Introduction
Esophageal cancer is the eighth most common cancer
worldwide, with 456,000 new cases and ~0.4 million mortalities
reported in 2012. Due to the geographical variation in the
occurrence of esophageal cancer, 49% of new cases occurred in China
(1), particularly in two counties
renowned for the high occurrence rates of esophageal cancer:
Linxian, Henan and Yanting, Sichuan (2). This is due to the tumors being
asymptomatic in early stages, meaning >50% of patients present
at an advanced stage that is too late for treatment by
esophagectomy. Chemotherapy is the main treatment for advanced
esophageal cancer (3). However,
chemotherapy is associated with drug resistance and side effects.
Therefore, the identification of novel and safe treatments for
esophageal cancer is necessary.
Autophagy is an intricate and conserved process.
Autophagic flux refers to the process by which damaged organelles
and unfolded proteins are sequestered into autophagosomes within
the cytoplasm, which fuse with lysosomes to form autolysosomes, to
induce the degradation of intracellular components (4). The induction of autophagy has been
identified as a drug resistance mechanism that promotes cancer cell
survival via self-digestion (5,6).
Therefore, targeting autophagy may be a promising approach for
cancer therapy.
Chloroquine (CQ) has been safely and widely used as
an anti-malarial for >60 years (7). A number of studies have suggested that
CQ can inhibit autophagy through the prevention of lysosome
acidification, and subsequently, the inhibition of
autophagosome-lysosome fusion, to block the degradation of
autolysosomes at the last step of autophagy (8,9). Previous
studies have indicated that CQ exhibited an antitumor effect on
several types of cancer, including glioblastoma (10,11),
hepatocellular carcinoma (12),
breast cancer (13), prostate cancer
(14) and pancreatic cancer (15), which prompts us to hypothesize that CQ
may be able to influence the growth of esophageal carcinoma through
the modulation of autophagy. Therefore, the aim of the present
study is to explore the antitumor effect of CQ on the esophageal
squamous carcinoma cell line EC109, and the potential mechanism for
this effect.
Materials and methods
Cells and reagents
EC109 human esophageal squamous cell carcinoma cells
were obtained from the Institute of Rheumatism Immunity, Affiliated
Hospital of North Sichuan Medical College (Nanchong, China). EC109
cells were grown in RPMI-1640 supplemented with 10% fetal bovine
serum (both from Hyclone; GE Healthcare Life Sciences, Logan, UT,
USA), penicillin (100 U/ml) and streptomycin (100 µg/ml;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Cells were
incubated in a humidified incubator in 5% CO2 at
37°C.
CQ was kindly provided by Dr Yibin Deng (Hormel
Institute, University of Minnesota, Austin, MN, USA). RPMI-1640 was
purchased from Hyclone; GE Healthcare Life Sciences. All primary
antibodies, including light chain (LC)3 (cat. no. 4108), p62/SQSTM1
(cat. no. 5114), phosphorylated (p)-mechanistic target of rapamycin
(mTOR; cat. no. 2971), p-protein kinase B (Akt; cat. no. 9271),
p-70S6K (cat. no. 9205), p-4E binding protein 1 (4EBP1; cat. no.
9451)and GAPDH (cat. no. 2118) were obtained from CST Biological
Reagents Co., Ltd. (Shanghai, China).
MTT viability assay
The cells were seeded at a density of
2×103 per well in a 96-well plate and incubated for 24
h. CQ was added to each well in a concentration series of 50, 100,
150 and 200 µmol/l and incubated for a further 12, 24 and 36 h. A
total of 20 µl MTT (5 mg/ml; Sigma-Aldrich) was added to each well
and incubated at 37°C for 4 h, then 100 µl DMSO was added to each
well to dissolve crystals and the plate was agitated for 10 min.
Absorbance values at 490 nm were detected by a multi-mode detection
platform (Molecular Devices Austria GmbH, Wals, Austria).
Inhibition rate was calculated as [(Acontrol -
Ablank) - (Atreated -
Ablank)]/(Acontrol - Ablank)x100.
Each experiment was assayed in triplicate.
Scratch assay
Cells were seeded in a 6-well plate at a density of
5×105 cells per well and cultured until the cells formed
a confluent monolayer. A 10 µl pipette tip was used to scratch the
cells and create a wound. The medium was removed and the cultures
were washed twice with PBS to remove non-adherent cells. The cells
were then incubated with complete RPMI medium, with and without 200
µmol/l. The migratory ability of the cells was determined according
to the extent of closure of the scratch, observed at 0 and 24 h
after incubation with CQ. Images were acquired using an inverted
microscope (DMI 3000B; Leica microsystems GmbH, Wetzlar,
Germany).
Transwell migration assay
Cells (6×104) in RPMI 1640 with 5% FBS
were seeded into the upper chamber of polycarbonate membrane
Transwell inserts (Corning Incorporated, Corning, NY, USA) with or
without 200 µmol/l CQ. RPMI-1640 supplemented with 20% FBS was
added to the lower chamber as a chemo-attractant solution.
Following a 48-h incubation, the remaining cells on the upper
chamber surface were removed, and the cells that had migrated to
the underside of the chamber were fixed in methanol and stained
with 10% Giemsa for 10 min at room temperature. The cell migration
ability was determined by the number of cells that had migrated to
the lower side of the filter. These experiments were repeated three
times, and the filters were photographed under an inverted
microscope.
Soft agar colony-forming assay
To assess the transformation ability of EC109 cells
upon CQ treatment, 8×103 cells were suspended in
complete RPMI medium containing 0.6% soft agar, seeded in
triplicate on 60 mm dishes pre-coated with 3.0% agar in complete
growth medium, and incubated at 37°C in a 5% CO2
humidified atmosphere with and without 200 µmol/l CQ. The culture
medium was replaced every 4 days. Following an incubation of 18
days, colonies were photographed using an inverted microscope.
Green fluorescent protein (GFP)-LC3
transfection
EC109 cells were transfected with a GFP-LC3 plasmid
using Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and incubated for 24 h and
treated with and without CQ. The images were obtained by
fluorescence microscope.
Western blot analysis
Cells treated with or without 200 µmol/l CQ were
lysed in radioimmunoprecipitation assay buffer (Beyotime Institute
of Biotechnology, Haimen, China) containing proteinase inhibitors,
and lysates were collected. The protein samples were detected using
SDS-PAGE (8–15% gel). A total of 40 µg of protein was loaded into
each lane of the SDS-PAGE gel and transferred onto a polyvinyl
difluoride membrane. Protein was quantified using a BCA assay.
Membranes were blocked in 5% fat-free milk for 1 h at room
temperature, and probed with the previously specified primary
antibodies (dilution 1:1,000) overnight at 4°C, followed by
horseradish peroxidase-conjugated secondary antibodies (1:5,000;
Boster Biological Technology, Pleasanton, CA, USA) and incubated
for 1 h at room temperature. Bands were visualized using an
enhanced chemiluminscence kit (Merck KGaA).
Cell death assessment by flow
cytometry
Cells were seeded at 5×105 cells per well
in a 6-well plate and incubated with or without 200 µmol/l CQ for
24 h. Cells were harvested by trypsinization and washed with PBS.
Subsequent to staining with Annexin V-fluorescein isothiocyanate
for 15 min at 4°C and propidium iodide for 2 min at room
temperature (Nanjing KeyGen BioTech, Co., Ltd., Nanjing, China),
cells were detected using flow cytometry using FACSComp (version
4.1; Beckman Coulter, Inc., Brea, CA, USA).
Statistical analysis
The results are expressed as the mean ± standard
deviation and t-tests or one-way analysis of variance followed by a
least significant difference t-test were performed using SPSS
(version 13.0; SPSS, Inc., Chicago, IL, USA) statistical software.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Proliferation, migration and colony
formation abilities of EC109 are inhibited by CQ
An increasing number of studies have described the
anti-tumor effect of CQ. Thus, the first aim of the present study
was to explore whether CQ affected the growth, migration or
colony-forming ability of EC109 cells. According to the results of
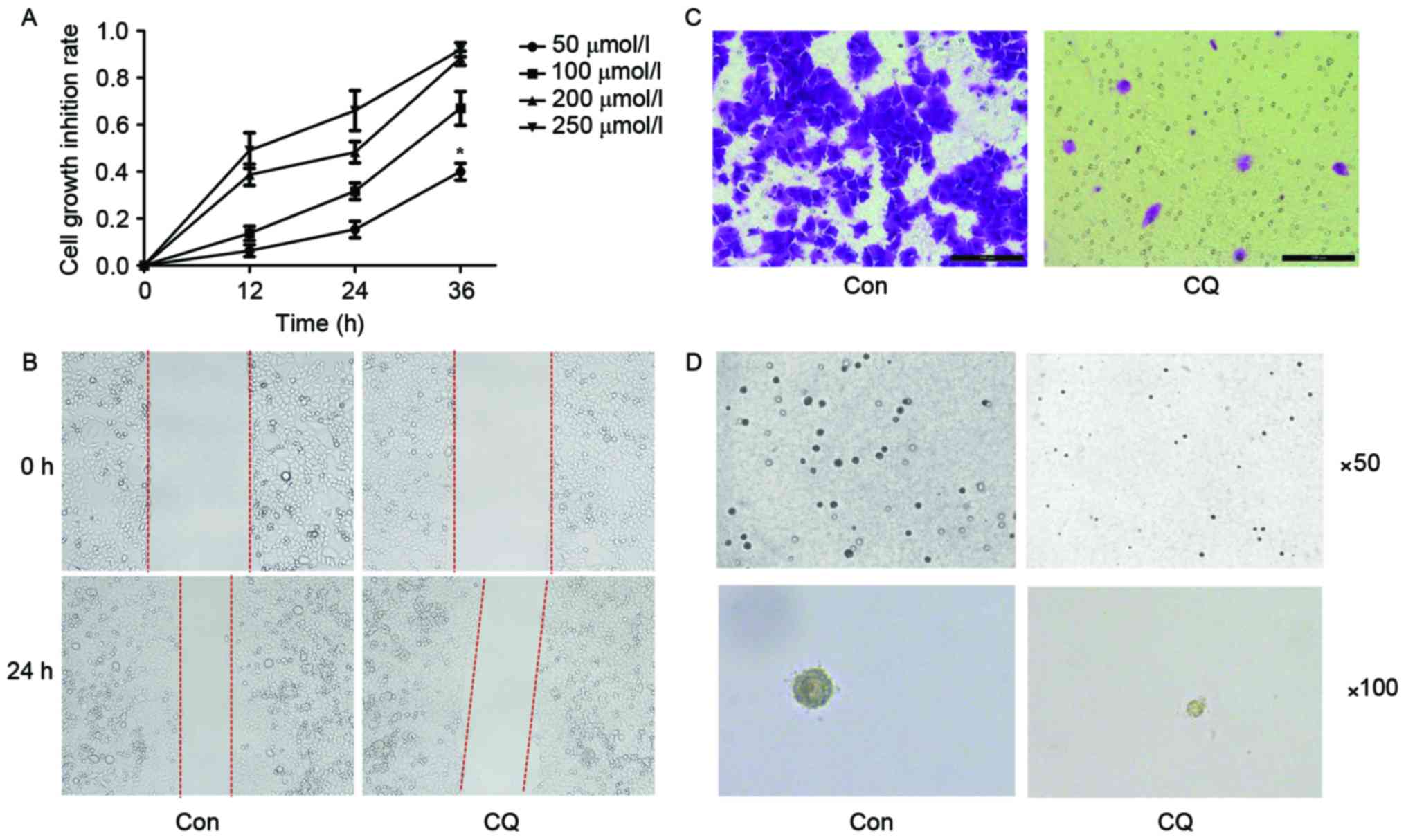
the MTT assay, CQ suppressed the viability of EC109 cells in a time
and dose-dependent manner (Fig. 1A).
The scratch assay indicated that the migration ability of EC109
cells was inhibited after a 24 h CQ treatment (Fig. 1B). A Transwell migration assay was
performed for the same purpose, and a similar result was obtained
(Fig. 1C). The colony formation
ability of EC109 cells was evaluated using a soft agar assay; it
was revealed that the number and size of colonies were reduced in
the CQ group compared with the control group (Fig. 1D).
Cell death is induced by CQ in EC109
cells
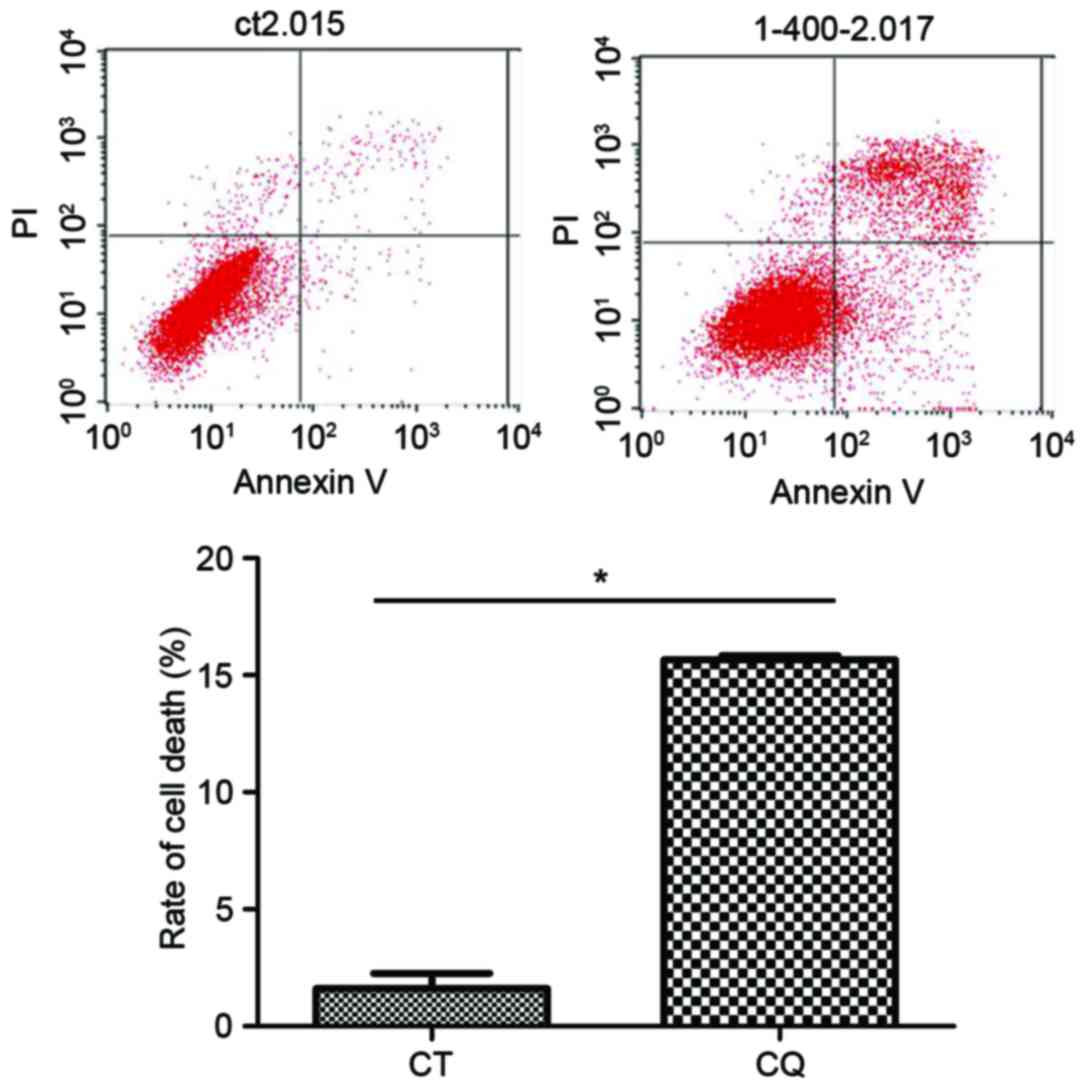
The results of flow cytometry demonstrated that the
cell death ratio in the CQ group was significantly increased
compared with the control group (P<0.05; Fig. 2).
CQ modulates autophagy through
inducing the formation of autophagosomes and preventing the
degradation of autophagosomes in EC109 cells
A number of studies have indicated that the
induction of autophagy promoted tumor cell survival, whereas others
have achieved the opposite result (16). Therefore, the present study aimed to
explore whether CQ could modulate autophagy and influence the
survival of EC109 cells.
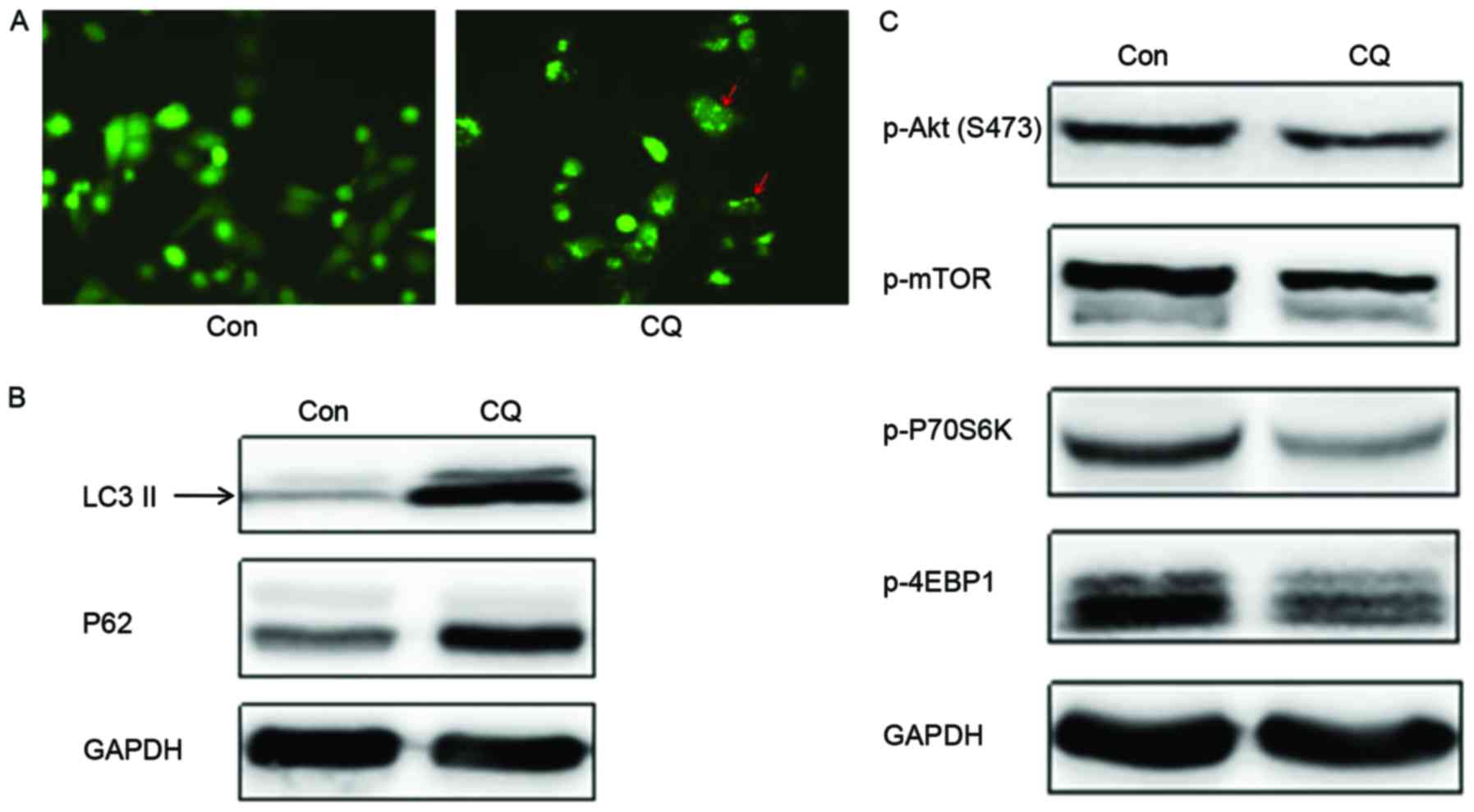
The GFP-LC3 fused protein translocates rapidly to
nascent autophagosomes in a punctate distribution during the
initiation of autophagy. Subsequent to the transfection of a
GFP-LC3 plasmid into EC109 cells to monitor autophagosome formation
by fluorescence microscopy, it was observed that the number of
autophagosomes was evidently increased following CQ treatment
(Fig. 3A). LC3 has two isoforms,
LC3-I and LC3-II; LC3-I is cleaved by autophagy associated protein
ATG4 and lipidated to form LC3-II during the initiation of
autophagy. Thus, the expression of LC3-II is often used as a marker
to evaluate the progression of autophagy (17). Western blot analysis revealed that CQ
treatment markedly enhanced LC3-II expression (Fig. 3B), which indicated that CQ could
induce the formation of autophagosomes in EC109 cells; this result
corroborated the conclusion drawn from the GFP-LC3 transfection
experiment.
Another autophagy marker, p62, is sequestered within
autophagosomes and then degraded by lysosomes during the last stage
of autophagy. The present study demonstrates that the expression of
p62 was upregulated following CQ treatment compared with the
control (Fig. 3B). Collectively,
these results demonstrate that CQ inhibited autophagy by preventing
autophagosome degradation at the last stage of autophagy, despite
inducing autophagosome formation during the initiation of
autophagy.
Based on the results outlined in the previous
section, it is hypothesized that CQ could suppress the growth and
proliferation of EC109 cells in a time- and dose-dependent manner
and reduce their migration and colony formation ability by
preventing autophagosome degradation. In order to elucidate the
mechanism for this process as induced by CQ, the expression of key
proteins in the Akt/mTOR signaling pathway, which serves an
important role in promoting cell growth and proliferation and is
associated with the regulation of autophagy, was assessed. Western
blotting demonstrated that the expression of autophagic negative
regulatory factors, p-Akt and p-mTOR, decreased on CQ treatment.
This suggests that autophagosome formation was initiated. The
phosphorylation levels of the downstream proteins, including the
cell translation regulation factors, p70S6K and 4EBP1, also
decreased with CQ treatment (Fig.
3C), and therefore, CQ may initiate the formation of
autophagosomes, but inhibit the degradation of the autophagosome in
the final stage of autophagy.
Discussion
Esophageal carcinoma is one of the most common types
of malignant neoplasm and a leading cause for cancer-associated
mortality worldwide (1). Radiotherapy
and chemotherapy are the most common treatments for patients with
advanced esophageal carcinoma, with cisplatin, fluorouracil,
oxaliplatin and capecitabine being the most commonly used
chemotherapy agents (18). These
drugs gradually induce chemoresistance and side effects due to the
higher and longer dosages required. Developing an effective drug
may cost $800 million and 15 years (19), therefore, a more effective way to
solve the problem may be to develop new chemotherapeutics from
existing drugs.
Over the past ten years, autophagy inhibition has
been a popular topic in the field of cancer therapy. Autophagy is
an important regulatory mechanism involved in cell growth,
maturation and death, functions which are associated with a number
of diseases, including cancer. Autophagic activity in the context
of cancer is a double-edged sword: It degrades damaged organelles
and recycles macromolecules to maintain a stable cellular
environment, preventing tumor formation (20), but autophagy may also contribute to
tumorigenesis by helping cancer cells to survive in response to
growth-limiting conditions, including nutrient deficiency, absence
of growth factors, hypoxia and exposure to cytotoxic drugs
(6). A recent study revealed that in
an increasing proportion of cases, chemotherapy failure is due to
drug-initiated autophagy, which eventually induces chemoresistance
(21).
As an inhibitor of autophagy, a number of studies
have suggested CQ as a promising approach for cancer therapy as it
is relatively safe and inexpensive (7,22,23). However, the specific anti-tumor
mechanisms of CQ remain uncharacterized. In order to explore
whether CQ could be used as a potential chemotherapeutic agent for
the treatment of esophageal carcinoma, it was used to treat EC109
esophageal squamous cell carcinoma cells. The MTT assay results
indicated that CQ was able to suppress the proliferation of EC109
cells in a time and dose-dependent manner, and the migration
ability of the cells was also inhibited, as demonstrated using
scratch and Transwell chamber tests. Furthermore, CQ suppressed
colony formation on soft agar, which indicated that the
transformation ability of EC109 cells was impaired. Collectively,
these findings demonstrated that CQ may have an antitumor effect on
EC109 cells.
In order to determine whether autophagy was involved
in the antitumor activity of CQ, autophagosome formation was
monitored by transfecting EC109 cells with a GFP-LC3 plasmid, and
observing the expression of LC3 by fluorescence, and LC3-II and p62
by western blot analysis. As the substrates of autophagic flux, LC3
and p62 are involved in the process of autophagosome-lysosome
fusion, which can be used to evaluate the formation and degradation
of the autophagosome (24). Results
demonstrate that CQ treatment increased the expression of LC3 and
p62, indicating that autophagosome formation was increased, and
autophagosome degradation was markedly inhibited, subsequent to CQ
treatment.
To investigate the signaling mechanism of
CQ-mediated autophagy in EC109 cells, the Akt/mTOR pathway, which
serves a central role in regulating cell growth, was studied. The
over-activation of this pathway contributes to cancer development
and progression; Akt positively regulates the phosphorylation and
activation of mTOR, which is a sensor of nutrient levels to promote
cell growth (25). Furthermore, the
Akt/mTOR pathway has been demonstrated to be involved in the
regulation of autophagy; the inhibition of mTOR induces autophagy
via the phosphorylation and inactivation of Unc-51-like autophagy
activating kinase 1, a serine/threonine kinase that initiates
autophagy (26). mTOR exists in two
cellular complexes, mTORC1 and mTORC2 (27). mTORC1 phosphorylates S6K1 and 4E-BP1,
which contribute to translation, protein synthesis and cell growth.
mTORC2 regulates the phosphorylation of Akt. In the present study,
the expression of p-mTOR, p-Akt, p-p70S6K1 and p-4EBP1 were
markedly downregulated following CQ treatment.
The increased LC3-II expression and the inhibition
of the Akt/mTOR pathway indicated that CQ induced autophagic flux
and facilitated the formation of autophagosomes. However, the
upregulation of p62 indicated that CQ also inhibited the
degradation of the autophagosomal contents. Taken together, these
results demonstrate that, as an inhibitor of autophagy, CQ induced
the formation of the autophagosome, but inhibited the degradation
of autophagosome in the last stage of autophagy. The overall effect
was autophagic cell death activation by CQ, as confirmed by flow
cytometry. Liu et al came to a similar conclusion regarding
cervical cancer by incubating SiHa cells with hydroxychloroquine,
the hydroxylated analog of CQ (28);
another study suggested that CQ suppressed pancreatic cancer growth
independent of TP53/TRP53 status by inhibiting autophagy (15).
Based on the data of the present study, CQ exerted
an antitumor effect on EC109 through the modulation of autophagy,
suggesting that CQ may exhibit promising therapeutic benefits for
the treatment of esophageal squamous cell carcinoma.
Acknowledgements
The authors would like to thank Professor Yibing
Deng (Hormel Institute, University of Minnesota, Austin, MN, USA)
and Professor Minghui Yang (the Institute of Rheumatism Immunity of
Affiliated Hospital of North Sichuan Medical College, Nanchong,
China). The present study was funded by the Natural Science
Foundation of the Education Department of Sichuan Province (grant
no. 16ZA0224) and the Sichuan Province Special Project for the
Youth Team of Science and Technology Innovation (grant no.
2015TD0029).
References
|
1
|
Stewart BW and Wild CP: World cancer
report 2014. lyon cedex. France: The international agency for
research on cancer;
|
|
2
|
Lin Y, Totsuka Y, Shan B, Wang C, Wei W,
Qiao Y, Kikuchi S, Inoue M, Tanaka H and He Y: Esophageal cancer in
high-risk areas of China: Research progress and challenges. Ann
Epidemiol. 27:215–221. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu L, Gu C, Zhong D, Shi L, Kong Y, Zhou Z
and Liu S: Induction of autophagy counteracts the anticancer effect
of cisplatin in human esophageal cancer cells with acquired drug
resistance. Cancer Lett. 355:34–45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang XJ, Chen S, Huang KX and Le WD: Why
should autophagic flux be assessed? Acta Pharmacol Sin. 34:595–599.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peng X, Gong F, Chen Y, Jiang Y, Liu J, Yu
M, Zhang S, Wang M, Xiao G and Liao H: Autophagy promotes
paclitaxel resistance of cervical cancer cells: Involvement of
Warburg effect activated hypoxia-induced factor 1-α-mediated
signaling. Cell Death Dis. 5:e13672014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
ODonovan TR, OSullivan GC and McKenna SL:
Induction of autophagy by drug-resistant esophageal cancer cells
promotes their survival and recovery following treatment with
chemotherapeutics. Autophagy. 7:509–524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Solomon VR and Lee H: Chloroquine and its
analogs: A new promise of an old drug for effective and safe
cancertherapies. Eur J Pharmacol. 625:220–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rabinowitz JD and White E: Autophagy and
metabolism. Science. 330:1344–1348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zanotto-Filho A, Braganhol E, Klafke K,
Figueiró F, Terra SR, Paludo FJ, Morrone M, Bristot IJ, Battastini
AM, Forcelini CM, et al: Autophagy inhibition improves the efficacy
of curcumin/temozolomide combination therapy in glioblastomas.
Cancer Lett. 358:220–231. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Golden EB, Cho HY, Jahanian A, Hofman FM,
Louie SG, Schönthal AH and Chen TC: Chloroquine enhances
temozolomide cytotoxicity in malignant gliomas by blocking
autophagy. Neurosurg Focus. 37:E122014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mei L, Chen Y, Wang Z, Wang J, Wan J, Yu
C, Liu X and Li W: Synergistic anti-tumour effects of tetrandrine
and chloroquine combination therapy in human cancer: A potential
antagonistic role for p21. Br J Pharmacol. 172:2232–2245. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lefort S, Joffre C, Kieffer Y, Givel AM,
Bourachot B, Zago G, Bieche I, Dubois T, Meseure D, Vincent-Salomon
A, et al: Inhibition of autophagy as a new means of improving
chemotherapy efficiency in high-LC3B triple-negative breast
cancers. Autophagy. 10:2122–2142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Farrow JM, Yang JC and Evans CP: Autophagy
as a modulator and target in prostate cancer. Nat Rev Urol.
11:508–516. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang A and Kimmelman AC: Inhibition of
autophagy attenuates pancreatic cancer growth independent of
TP53/TRP53 status. Autophagy. 10:1683–1684. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
White E, Mehnert JM and Chan CS:
Autophagy, metabolism, and cancer. Clin Cancer Res. 21:5037–5046.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rustgi AK and El-Serag HB: Esophageal
carcinoma. N Engl J Med. 371:2499–2509. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
DiMasi JA, Hansen RW and Grabowski HG: The
price of innovation: New estimates of drug development costs. J
Health Econ. 22:151–185. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu S and Li X: Autophagy inhibition
enhances sensitivity of endometrial carcinoma cells to paclitaxel.
Int J Oncol. 46:2399–2408. 2015.PubMed/NCBI
|
|
22
|
Balic A, Sørensen MD, Trabulo SM, Sainz B
Jr, Cioffi M, Vieira CR, Miranda-Lorenzo I, Hidalgo M, Kleeff J,
Erkan M and Heeschen C: Chloroquine targets pancreatic cancer stem
cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer
Ther. 13:1758–1771. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fan C, Wang W, Zhao B, Zhang S and Miao J:
Chloroquine inhibits cell growth and induces cell death in A549
lung cancer cells. Bioorg Med Chem. 14:3218–3222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang L, Gao C, Yao S and Xie B: Blocking
autophagic flux enhances matrine-induced apoptosis in human
hepatoma cells. Int J Mol Sci. 14:23212–23230. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xie X, White EP and Mehnert JM: Coordinate
autophagy and mTOR pathway inhibition enhances cell death in
melanoma. PLoS One. 8:e550962013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fan QW, Cheng C, Hackett C, Feldman M,
Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA,
Debnath J, et al: Akt and autophagy cooperate to promote survival
of drug-resistant glioma. Sci Signal. 3:ra812010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu Q, Luo XY, Jiang H, Yang MH, Yuan GH,
Tang Z and Wang H: Hydroxychloroquine facilitates autophagosome
formation but not degradation to suppress the proliferation of
cervical cancer SiHa cells. Oncol Lett. 7:1057–1062.
2014.PubMed/NCBI
|