Introduction
Helicobacter pylori is a gram-negative
bacterial species that represents the most common bacterial
infection of the gastric epithelium worldwide (1,2). H.
pylori colonization is the most prominent identified risk
factor for malignancies that arise within the stomach, and it
increases the risk of atrophic gastritis, intestinal metaplasia,
and distal gastric adenocarcinoma (3–6). H.
pylori infection has been classified by the World Health
Organization as a carcinogen for the development of gastric
carcinoma (7); this cancer type is
the second most common cause of cancer-associated mortality
worldwide, accounting for ~989,600 new cases and ~738,000
mortalities per year, half of which occur in eastern Asia (8,9).
Since the initial description of H. pylori by
Marshall and Warren in 1984 (10),
significant advances towards understanding gastric carcinogenesis
have been achieved. Gastric carcinogenesis is a multifactorial and
multistep process that involves the activation of oncogenes and the
inactivation of tumor suppressor genes (11–16). One
such tumor suppressor gene is phosphatase and tensin homolog (PTEN)
(17–19), which has lipid and protein phosphatase
activities. PTEN inactivation has been observed in glioblastomas,
endometrial carcinomas and skin, prostate and breast cancers
(17–19). Previous studies have indicated that
PTEN inactivation in gastric cancers may be due to genetic or
epigenetic changes, including mutation, loss of heterozygosity,
promoter hypermethylation, regulation of microRNA and
post-translational phosphorylation (14,20–25).
Phosphorylation of PTEN on sites within the C2 domain (Ser380,
Thr382 and Thr383) decreases phosphatase activity and increases
stability (26,27), which may cause loss of tumor
suppressor function and increased susceptibility to cancer. In our
previous study, it was demonstrated that PTEN phosphorylation at
residues Ser380/Thr382/Thr383 is a novel mechanism underlying PTEN
inactivation in gastric carcinogenesis (14,28). This
was observed to be triggered by H. pylori infection and
required for the activation of the
phosphatidylinositol-4,5-bisphosphate 3-kinase/protein kinase B
signaling pathway and the subsequent promotion of cell survival
(14).
PTEN regulates a variety of other downstream
signaling pathways, including the regulation of cell migration,
invasion and growth by focal adhesion kinase (FAK) (29–32). FAK
is a key molecule that is implicated in integrin signaling, which
contributes to cancer progression, invasion and metastasis
(29,33,34).
Previous studies have identified that H. pylori may induce
FAK activation and cytoskeletal reorganization (35,36).
Therefore, the aim of the present study was to determine the effect
of H. pylori-induced PTEN phosphorylation on the activation
of FAK in vivo and in vitro, in order to identify the
mechanisms underlying gastric cancer development.
Materials and methods
H. pylori strain
As previously described (14,28), the
CagA+ and VagA+ H. pylori strain
ATCC43504 (National Institute for Communicable Diseases and
Prevention of Chinese Center for Disease Control and Prevention,
Beijing, China) was cultured on Campylobacter agar plates
(Shanghai Municipal Center For Disease Control and Prevention,
Shanghai, China) supplemented with 10% sheep blood (Shanghai
Kangfeng, Biological Technology Co., Ltd, Shanghai, China) and
incubated at 37°C under microaerophilic conditions (5%
O2, 10% CO2 and 85% N2) for 24 h,
then subcultured in Brucella broth (Shanghai Municipal Center For
Disease Control and Prevention) supplemented with 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific Inc., Waltham, MA, USA)
at 37°C under a microaerophilic atmosphere for 16–18 h. Bacterial
density was estimated spectrophotometrically as the absorbance at
600 nm [optical density (OD)600], and viable counts were
determined as colony-forming units (CFU)/ml (1
OD600=109 CFU/ml).
Mongolian gerbils
A total of 79 specific pathogen-free male Mongolian
gerbils (age, 5–8 weeks; weight, 30–50 g) purchased from the
Zhejiang Academy of Medical Sciences (Hangzhou, China), were
maintained in an isolated clean room with regulated temperature
(20–22°C), humidity (~55%) and a 12/12-h light/dark cycle with
ad libitum rodent diet and water. As previously described
(14,28), following one week of observation, the
gerbils were administered 1 ml orogastric infusions of sterile
Brucella broth (n=25; controls) or 1×109 CFU of H.
pylori (n=54) once every three days, for a total of 10
infusions. Gerbils were fasted for 12 h prior to H. pylori
inoculation and drinking water was withheld following the
inoculation. Food and water were freely available to the gerbils at
4 h post-inoculation. The animals were euthanized using sodium
pentobarbital (150 mg/kg) and cervical dislocation at 6 months
(H. pylori-infected, n=30; controls, n=15) or 12 months
(H. pylori-infected, n=24; controls, n=10). Then gastric
tissue was separated from the animals, which were cut along the
greater curvature of stomach, the gastric tissue was smoothed out
and linear strips of gastric tissue extending from the
squamocolumnar junction through the proximal duodenum were
collected. Detection of H. pylori infection was carried out
using Giemsa staining (14). All
animal experiments and procedures were approved by the Ethics
Committee of The First Affiliated Hospital of Nanchang University
(Nanchang, China).
Cell line and lentivirus
infection
The GES-1 immortalized human gastric epithelial
mucosa cell line from the Beijing Institute for Cancer Research
(Beijing, China) was cultured in Dulbecco's modified Eagle's medium
(DMEM; Hyclone, Logan, Utah, USA) supplemented with 10% FBS, 100 U
penicillin and 100 µg/ml streptomycin (Gibco; Thermo Fisher
Scientific Inc., Waltham, MA, USA) at 37°C in an atmosphere
containing 5% CO2. DMEM supplemented with 10% FBS to
suspend the H. pylori was added to infect GES-1 cells with
various multiplicity of infection (MOI) for 0, 0.5, 1, 3 and 6 h.
The same volume of DMEM (5 ml) supplemented with 10% FBS was added
to GES-1 cells in the control group.
Wild-type PTEN, dominant-negative mutant PTEN
(C124S) and empty lentiviral supernatants were purchased from
Invitrogen (Thermo Fisher Scientific, Inc.). For lentiviral
infection, GES-1 cells were cultured to ~60% confluence and
incubated with the aforementioned three lentiviral supernatants,
respectively, and hexadimethrine bromide (Sigma-Aldrich, St. Louis,
MO, USA) for 6 h as aforementioned. After 48 h, the cells were
split and cultured in selection media supplemented with blasticidin
(6 µg/ml; Sigma-Aldrich) for a further 2 weeks at 37°C in an
atmosphere containing 5% CO2. The stable cell lines
expressing a wild type or dominant-negative mutant PTEN were then
established. As previously described, PTEN was overexpressed in
stable cell lines carried with wild-type PTEN (28). Dominant-negative mutant PTEN (C124S)
contributed to the phosphorylation and inactivation of PTEN at
residues Ser380/Thr382/383. In addition, the empty vector did not
contain exogenous gene.
Immunohistochemistry
Immunohistochemistry was performed on
paraffin-embedded sections of Mongolian gerbil gastric tissues
using anti-PTEN (cat. no. ab31392; dilution 1:150),
anti-phosphorylated (p)-PTEN (Ser380/Thr382/Thr383; cat. no.
ab47332; 1:800), anti-FAK (cat. no. ab40794; 1:400), and anti-p-FAK
(Tyr397; cat. no. ab4803; 1:400) antibodies (all from Abcam,
Cambridge, UK) and polyperoxidase-conjugated anti-mouse or rabbit
immunoglobulin G antibody as provided in PV-9000 Polymer Detection
System (Zhongshan Goldenbridge, Beijing, China) using previously
described methods (12,14,15). The
stained sections were selected, reviewed and scored from five
randomly selected high-power fields (40× objective lens) by two
pathologists blinded to the histopathological data. Grading
discrepancies were re-reviewed and discussed to obtain a final
score. Epithelial cells with yellow or brown staining in the
nucleus and/or cytoplasm were defined as positive for
immunoreactivity. The percentage of immunoreactive cells from 100
cells selected for each field, were averaged from the five fields
and scored as follows: 0, ≤5.0%; 1, 5.1–25.0%; 2, 25.1–50.0%; 3,
50.1–75.0%; and 4, >75.1% immunoreactivity. Furthermore, the
staining intensity was also semi-quantitatively assessed as
follows: 0, no staining; 1, weak staining; 2, moderate staining;
and 3, intense staining. The overall protein expression level was
presented as a grade calculated from the integrated scores of the
‘area × intensity’, as follows: Grade 1, score 0–2 (negative);
grade 2, score 3–5 (weakly positive); grade 3, score 6–8
(moderately positive); and grade 4, score 9–12 (intensely
positive).
Immunoblotting
Western blotting was performed according to standard
methods as described previously (13)
using anti-PTEN (cat. no. 9559; Cell Signaling Technology, Inc.,
Danvers, MA, USA; dilution 1:10,000), anti-p-PTEN
(Ser380/Thr382/Thr383; cat. no. 9554; Cell Signaling Technology,
Inc.; dilution 1:10,000), anti-FAK (cat. no. ab40794; Abcam;
1:1,000), anti-p-FAK (Tyr397; cat. no. ab4803; Abcam; 1:1,000) and
anti-β-actin (sc-1615-R; 1:1,000; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) antibodies. Quantity One software (Bio-Rad
Laboratories, Inc., Hercules, CA, USA; version 4.5.1) was used for
densitometric measurements of band intensities.
Invasion assays
GES-1 cells were seeded at a density of
1×104 cells/well in Transwell chambers (8.0 µm pore
size; Corning Inc., Corning, NY, USA) coated with Matrigel (BD
Biosciences, Franklin Lakes, NJ, USA), with medium supplemented
with 10% FBS in the lower chamber as the chemoattractant and medium
supplemented with 0.2% BSA in the upper chamber, then incubated for
20 h at 37°C with 5% CO2. Cells that did not migrate
through the pores were manually removed with a cotton swab. Cells
adhered to the bottom of the chamber membrane were fixed in
methanol at room temperature for 5 min and stained with
hematoxylin. In three independent experiments, the number of
migrated cells on the lower surface of the membrane was counted in
10 visual fields per well using a light microscope at ×400
magnification and means of these counts were analyzed for
statistical significance.
Statistical analysis
The data are presented as the mean ± standard
deviation or the fold of the control. Statistical analysis was
performed using SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA).
Student's t-tests or Mann-Whitney U tests were used to compare the
differences in the mean value between various groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
H. pylori infection increases PTEN and
FAK phosphorylation in vivo
The Mongolian gerbils were successfully infected
with H. pylori, which was confirmed by Giemsa staining. No
animals challenged with Brucella broth alone exhibited detectable
evidence of H. pylori infection.
Immunohistochemical analysis revealed no significant
difference in PTEN expression levels from H. pylori
infection at 6 or 12 months following inoculation (Fig. 1). Inhibitory p-PTEN demonstrated
variable protein expression levels in all groups, with the
exception of the control group after 6 months. The expression
levels of p-PTEN were significantly higher in the gastric mucosae
of Mongolian gerbils at 12 months following H. pylori
infection (P=0.000) compared with the control, but not in the
corresponding groups after 6 months. The PTEN expression levels
were higher at 12 months than at 6 months with or without H.
pylori infection. Immunohistochemical analysis of FAK revealed
no difference in expression levels from H. pylori infection
after 6 or 12 months compared with the controls; however, the
expression levels were significantly higher at 12 months compared
with at 6 months in the groups with or without H. pylori
infection (P=0.000; Fig. 1). By
contrast, after 6 months, active p-FAK expression levels were not
observed to be altered by H. pylori infection compared with
the control group, whereas the expression levels were significantly
higher compared with the controls after 12 months (P=0.041).
Furthermore, statistical analysis demonstrated that p-FAK
expression levels in H. pylori-infected gerbils were
significantly higher after 12 months compared with 6 months
infection (P=0.000).
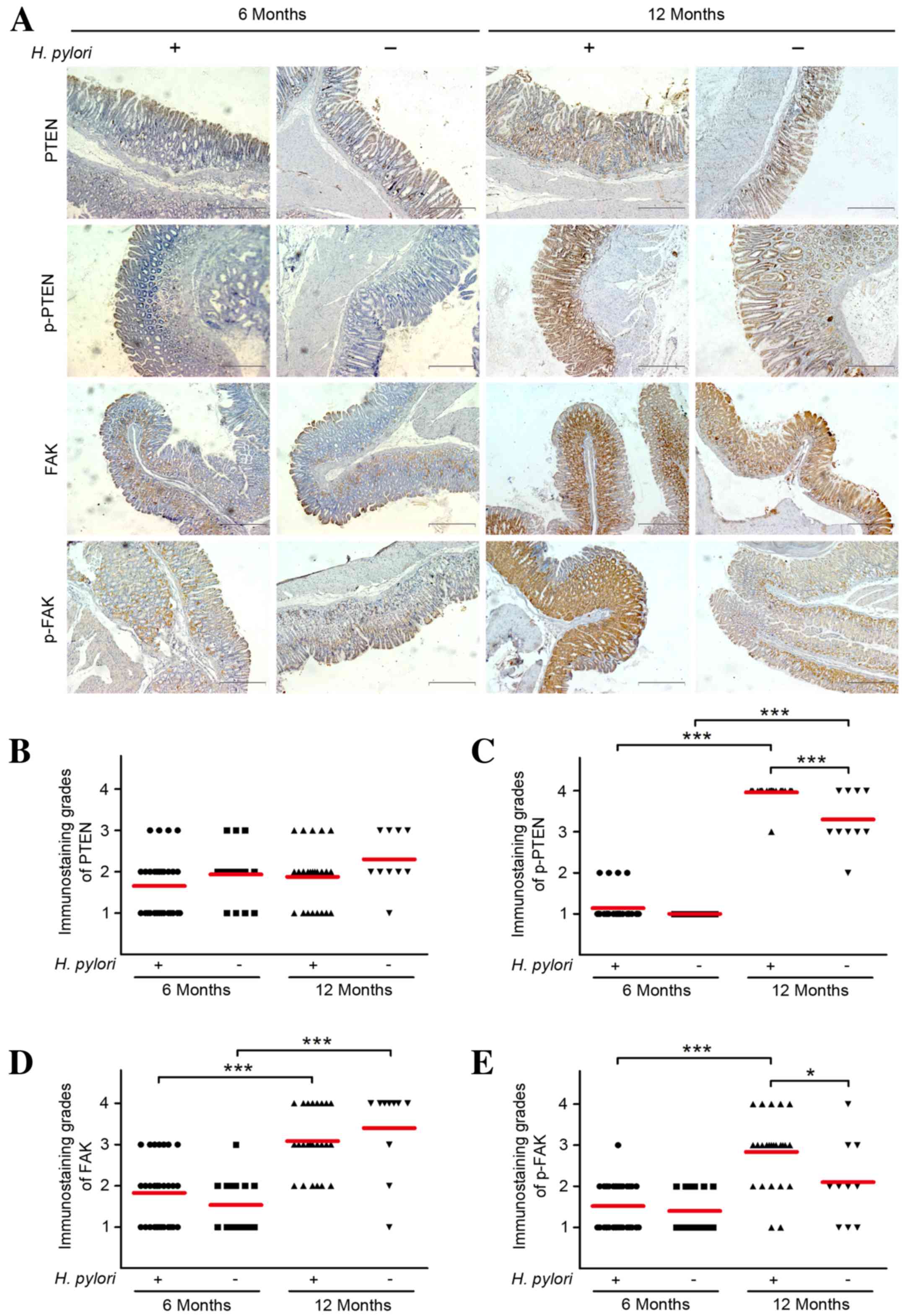 | Figure 1.Immunohistochemical staining of PTEN,
p-PTEN, FAK and p-FAK in the gastric mucosal regions of Mongolian
gerbils. Gastric tissue sections from H. pylori-infected
gerbils were collected at 6 or 12 months following inoculation and
stained using antibodies against PTEN, p-PTEN, FAK and p-FAK. (A)
Representative images of immunohistochemical staining are shown
(scale bars, 200 µm). Immunoreactivity levels of (B) PTEN, (C)
p-PTEN, (D) FAK and (E) p-FAK in the tissues were
semi-quantitatively assessed, and the protein expression levels
were expressed as grades 1–4. Mean grades (red lines) for each
protein in each group are shown. *P<0.05; ***P<0.001. PTEN,
phosphatase and tensin homolog; p-, phosphorylated; FAK, focal
adhesion kinase; H. pylori, Helicobacter pylori. |
H. pylori increases PTEN and FAK
phosphorylation in vitro
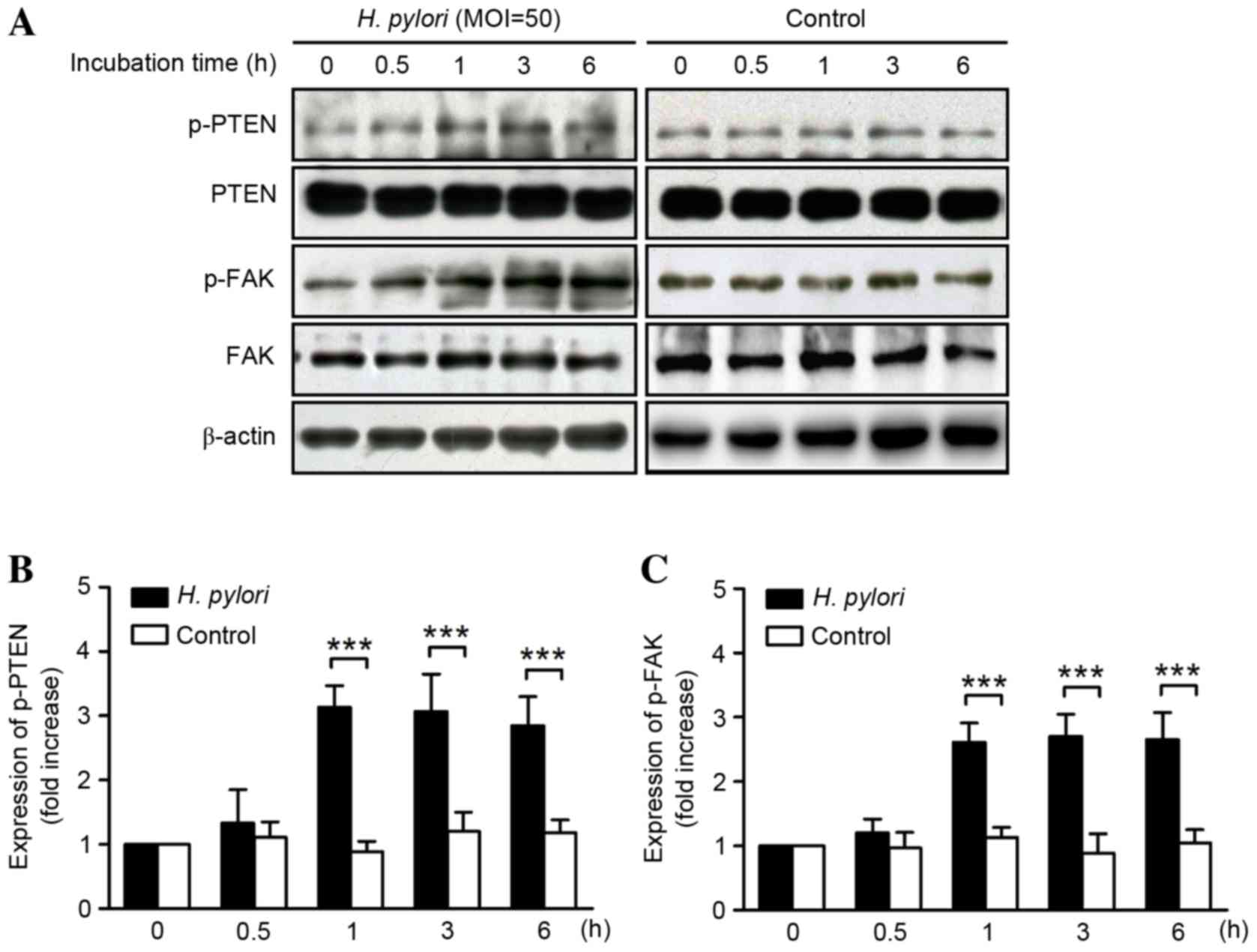
Western blot analysis of protein lysates from GES-1
cells demonstrated that incubation with H. pylori increased
the phosphorylation of PTEN from 1 h, which was observed to be
maintained for ≤6 h (P<0.001) without altering the total PTEN
protein expression levels (Fig. 2A).
Relative to 0 h, the expression levels of p-PTEN were 1.33±0.62,
3.13±0.34, 3.07±0.58, and 2.85±0.45 at 0.5, 1, 3 and 6 h,
respectively (Fig. 2B). Similarly,
the expression levels of p-FAK were 1.22±0.22, 2.62±0.30, 2.70±0.35
and 2.65±0.62 relative to 0 h, after 0.5, 1, 3 and 6 h,
respectively (Fig. 2C). The increase
was determined to be significant after 1–6 h of GES-1 cell
incubation with H. pylori (P<0.001) without affecting the
total FAK protein expression.
Role of PTEN in H. pylori-induced
phosphorylation of FAK
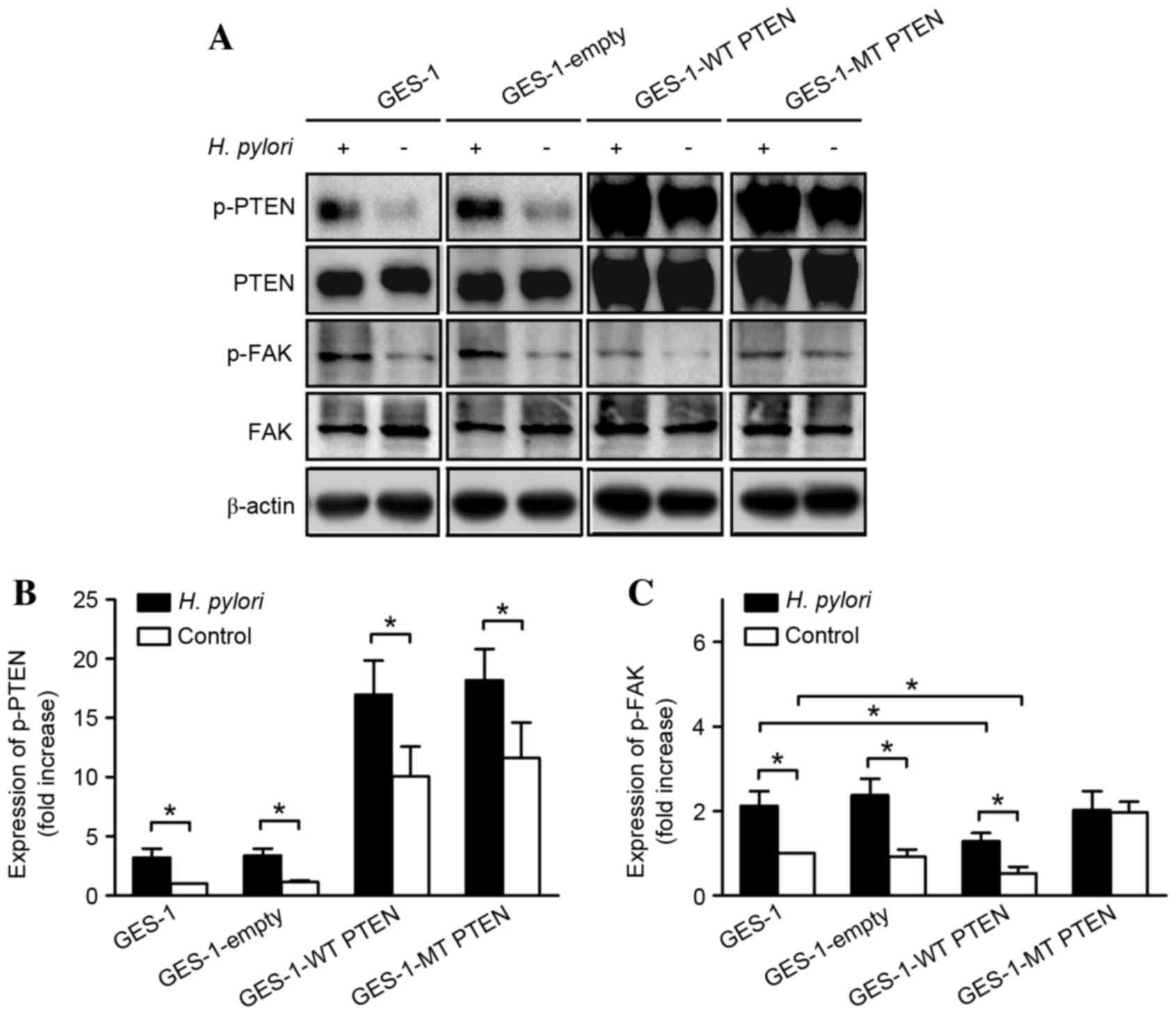
Analysis of the western blots revealed that the
relative p-PTEN/PTEN expression levels in GES-1 cells transfected
with an empty vector were equal to those in the untransfected
control GES-1 cells, while the relative expression levels were
significantly increased by H. pylori infection in all groups
(P=0.025) (Fig. 3). Overexpression of
wild type PTEN markedly inhibited FAK phosphorylation in the
GES-1-wild type PTEN group compared with that in the empty vector
and untransfected group; although H. pylori increased the
levels of p-FAK, this increase was significantly suppressed by
overexpression of wild type PTEN, compared with the untransfected
group (P=0.023). However, overexpression of an inactive
mutant PTEN construct (which mimics phosphorylation at
Ser380/Thr382/Thr383) blocked the H. pylori-induced increase
in p-FAK, without affecting total FAK expression levels. In
addition, this overexpression of mutant PTEN increased basal
expression levels of p-FAK, thereby mimicking the effects of H.
pylori infection.
Role of PTEN in H. pylori-induced cell
invasion
Incubation of GES-1 cells with H. pylori
increased the number of cells that migrated through a Transwell
membrane (159.03±15.96 vs. 113.67±7.23; P=0.002; Fig. 4). Similarly, invasion was enhanced by
H. pylori infection in GES-1 cells transfected with an empty
control vector (179.67±6.02 vs. 118.10±9.00; P=0.001), wild type
PTEN (138.97±5.00 vs. 85.06±10.00; P=0.001) and mutant PTEN
(185.87±4.93 vs. 150.32±7.57; P=0.002). Following H. pylori
infection, the number of invading cells overexpressing wild type
PTEN was decreased compared with the empty vector group (P=0.001).
In addition, there was a significant difference among the
GES-1-wild type PTEN, mutant PTEN and the untransfected group, as
wild type PTEN overexpression decreased the number of invading
cells, whereas mutant PTEN expression levels increased the number
of invading cells compared with the untransfected group
(P=0.004).
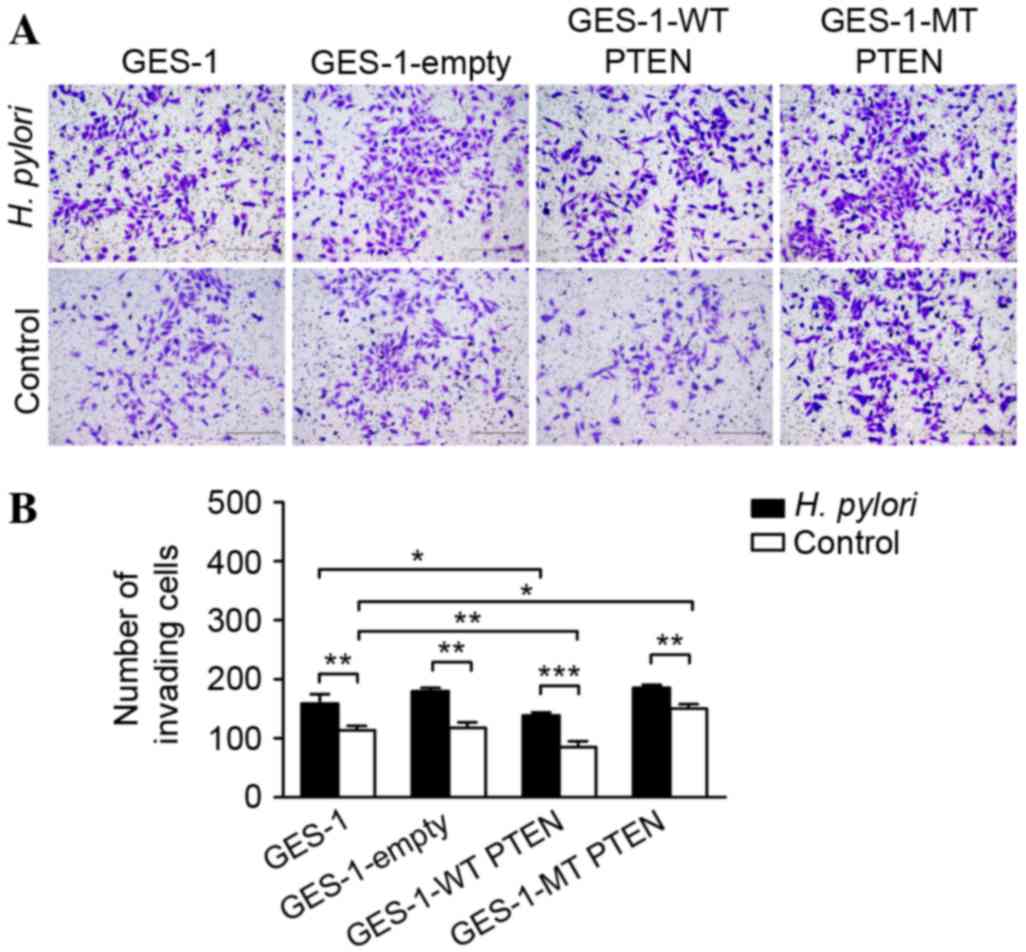 | Figure 4.Invasion assay of GES-1 gastric
epithelial cells overexpressing mt or wt PTEN constructs following
incubation with H. pylori. (A) Representative micrographs
showing the Transwell invasion of hematoxylin-stained GES-1 cells
expressing wt or mt PTEN constructs (GES-1-WT PTEN and GES-1-MT
PTEN, respectively), with and without incubation with H.
pylori (multiplicity of infection, 50; scale bar, 200 µm). (B)
Numbers of cells invaded through the membranes are presented as the
mean ± standard deviation of triplicated experiments. *P<0.05;
**P<0.01; ***P<0.001. PTEN, phosphatase and tensin homolog;
p-, phosphorylated; FAK, focal adhesion kinase; H. pylori,
Helicobacter pylori; mt, mutant; wt, wild type. |
Discussion
PTEN is recognized as a frequently inactivated tumor
suppressor in a variety of human cancers, and several lines of
evidence suggest that abnormal activation of FAK is associated with
tumor development (37–39). Continuing from our previous study
(28), which described the H.
pylori-induced phosphorylation of PTEN, the results of the
present study demonstrate that gastric cell invasion is promoted by
this process via the activation of FAK.
H. pylori infection is one of the most
significant identified risk factors for gastric carcinogenesis.
Numerous studies use animal models to investigate H. pylori
infection-associated diseases. The use of various animal species
and bacterial strains results in a large variability in the
reported results (40). Mongolian
gerbils are a unique model due to their low incidence of naturally
occurring gastritis and bacterial disease, and have, therefore,
commonly been used for studying infection with a standard strain of
H. pylori (ATCC43504) (41).
The results of the current study demonstrate that, concordant with
previous results from patients (28),
6–12 months of infection in these gerbils did not significantly
alter PTEN expression levels. However, H. pylori infection
did enhance the expression levels of p-PTEN and p-FAK in gerbils.
Although FAK expression levels increased with age, they were not
affected by H. pylori infection.
The in vitro experiments confirmed that H.
pylori-induced PTEN inactivation promotes FAK phosphorylation,
which is consistent with the results of previous studies (34,35). By
overexpressing an inactive form of PTEN, where phosphatase activity
is eliminated by the mutation of a cysteine to a serine at position
124 (42), the present study
demonstrated that the H. pylori-induced phosphorylation of
FAK is dependent on PTEN activity. In addition, the overexpression
of wild type PTEN reduced basal p-FAK levels, indicating enhanced
phosphatase activity. These results are concordant with those of a
previous study (39) which indicated
that overexpression of PTEN inhibits the invasion and metastasis of
gastric cancer. However, FAK expression in that study was reported
to be downregulated, which was not observed in the current
study.
PTEN is known to be crucially involved in the
regulation of cell proliferation, growth and survival, and in
tumorigenesis (24); however, the
role of PTEN in cancer invasion and metastasis is not well
understood. The results of the current study demonstrate that H.
pylori infection significantly increases gastric epithelial
cell invasion. Furthermore, cell invasiveness was found to be
dependent on PTEN activity, as it was suppressed by wild type, and
enhanced by mutant, PTEN overexpression. However, H. pylori
infection was still able to increase the invasion of cells
overexpressing mutant PTEN, suggesting the involvement of an
additional signaling pathway, including one that may act directly
on the cell cytoskeleton, matrix metalloproteinase-7 or integrins
(43–45).
Previous reports have suggested that gastric
carcinogenesis is associated with dysregulation of the PTEN
signaling pathway (13,14,24,25). The
results of the present study demonstrate that H. pylori
infection enhances this dysregulation by promoting the
phosphorylation of PTEN, which subsequently increases FAK
activation and gastric epithelial cell invasion. Therefore, we
propose that early H. pylori infection may influence gastric
cancer development by altering signaling pathways important for the
morphological changes, adhesion, migration and proliferation of
gastric mucosal cells.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81060038 and
81460377), the Natural Science Foundation of Jiangxi Province,
China (grant nos. 20142BAB215036 and 20151BAB205041), the Science
and Technology Foundation of Department of Education of Jiangxi
Province, China (grant no, GJJ14169), the National Science and
Technology Major Projects program for ‘Major New Drugs Innovation
and Development’ of China (grant no. 2011ZX09302-007-03) and the
‘Talent 555 Project’ of Jiangxi Province, China. The authors would
like to thank Professor Ke Yang at the Beijing Institute for Cancer
Research for providing the GES-1 immortalized gastric epithelial
mucosa cell line, and Professor Zhang Jianzhong of the National
Institute for Communicable Diseases and Prevention of Chinese
Center for Disease Control and Prevention for providing the H.
pylori type strain ATCC43504.
Glossary
Abbreviations
Abbreviations:
|
PTEN
|
phosphatase and tensin homolog
|
|
FAK
|
focal adhesion kinase
|
|
FBS
|
fetal bovine serum
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
References
|
1
|
Moss SF and Sood S: Helicobacter pylori.
Curr Opin Infect Dis. 16:445–451. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peek RM Jr and Crabtree JE: Helicobacter
infection and gastric neoplasia. J Pathol. 208:233–248. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Uemura N, Okamoto S, Yamamoto S, Matsumura
N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N and Schlemper RJ:
Helicobacter pylori infection and the development of gastric
cancer. N Engl J Med. 345:784–789. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nomura A, Stemmermann GN, Chyou PH, Kato
I, Perez-Perez GI and Blaser MJ: Helicobacter pylori infection and
gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med.
325:1132–1136. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Parsonnet J, Friedman GD, Vandersteen DP,
Chang Y, Vogelman JH, Orentreich N and Sibley RK: Helicobacter
pylori infection and the risk of gastric carcinoma. N Engl J Med.
325:1127–1131. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Correa P: Helicobacter pylori and gastric
cancer: State of the art. Cancer Epidemiol Biomarkers Prev.
5:477–481. 1996.PubMed/NCBI
|
|
7
|
No authors listed: Schistosomes, liver
flukes and Helicobacter pylori. IARC Working Group on the
Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994.
IARC Monogr Eval Carcinog Risks Hum. 61:1–241. 1994.PubMed/NCBI
|
|
8
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marshall BJ and Warren JR: Unidentified
curved bacilli in the stomach of patients with gastritis and peptic
ulceration. Lancet. 1:1311–1315. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu Y, Shu X, Chen J, Xie Y, Xu P, Huang
DQ and Lu NH: Effect of Helicobacter pylori eradication on
oncogenes and cell proliferation. Eur J Clin Invest. 38:628–633.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang Z, Shu X, Chen L, Chen J, Xie Y and
Lu NH: Expression of p53-MDM2 feedback loop related proteins in
various gastric pathologies in relation to Helicobacter pylori
infection: Implications in gastric carcinogenesis. Clin Res Hepatol
Gastroenterol. 36:235–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang Z, Yuan XG, Chen J and Lu NH: Is
NEDD4-1 a negative regulator of phosphatase and tensin homolog in
gastric carcinogenesis? World J Gastroenterol. 18:6345–6348. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang Z, Yuan XG, Chen J, Luo SW, Luo ZJ
and Lu NH: Reduced expression of PTEN and increased PTEN
phosphorylation at residue Ser380 in gastric cancer tissues: A
novel mechanism of PTEN inactivation. Clin Res Hepatol
Gastroenterol. 37:72–79. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li W, Xie C, Yang Z, Chen J and Lu NH:
Abnormal DNA-PKcs and Ku 70/80 expression may promote malignant
pathological processes in gastric carcinoma. World J Gastroenterol.
19:6894–6901. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xie C, Xu LY, Yang Z, Cao XM, Li W and Lu
NH: Expression of gammaH2AX in various gastric pathologies and its
association with Helicobacter pylori infection. Oncol Lett.
7:159–163. 2014.PubMed/NCBI
|
|
17
|
Li J, Yen C, Liaw D, Podsypanina K, Bose
S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al:
PTEN, a putative protein tyrosine phosphatase gene mutated in human
brain, breast, and prostate cancer. Science. 275:1943–1947. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Steck PA, Pershouse MA, Jasser SA, Yung
WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T,
et al: Identification of a candidate tumour suppressor gene, MMAC1,
at chromosome 10q23.3 that is mutated in multiple advanced cancers.
Nat Genet. 15:356–362. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li DM and Sun H: TEP1, encoded by a
candidate tumor suppressor locus, is a novel protein tyrosine
phosphatase regulated by transforming growth factor beta. Cancer
Res. 57:2124–2129. 1997.PubMed/NCBI
|
|
20
|
Sato K, Tamura G, Tsuchiya T, Endoh Y,
Sakata K, Motoyama T, Usuba O, Kimura W, Terashima M, Nishizuka S,
et al: Analysis of genetic and epigenetic alterations of the PTEN
gene in gastric cancer. Virchows Arch. 440:160–165. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Byun DS, Cho K, Ryu BK, Lee MG, Park JI,
Chae KS, Kim HJ and Chi SG: Frequent monoallelic deletion of PTEN
and its reciprocal associatioin with PIK3CA amplification in
gastric carcinoma. Int J Cancer. 104:318–327. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kang YH, Lee HS and Kim WH: Promoter
methylation and silencing of PTEN in gastric carcinoma. Lab Invest.
82:285–291. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chun-Zhi Z, Lei H, An-Ling Z, Yan-Chao F,
Xiao Y, Guang-Xiu W, Zhi-Fan J, Pei-Yu P, Qing-Yu Z and Chun-Sheng
K: MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell
proliferation and radioresistance by targeting PTEN. BMC Cancer.
10:3672010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu WT, Yang Z and Lu NH: Roles of PTEN
(Phosphatase and Tensin Homolog) in gastric cancer development and
progression. Asian Pac J Cancer Prev. 15:17–24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang Z, Xie C, Liu G, Cao X, Li W, Xu W
and Lu N: Helicobacter pylori phosphorylates PTEN tumor suppressor
to activate PI3K/Akt signaling pathway. J Gastroenterol Hepatol.
28:1012013.
|
|
26
|
Vazquez F, Ramaswamy S, Nakamura N and
Sellers WR: Phosphorylation of the PTEN tail regulates protein
stability and function. Mol Cell Biol. 20:5010–5018. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Torres J and Pulido R: The tumor
suppressor PTEN is phosphorylated by the protein kinase CK2 at its
C terminus. Implications for PTEN stability to proteasome-mediated
degradation. J Biol Chem. 276:993–998. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang Z, Xie C, Xu W, Liu G, Cao X, Li W,
Chen J, Zhu Y, Luo S, Luo Z, et al: Phosphorylation and
inactivation of PTEN at residues Ser380/Thr382/383 induced by
Helicobacter pylori promotes gastric epithelial cell survival
through PI3K/Akt pathway. Oncotarget. 6:31916–31926. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tamura M, Gu J, Matsumoto K, Aota S,
Parsons R and Yamada KM: Inhibition of cell migration, spreading,
and focal adhesions by tumor suppressor PTEN. Science.
280:1614–1617. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tamura M, Gu J, Takino T and Yamada KM:
Tumor suppressor PTEN inhibition of cell invasion, migration, and
growth: Differential involvement of focal adhesion kinase and
p130Cas. Cancer Res. 59:442–449. 1999.PubMed/NCBI
|
|
31
|
Tamura M, Gu J, Danen EH, Takino T,
Miyamoto S and Yamada KM: PTEN interactions with focal adhesion
kinase and suppression of the extracellular matrix-dependent
phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol
Chem. 274:20693–20703. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gu J, Tamura M, Pankov R, Danen EH, Takino
T, Matsumoto K and Yamada KM: Shc and FAK differentially regulate
cell motility and directionality modulated by PTEN. J Cell Biol.
146:389–403. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kornberg LJ: Focal adhesion kinase and its
potential involvement in tumor invasion and metastasis. Head Neck.
20:745–752. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
McLean GW, Carragher NO, Avizienyte E,
Evans J, Brunton VG and Frame MC: The role of focal-adhesion kinase
in cancer-a new therapeutic opportunity. Nat Rev Cancer. 5:505–515.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tabassam FH, Graham DY and Yamaoka Y: OipA
plays a role in Helicobacter pylori-induced focal adhesion kinase
activation and cytoskeletal re-organization. Cell Microbiol.
10:1008–1020. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tegtmeyer N, Wittelsberger R, Hartig R,
Wessler S, Martinez-Quiles N and Backert S: Serine phosphorylation
of cortactin controls focal adhesion kinase activity and cell
scattering induced by Helicobacter pylori. Cell Host Microbe.
9:520–531. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang C, Yang R, Yue D and Zhang Z:
Expression of FAK and PTEN in bronchioloalveolar carcinoma and lung
adenocarcinoma. Lung. 187:104–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang SY, Hao HL, Deng K, Li Y, Cheng ZY,
Lv C, Liu ZM, Yang J and Pan L: Expression levels of phosphatase
and tensin homolog deleted on chromosome 10 (PTEN) and focal
adhesion kinase in patients with multiple myeloma and their
relationship to clinical stage and extramedullary infiltration.
Leuk Lymphoma. 53:1162–1168. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang LL, Liu J, Lei S, Zhang J, Zhou W
and Yu HG: PTEN inhibits the invasion and metastasis of gastric
cancer via downregulation of FAK expression. Cell Signal.
26:1011–1020. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kodama M, Murakami K, Nishizono A and
Fujioka T: Animal models for the study of Helicobacter-induced
gastric carcinoma. J Infect Chemother. 10:316–325. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yan J, Luo YH and Mao YF: Establishment of
Helicobacter pylori infection model in Mongolian gerbils. World J
Gastroenterol. 10:852–855. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Myers MP, Pass I, Batty IH, van der Kaay
J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP and Tonks NK: The
lipid phosphatase activity of PTEN is critical for its tumor
supressor function. Proc Natl Acad Sci USA. 95:pp. 13513–13518.
1998; View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wroblewski LE, Noble PJ, Pagliocca A,
Pritchard DM, Hart CA, Campbell F, Dodson AR, Dockray GJ and Varro
A: Stimulation of MMP-7 (matrilysin) by Helicobacter pylori in
human gastric epithelial cells: Role in epithelial cell migration.
J Cell Sci. 116:3017–3026. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Al-Ghoul L, Wessler S, Hundertmark T,
Kruger S, Fischer W, Wunder C, Haas R, Roessner A and Naumann M:
Analysis of the type IV secretion system-dependent cell motility of
Helicobacter pylori-infected epithelial cells. Biochem Biophys Res
Commun. 322:860–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schneider S, Weydig C and Wessler S:
Targeting focal adhesions: Helicobacter pylori-host communication
in cell migration. Cell Commun Signal. 6:22008. View Article : Google Scholar : PubMed/NCBI
|