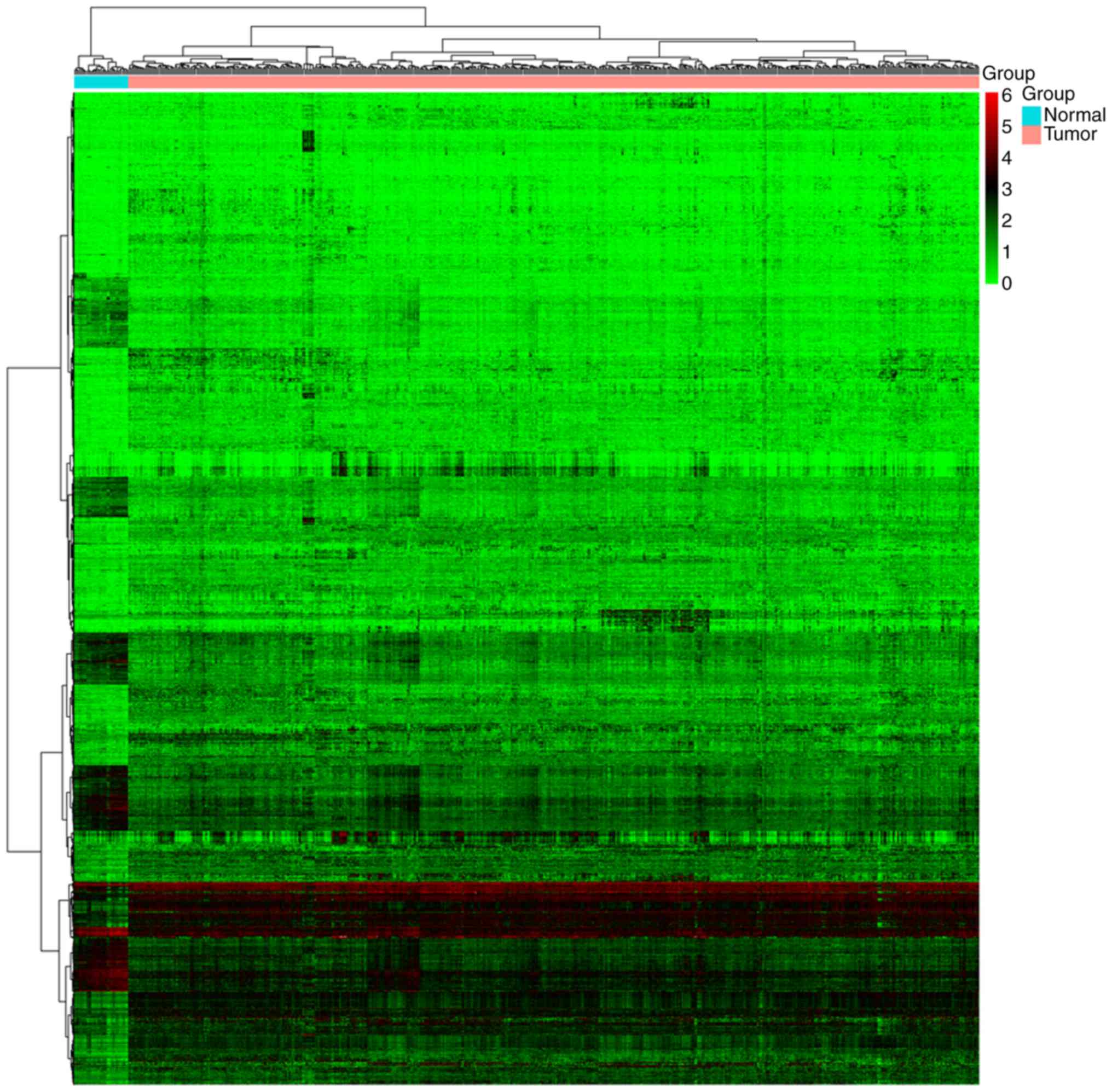
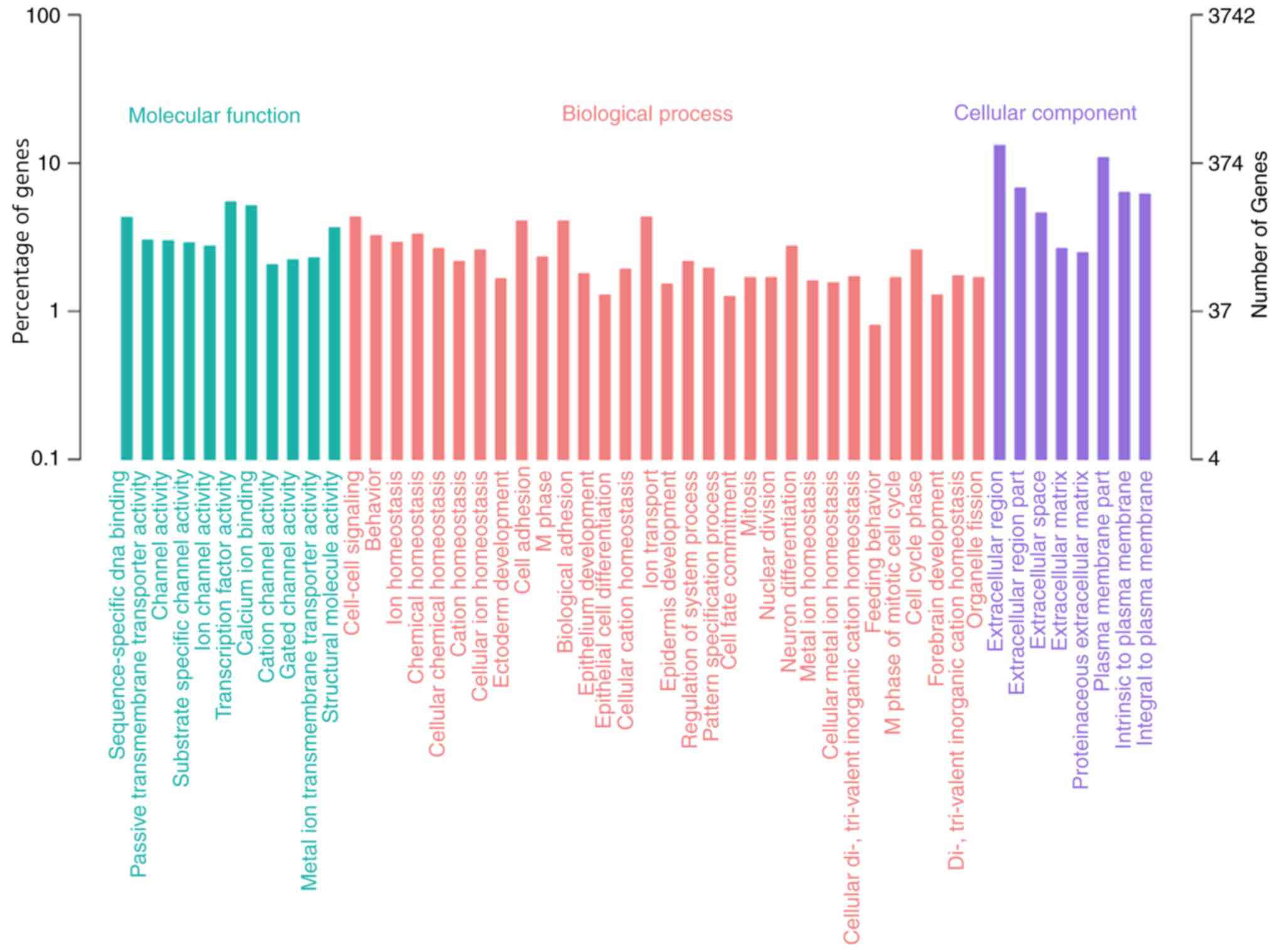
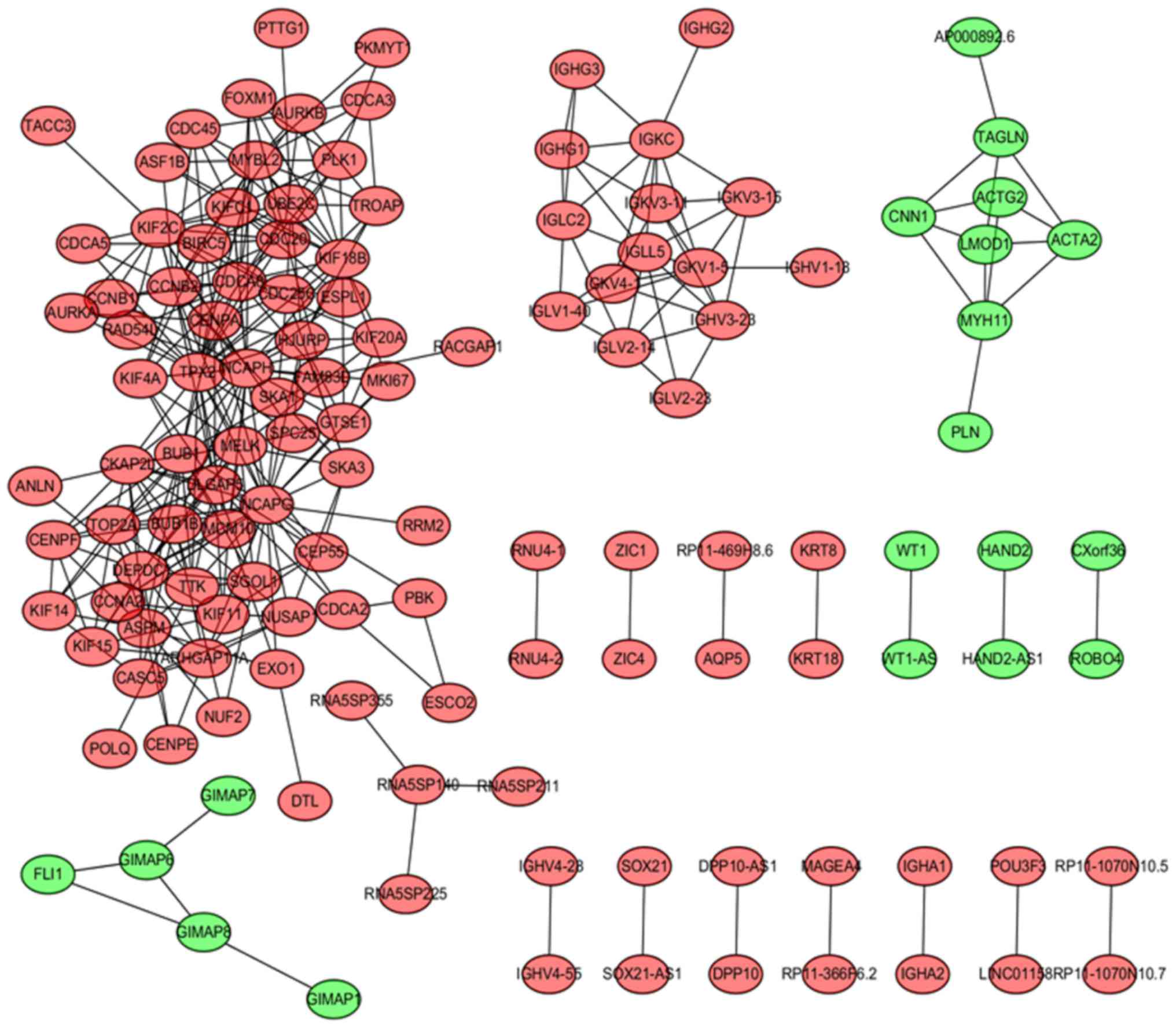
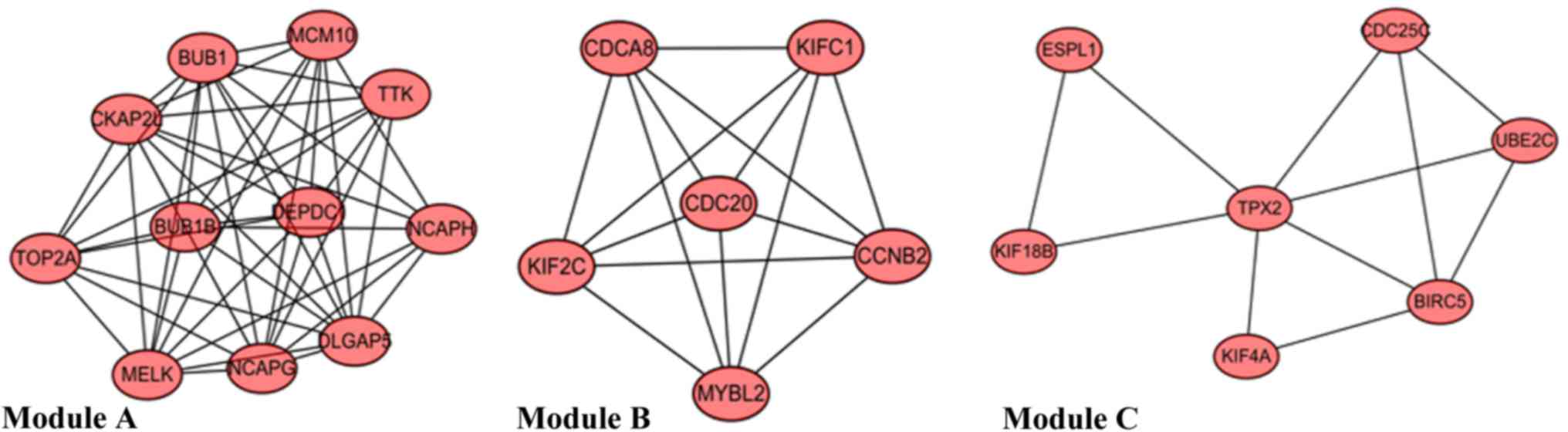
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rutgers JK: Update on pathology, staging
and molecular pathology of endometrial (uterine corpus)
adenocarcinoma. Future Oncol. 11:3207–3218. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Devis L, Moiola CP, Masia N,
Martinez-Garcia E, Santacana M, Stirbat TV, Brochard-Wyart F,
García Á, Alameda F, Cabrera S, et al: Activated leukocyte cell
adhesion molecule (ALCAM) is a marker of recurrence and promotes
cell migration, invasion, and metastasis in early-stage
endometrioid endometrial cancer. J Pathol. 241:475–487. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baser E, Togrul C, Ozgu E, Ayhan S, Caglar
M, Erkaya S and Gungor T: Sperm-associated antigen 9 is a promising
marker for early diagnosis of endometrial cancer. Asian Pac J
Cancer Prev. 14:7635–7638. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smogeli E, Davidson B, Cvancarova M, Holth
A, Katz B, Risberg B, Kristensen G and Lindemann K: L1CAM as a
prognostic marker in stage I endometrial cancer: A validation
study. BMC Cancer. 16:5962016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lenhard M, Heublein S, Kunert-Keil C,
Vrekoussis T, Lomba I, Ditsch N, Mayr D, Friese K and Jeschke U:
Immunosuppressive Glycodelin A is an independent marker for poor
prognosis in endometrial cancer. BMC Cancer. 13:6162013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Matsuo K, Gray MJ, Yang DY, Srivastava SA,
Tripathi PB, Sonoda LA, Yoo EJ, Dubeau L, Lee AS and Lin YG: The
endoplasmic reticulum stress marker, glucose-regulated protein-78
(GRP78) in visceral adipocytes predicts endometrial cancer
progression and patient survival. Gynecol Oncol. 128:552–559. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Voss MA, Gordon N, Maloney S, Ganesan R,
Ludeman L, McCarthy K, Gornall R, Schaller G, Wei W, Berditchevski
F and Sundar S: Tetraspanin CD151 is a novel prognostic marker in
poor outcome endometrial cancer. Br J Cancer. 104:1611–1618. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu Y, Zhu Y, Müller P, Mitra R and Ji Y:
Characterizing cancer-specific networks by integrating TCGA data.
Cancer Inform. 13 Suppl 2:S125–S131. 2014.
|
|
11
|
Cancer Genome Atlas Research Network, .
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang L, Cao C, Ma Q, Zeng Q, Wang H, Cheng
Z, Zhu G, Qi J, Ma H, Nian H and Wang Y: RNA-seq analyses of
multiple meristems of soybean: Novel and alternative transcripts,
evolutionary and functional implications. BMC Plant Biol.
14:1692014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dawson JA, Ye S and Kendziorski C:
R/EBcoexpress: An empirical Bayesian framework for discovering
differential co-expression. Bioinformatics. 28:1939–1940. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hulsegge I, Kommadath A and Smits MA:
Globaltest and GOEAST: Two different approaches for Gene Ontology
analysis. BMC Proc. 3 Suppl 4:pp. S102009; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI
|
|
16
|
Altermann E and Klaenhammer TR:
PathwayVoyager: Pathway mapping using the Kyoto Encyclopedia of
Genes and Genomes (KEGG) database. BMC Genomics. 6:602005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu J, Mao X, Cai T, Luo J and Wei L: KOBAS
server: A web-based platform for automated annotation and pathway
identification. Nucleic Acids Res. 34(Web Server Issue): W720–W724.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lamb J, Crawford ED, Peck D, Modell JW,
Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et
al: The Connectivity Map: Using gene-expression signatures to
connect small molecules, genes, and disease. Science.
313:1929–1935. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Knapinska AM, Estrada CA and Fields GB:
The roles of matrix metalloproteinases in pancreatic cancer. Prog
Mol Biol Transl Sci. 148:339–354. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Karahan N, Güney M, Baspinar S, Oral B,
Kapucuoglu N and Mungan T: Expression of gelatinase (MMP-2 and
MMP-9) and cyclooxygenase-2 (COX-2) in endometrial carcinoma. Eur J
Gynaecol Oncol. 28:184–188. 2007.PubMed/NCBI
|
|
21
|
Yu F, Jiang Q, Zhou Y, Yang Z, Yu X, Wang
H, Liu Z, Wang L, Fang W and Guo S: Abnormal expression of matrix
metalloproteinase-9 (MMP9) correlates with clinical course in
Chinese patients with endometrial cancer. Dis Markers. 32:321–327.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ito F, Furukawa N and Nakai T: Evaluation
of TOP2A as a predictive marker for endometrial cancer with
taxane-containing adjuvant chemotherapy. Int J Gynecol Cancer.
26:325–330. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen L, Cui J, Liang S, Pang Y and Liu P:
Update of research on the role of EZH2 in cancer progression. Onco
Targets Ther. 6:321–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yamaguchi H, Du Y, Nakai K, Ding M, Chang
SS, Hsu JL, Yao J, Wei Y, Nie L, Jiao S, et al: EZH2 contributes to
the response to PARP inhibitors through its PARP-mediated poly-ADP
ribosylation in breast cancer. Oncogene. 2017. View Article : Google Scholar
|
|
25
|
Lian Y, Yan C, Ding J, Xia R, Ma Z, Hui B,
Ji H, Zhou J and Wang K: A novel lncRNA, LL22NC03-N64E9.1,
represses KLF2 transcription through binding with EZH2 in
colorectal cancer. Oncotarget. 8:59435–59445. 2017.PubMed/NCBI
|
|
26
|
Zhou F, Chen J and Wang H: MicroRNA-298
inhibits malignant phenotypes of epithelial ovarian cancer by
regulating the expression of EZH2. Oncol Lett. 12:3926–3932.
2016.PubMed/NCBI
|
|
27
|
Yu L, Lu J, Zhang B, Liu X, Wang L, Li SY,
Peng XH, Xu X, Tian WD and Li XP: miR-26a inhibits invasion and
metastasis of nasopharyngeal cancer by targeting EZH2. Oncol Lett.
5:1223–1228. 2013.PubMed/NCBI
|
|
28
|
Jia N, Li Q, Tao X, Wang J, Hua K and Feng
W: Enhancer of zeste homolog 2 is involved in the proliferation of
endometrial carcinoma. Oncol Lett. 8:2049–2054. 2014.PubMed/NCBI
|
|
29
|
Ma F, Zhang H, Zhai Y, Huang W, Zhao C, Ou
S, Zhou H, Yuan W, Wang Z, Wang H, et al: Functional polymorphism
−31C/G in the promoter of BIRC5 gene and risk of nasopharyngeal
carcinoma among chinese. PLoS One. 6:e167482011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chuwa AH, Sone K, Oda K, Ikeda Y, Fukuda
T, Wada-Hiraike O, Inaba K, Makii C, Takeuchi M, Oki S, et al:
Significance of survivin as a prognostic factor and a therapeutic
target in endometrial cancer. Gynecol Oncol. 141:564–569. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li M, York JP and Zhang P: Loss of Cdc20
causes a securin-dependent metaphase arrest in two-cell mouse
embryos. Mol Cell Biol. 27:3481–3488. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hadjihannas MV, Bernkopf DB, Brückner M
and Behrens J: Cell cycle control of Wnt/b-catenin signalling by
conductin/axin2 through CDC20. EMBO Rep. 13:347–354. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Harley ME, Allan LA, Sanderson HS and
Clarke PR: Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its
Cdc20-dependent destruction during mitotic arrest. EMBO J.
29:2407–2420. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xie Q, Wu Q, Mack SC, Yang K, Kim L,
Hubert CG, Flavahan WA, Chu C, Bao S and Rich JN: CDC20 maintains
tumor initiating cells. Oncotarget. 6:13241–13254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gayyed MF, El-Maqsoud NM, Tawfiek ER, El
Gelany SA and Rahman MF: A comprehensive analysis of CDC20
overexpression in common malignant tumors from multiple organs: Its
correlation with tumor grade and stage. Tumour Biol. 37:749–762.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chang HT, Chou CT, Lin YS, Shieh P, Kuo
DH, Jan CR and Liang WZ: Esculetin, a natural coumarin compound,
evokes Ca(2+) movement and activation of Ca(2+)-associated
mitochondrial apoptotic pathways that involved cell cycle arrest in
ZR-75-1 human breast cancer cells. Tumour Biol. 37:4665–4678. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim AD, Han X, Piao MJ, Hewage SR, Hyun
CL, Cho SJ and Hyun JW: Esculetin induces death of human colon
cancer cells via the reactive oxygen species-mediated mitochondrial
apoptosis pathway. Environ Toxicol Pharmacol. 39:982–989. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim AD, Hewage SR Madduma, Piao MJ, Kang
KA, Cho SJ and Hyun JW: Esculetin induces apoptosis in human colon
cancer cells by inducing endoplasmic reticulum stress. Cell Biochem
Funct. 33:487–494. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pan H, Wang BH, Lv W, Jiang Y and He L:
Esculetin induces apoptosis in human gastric cancer cells through a
cyclophilin D-mediated mitochondrial permeability transition pore
associated with ROS. Chem Biol Interact. 242:51–60. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jeon YJ, Jang JY, Shim JH, Myung PK and
Chae JI: Esculetin, a coumarin derivative, exhibits
anti-proliferative and pro-apoptotic activity in G361 human
malignant melanoma. J Cancer Prev. 20:106–112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang J, Lu ML, Dai HL, Zhang SP, Wang HX
and Wei N: Esculetin, a coumarin derivative, exerts in vitro and in
vivo antiproliferative activity against hepatocellular carcinoma by
initiating a mitochondrial-dependent apoptosis pathway. Braz J Med
Biol Res. 48:245–253. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kuo HC, Lee HJ, Hu CC, Shun HI and Tseng
TH: Enhancement of esculetin on Taxol-induced apoptosis in human
hepatoma HepG2 cells. Toxicol Appl Pharmacol. 210:55–62. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cho JH, Shin JC, Cho JJ, Choi YH, Shim JH
and Chae JI: Esculetin (6,7-dihydroxycoumarin): A potential cancer
chemopreventive agent through suppression of Sp1 in oral squamous
cancer cells. Int J Oncol. 46:265–271. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kok SH, Yeh CC, Chen ML and Kuo MY:
Esculetin enhances TRAIL-induced apoptosis through DR5 upregulation
in human oral cancer SAS cells. Oral Oncol. 45:1067–1072. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Arora R, Sawney S, Saini V, Steffi C,
Tiwari M and Saluja D: Esculetin induces antiproliferative and
apoptotic response in pancreatic cancer cells by directly binding
to KEAP1. Mol Cancer. 15:642016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jeon YJ, Cho JH, Lee SY, Choi YH, Park H,
Jung S, Shim JH and Chae JI: Esculetin induces apoptosis through
EGFR/PI3K/Akt signaling pathway and nucleophosmin relocalization. J
Cell Biochem. 117:1210–1221. 2016. View Article : Google Scholar : PubMed/NCBI
|