an important clinical problem
in oncology. In the present study, PP2A was demonstrated to be an
important inhibitory factor for signal activation via
phosphorylation of ERα Ser167 in breast cancer MCF-7 cells. PP2A
activation by FSK increased the appearance of LTED the effect of
Eve. On the basis of these results, it was suggested that FSK may
serve an auxiliary role in treating AIs-resistant breast
cancer.
In a typical cell, the functions of nearly one-third
of proteins are regulated via phosphorylation, and this process
controls various biological functions, including cell division,
growth, proliferation, and apoptosis (29,30).
Depending upon the physiological requirements of the cell, proteins
transiently shift from a phosphorylated to a dephosphorylated
state, with the balance controlled by protein kinases and
phosphatases (30,31). PP2A, a serine/threonine protein
phosphatase, has been previously suggested to be a tumor suppressor
protein in AIs-resistant ER-positive breast cancer cells (17,28,32). In
the present study, PP2A tumor suppressor activity was first
observed upon treatment with OA, a selective but not specific
inhibitor of PP2A, which potently promoted resistance to
E2 deprivation in MCF-7 cells. It was subsequently
demonstrated that the combination of the PP2A activator FSK and Eve
significantly decreased LTED cell viability. The only known targets
of OA are the catalytic subunits PP1 and PP2A, which are essential
components of two basic cellular functions: Growth and cell
division (31,33). Previously, co-immunoprecipitation and
in vitro pull-down assays revealed a direct association
between the PP2A-B55 holoenzyme, and Akt; the selectivity of the
holoenzyme regulates Akt Thr308 phosphorylation (34).
Our previous study indicated that inhibition of PP2A
significantly increases ERα phosphorylation (17). Furthermore, the present study
demonstrated that expression of PP2A decreased in response to
E2 depletion. As presented in Fig. 1, the responsiveness of Erα
phosphorylation to rapamycin in MCF-7 and LTED cells after 5 days
of E2 depletion was poor. This result indicates that
inactivation of S6K was slowed by the reduction of PP2A expression
(35). Owing to its substantial
effect on ERα phosphorylation, the reduction in PP2A levels is
considered to contribute to the abnormal activation of IGF-I
receptor/insulin receptor substrate-2 or promote AIs-acquired
resistance (36). Cancerous inhibitor
of PP2A is a novel oncogene that is frequently overexpressed in
breast cancer, and has been reported to be downregulated by the
phytoestrogen genistein, which has a high affinity for the estrogen
receptor (32).
Eve induces Akt activation. PP2A is an important
molecule for Akt suppression, however is downregulated in the
E2 depleted state. The present study suggested that PP2A
activation by FSK is a means to eliminate the effect of the
decrease in PP2A levels, and it may be effective to use FSK in
addition to the AIs and Eve combination (Fig. 5B). The present study supports the
previous implication of PP2A (37) as
a therapeutic target in AI-resistant breast cancer.
Acknowledgements
The present study was supported in part by
Grants-in-Aid for Scientific Research from the Ministry of
Education, Science, Sports, and Culture of Japan (grant no.
25461398), Aichi Cancer Research Foundation (grant no. 673) and
Grants-in-Aid for Research from Fujita Health University (grant no.
0).
References
|
1
|
Klinge CM: Estrogen receptor interaction
with estrogen response elements. Nucleic Acids Res. 29:2905–2919.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Platet N, Cathiard AM, Gleizes M and
Garcia M: Estrogens and their receptors in breast cancer
progression: A dual role in cancer proliferation and invasion. Crit
Rev Oncol Hematol. 51:55–67. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lin NU and Winer EP: Advances in adjuvant
endocrine therapy for postmenopausal women. J Clin Oncol.
26:798–805. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cuzick J, Sestak I, Baum M, Buzdar A,
Howell A, Dowsett M and Forbes JF: ATAC/LATTE investig: Effect of
anastrozole and tamoxifen as adjuvant treatment for early-stage
breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol.
11:1135–1141. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
BIG 1–98 Collaborative Group, . Mouridsen
H, Giobbie-Hurder A, Goldhirsch A, Thürlimann B, Paridaens R, Smith
I, Mauriac L, Forbes J, Price KN, et al: Letrozole therapy alone or
in sequence with tamoxifen in women with breast cancer. N Engl J
Med. 361:766–776. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Regan MM, Neven P, Giobbie-Hurder A,
Goldhirsch A, Ejlertsen B, Mauriac L, Forbes JF, Smith I, Láng I,
Wardley A, et al: Assessment of letrozole and tamoxifen alone and
in sequence for postmenopausal women with steroid hormone
receptor-positive breast cancer: The BIG 1–98 randomised clinical
trial at 8.1 years median follow-up. Lancet Oncol. 12:1101–1108.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Becker MA, Ibrahim YH, Cui X, Lee AV and
Yee D: The IGF pathway regulates ERα through a S6K1-dependent
mechanism in breast cancer cells. Mol Endocrinol. 25:516–528. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yamnik RL, Digilova A, Davis DC, Brodt ZN,
Murphy CJ and Holz MK: S6 kinase 1 regulates estrogen receptor
alpha in control of breast cancer cell proliferation. J Biol Chem.
284:6361–6369. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boulay A, Rudloff J, Ye J, Zumstein-Mecker
S, O'Reilly T, Evans DB, Chen S and Lane HA: Dual inhibition of
mTOR and estrogen receptor signaling in vitro induces cell death in
models of breast cancer. Clin Cancer Res. 11:5319–5328. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miller TW, Hennessy BT, González-Angulo
AM, Fox EM, Mills GB, Chen H, Higham C, García-Echeverría C, Shyr Y
and Arteaga CL: Hyperactivation of phosphatidylinositol-3 kinase
promotes escape from hormone dependence in estrogen
receptor-positive human breast cancer. J Clin Invest.
120:2406–2413. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Orti E, Bodwell JE and Munck A:
Phosphorylation of steroid hormone receptors. Endocr Rev.
13:105–128. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fox EM, Kuba MG, Miller TW, Davies BR and
Arteaga CL: Autocrine IGF-I/insulin receptor axis compensates for
inhibition of AKT in ER-positive breast cancer cells with
resistance to estrogen deprivation. Breast Cancer Res. 15:R552013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Le Goff P, Montano MM, Schodin DJ and
Katzenellenbogen BS: Phosphorylation of the human estrogen
receptor. Identification of hormone-regulated sites and examination
of their influence on transcriptional activity. J Biol Chem.
269:4458–4466. 1994.PubMed/NCBI
|
|
14
|
Arnold SF, Obourn JD, Jaffe H and Notides
AC: Serine 167 is the major estradiol-induced phosphorylation site
on the human estrogen receptor. Mol Endocrinol. 8:1208–1214. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Haystead TA, Sim AT, Carling D, Honnor RC,
Tsukitani Y, Cohen P and Hardie DG: Effects of the tumour promoter
okadaic acid on intracellular protein phosphorylation and
metabolism. Nature. 337:78–81. 1989. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suganuma M, Fujiki H, Suguri H, Yoshizawa
S, Hirota M, Nakayasu M, Ojika M, Wakamatsu K, Yamada K and
Sugimura T: Okadaic acid: An additional
non-phorbol-12-tetradecanoate-13-acetate-type tumor promoter. Proc
Natl Acad Sci USA. 85:pp. 1768–1771. 1988, View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hayashi T, Hikichi M, Utsumi T, Harada N
and Yukitake J: Inhibition of PP2A in MCF-7 cells leads to
hormone-independent growth. Int J Anal Bio-Sci. 4:1–5. 2016.
View Article : Google Scholar
|
|
18
|
Mumby M: PP2A: Unveiling a reluctant tumor
suppressor. Cell. 130:21–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Westermarck J and Hahn WC: Multiple
pathways regulated by the tumor suppressor PP2A in transformation.
Trends Mol Med. 14:152–160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kong M, Fox CJ, Mu J, Solt L, Xu A,
Cinalli RM, Birnbaum MJ, Lindsten T and Thompson CB: The
PP2A-associated protein alpha4 is an essential inhibitor of
apoptosis. Science. 306:695–698. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sontag E: Protein phosphatase 2A: The
trojan horse of cellular signaling. Cell Signal. 13:7–16. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Janssens V and Goris J: Protein
phosphatase 2A: A highly regulated family of serine/threonine
phosphatases implicated in cell growth and signalling. Biochem J.
353:417–439. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eichhorn PJ, Creyghton MP and Bernards R:
Protein phosphatase 2A regulatory subunits and cancer. Biochim
Biophys Acta. 1795:1–15. 2009.PubMed/NCBI
|
|
24
|
Shim WS, Conaway M, Masamura S, Yue W,
Wang JP, Kmar R and Santen RJ: Estradiol hypersensitivity and
mitogen-activated protein kinase expression in long-term estrogen
deprived human breast cancer cells in vivo. Endocrinology.
141:396–405. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Santen RJ, Song RX, Zhang Z, Kumar R, Jeng
MH, Masamura A, Lawrence J Jr, Berstein L and Yue W: Long-term
estradiol deprivation in breast cancer cells up-regulates growth
factor signaling and enhances estrogen sensitivity. Endocr Relat
Cancer. 12 Suppl 1:S61–S73. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Walter P, Green S, Greene G, Krust A,
Bornert JM, Jeltsch JM, Staub A, Jensen E, Scrace G, Waterfield M,
et al: Cloning of the human estrogen receptor cDNA. Proc Natl Acad
Sci USA. 82:pp. 7889–7893. 1985, View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Levenson AS and Jordan VC: MCF-7: The
first hormone-responsive breast cancer cell line. Cancer Res.
57:3071–3078. 1997.PubMed/NCBI
|
|
28
|
Baldacchino S, Saliba C, Petroni V, Fenech
AG, Borg N and Grech G: Deregulation of the phosphatase, PP2A is a
common event in breast cancer, predicting sensitivity to FTY720.
EPMA J. 5:32014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Duronio RJ and Xiong Y: Signaling pathways
that control cell proliferation. Cold Spring Harb Perspect Biol.
5:a0089042013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bononi A, Agnoletto C, De Marchi E, Marchi
S, Patergnani S, Bonora M, Giorgi C, Missiroli S, Poletti F,
Rimessi A and Pinton P: Protein kinases and phosphatases in the
control of cell fate. Enzyme Res. 2011:3290982011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mumby MC and Walter G: Protein
serine/threonine phosphatases: Structure, regulation, and functions
in cell growth. Physiol Rev. 73:673–699. 1993.PubMed/NCBI
|
|
32
|
Zhao Q, Zhao M, Parris AB, Xing Y and Yang
X: Genistein targets the cancerous inhibitor of PP2A to induce
growth inhibition and apoptosis in breast cancer cells. Int J
Oncol. 49:1203–1210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Holmes CF, Luu HA, Carrier F and Schmitz
FJ: Inhibition of protein phosphatases-1 and −2A with
acanthifolicin. Comparison with diarrhetic shellfish toxins and
identification of a region on okadaic acid important for
phosphatase inhibition. FEBS Lett. 270:216–218. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY
and Chiang CW: Regulation of phosphorylation of Thr-308 of Akt,
cell proliferation, and survival by the B55alpha regulatory subunit
targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol
Chem. 283:1882–1892. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peterson RT, Desai BN, Hardwick JS and
Schreiber SL: Protein phosphatase 2A interacts with the 70-kDa S6
kinase and is activated by inhibition of FKBP12-rapamycinassociated
protein. Proc Natl Acad Sci USA. 96:pp. 4438–4442. 1999, View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hurvitz SA and Pietras RJ: Rational
management of endocrine resistance in breast cancer: A
comprehensive review of estrogen receptor biology, treatment
options, and future directions. Cancer. 113:2385–2397. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li L, Zhang J, Xiong N, Li S, Chen Y, Yang
H, Wu C, Zeng H and Liu Y: Notch-1 signaling activates NF-κB in
human breast carcinoma MDA-MB-231 cells via PP2A-dependent AKT
pathway. Med Oncol. 33:332016. View Article : Google Scholar : PubMed/NCBI
|