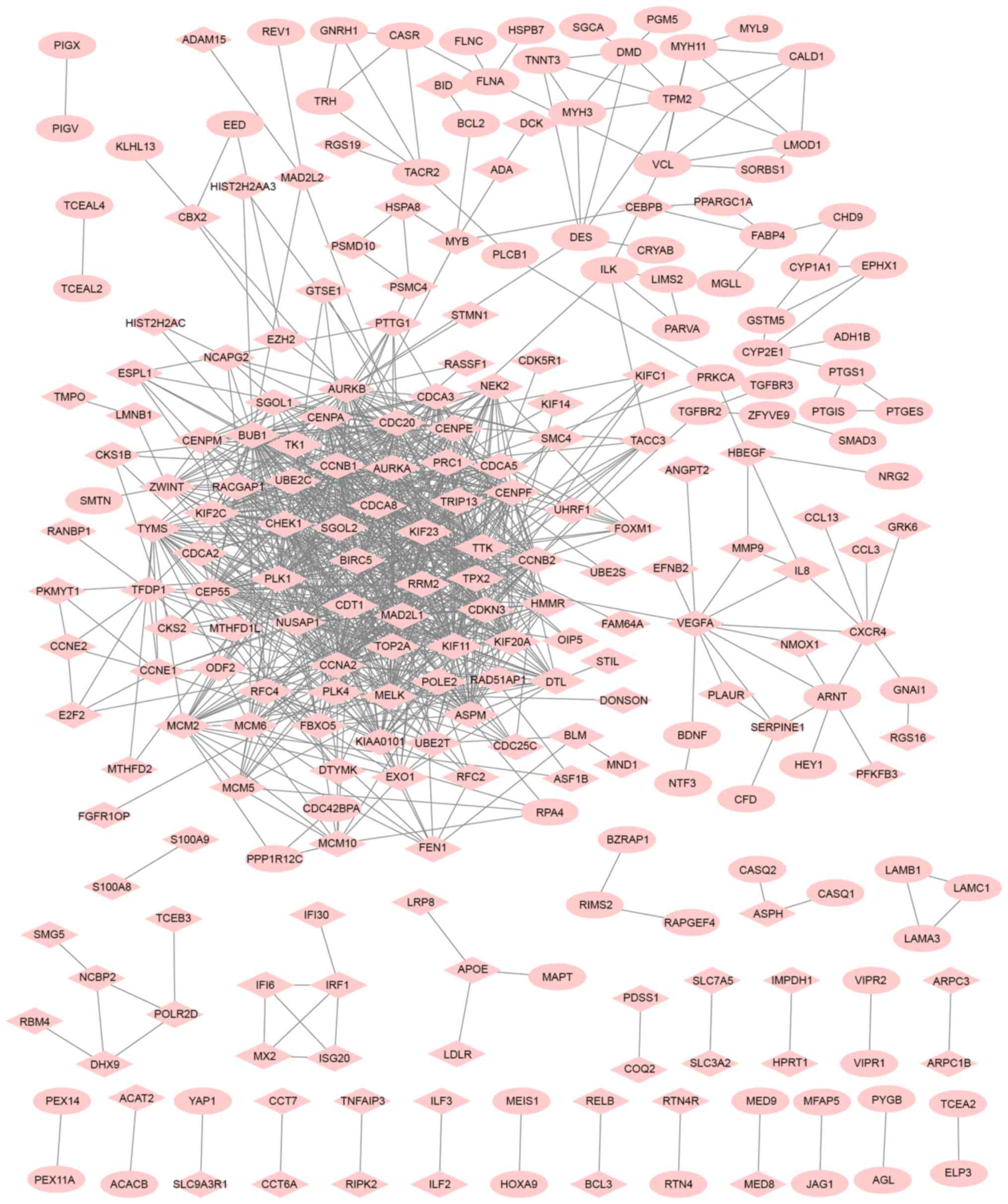
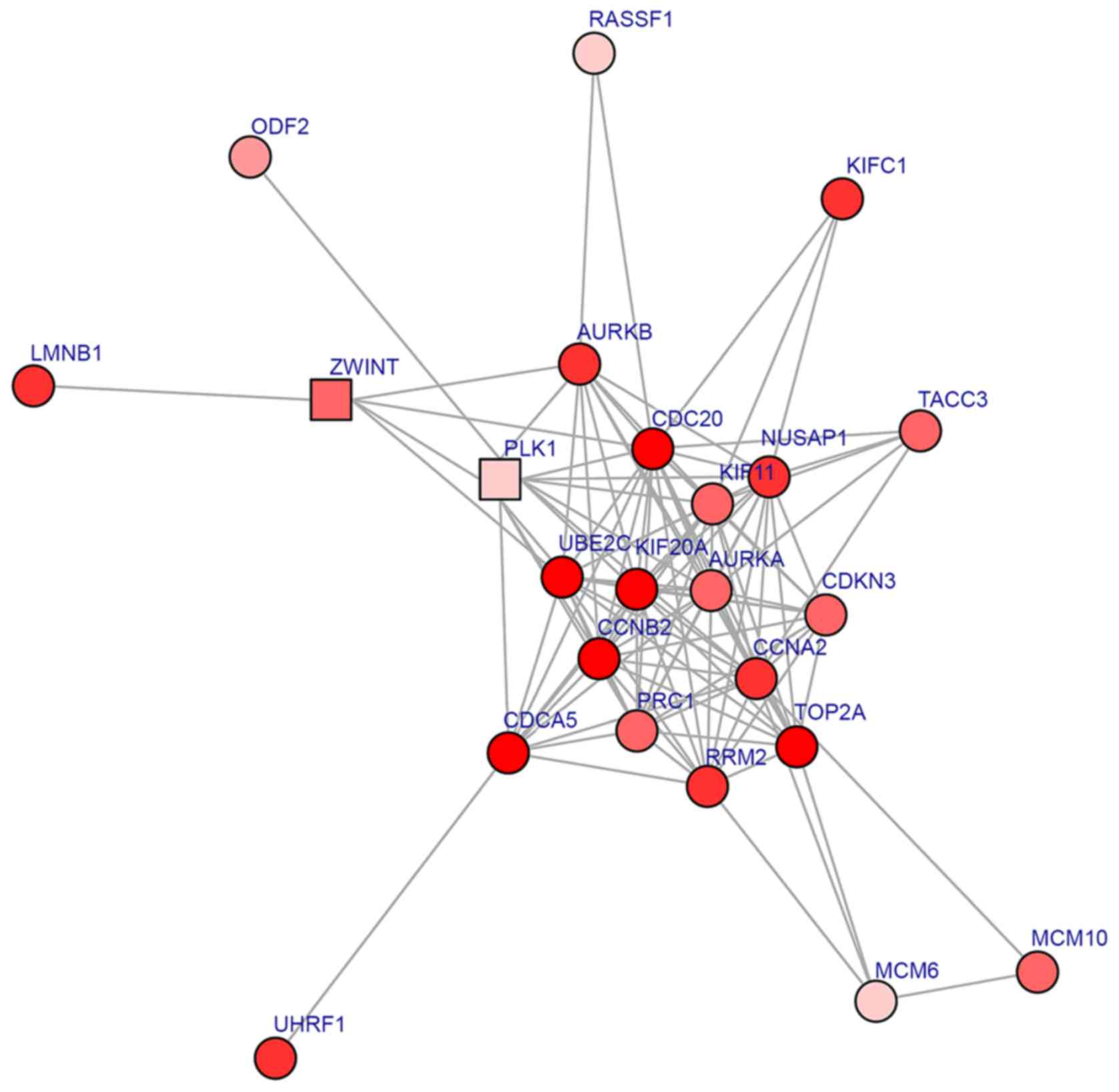
|
1
|
Di Pierro GB, Gulia C, Cristini C,
Fraietta G, Marini L, Grande P, Gentile V and Piergentili R:
Bladder cancer: A simple model becomes complex. Curr Genomics.
13:3952012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ye F, Wang L, Castillo-Martin M, McBride
R, Galsky MD, Zhu J, Boffetta P, Zhang DY and Cordon-Cardo C:
Biomarkers for bladder cancer management: Present and future. Am J
Clin Exp Urol. 2:1–14. 2014.PubMed/NCBI
|
|
3
|
Sanchez-Carbayo M, Socci ND, Lozano J,
Saint F and Cordon-Cardo C: Defining molecular profiles of poor
outcome in patients with invasive bladder cancer using
oligonucleotide microarrays. J Clin Oncol. 24:778–789. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Morrison CD, Liu P, Woloszynska-Read A,
Zhang J, Luo W, Qin M, Bshara W, Conroy JM, Sabatini L, Vedell P,
et al: Whole-genome sequencing identifies genomic heterogeneity at
a nucleotide and chromosomal level in bladder cancer. Proc Natl
Acad Sci USA. 111:pp. E672–E681. 2014; View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu JY, Qian D, He LR, Li YH, Liao YJ, Mai
SJ, Tian XP, Liu YH, Zhang JX, Kung HF, et al: PinX1 suppresses
bladder urothelial carcinoma cell proliferation via the inhibition
of telomerase activity and p16/cyclin D1 pathway. Mol Cancer.
12:1482013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Florl AR and Schulz WA: Chromosomal
instability in bladder cancer. Arch Toxicol. 82:173–182. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zieger K, Wiuf C, Jensen KM, Ørntoft TF
and Dyrskjøt L: Chromosomal imbalance in the progression of
high-risk non-muscle invasive bladder cancer. BMC Cancer.
9:1492009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Diboun I, Wernisch L, Orengo CA and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7:2522006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dai M, Wang P, Boyd AD, Kostov G, Athey B,
Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al: Evolving
gene/transcript definitions significantly alter the interpretation
of GeneChip data. Nucleic Acids Res. 33:e1752005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
da W Huang, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI
|
|
11
|
da W Huang, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Croft D, Mundo AF, Haw R, Milacic M,
Weiser J, Wu G, Caudy M, Garapati P, Gillespie M, Kamdar MR, et al:
The Reactome pathway knowledgebase. Nucleic Acids Res. 42(Database
Issue): D472–D477. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matys V, Fricke E, Geffers R, Gößling E,
Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis
OV, et al: TRANSFAC: Transcriptional regulation, from patterns to
profiles. Nucleic Acids Res. 31:374–378. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao M, Sun J and Zhao Z: TSGene: A web
resource for tumor suppressor genes. Nucleic Acids Res. 41(Database
Issue): D970–D976. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen JS, Hung WS, Chan HH, Tsai SJ and Sun
HS: In silico identification of oncogenic potential of fyn-related
kinase in hepatocellular carcinoma. Bioinformatics. 29:420–427.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Franceschini A, Szklarczyk D, Frankild S,
Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C
and Jensen LJ: STRING v9. 1: Protein-protein interaction networks,
with increased coverage and integration. Nucleic Acids Res.
41(Database Issue): D808–D815. 2013.PubMed/NCBI
|
|
19
|
Saito R, Smoot ME, Ono K, Ruscheinski J,
Wang PL, Lotia S, Pico AR, Bader GD and Ideker T: A travel guide to
Cytoscape plugins. Nat Methods. 9:1069–1076. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Beisser D, Klau GW, Dandekar T, Müller T
and Dittrich MT: BioNet: An R-Package for the functional analysis
of biological networks. Bioinformatics. 26:1129–1130. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Ji P, Liu J, Broaddus RR, Xue F
and Zhang W: Centrosome-associated regulators of the G2/M
checkpoint as targets for cancer therapy. Mol Cancer. 8:82009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Choi JW, Kim Y, Lee JH and Kim YS: High
expression of spindle assembly checkpoint proteins CDC20 and MAD2
is associated with poor prognosis in urothelial bladder cancer.
Virchows Arch. 463:681–687. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kidokoro T, Tanikawa C, Furukawa Y,
Katagiri T, Nakamura Y and Matsuda K: CDC20, a potential cancer
therapeutic target, is negatively regulated by p53. Oncogene.
27:1562–1571. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu Y, Liu P, Wen W, Grubbs CJ, Townsend
RR, Malone JP, Lubet RA and You M: Cross-species comparison of
orthologous gene expression in human bladder cancer and
carcinogen-induced rodent models. Am J Transl Res. 3:8–27.
2010.PubMed/NCBI
|
|
25
|
Lee SJ, Lee EJ, Kim SK, Jeong P, Cho YH,
Yun SJ, Kim S, Kim GY, Choi YH, Cha EJ, et al: Identification of
pro-inflammatory cytokines associated with muscle invasive bladder
cancer; the roles of IL-5, IL-20, and IL-28A. PLoS One.
7:e402672012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Blaveri E, Simko JP, Korkola JE, Brewer
JL, Baehner F, Mehta K, Devries S, Koppie T, Pejavar S, Carroll P
and Waldman FM: Bladder cancer outcome and subtype classification
by gene expression. Clin Cancer Res. 11:4044–4055. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Puzio-Kuter AM, Castillo-Martin M, Kinkade
CW, Wang X, Shen TH, Matos T, Shen MM, Cordon-Cardo C and
Abate-Shen C: Inactivation of p53 and Pten promotes invasive
bladder cancer. Genes Dev. 23:675–680. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Medina-Aguilar R, Marchat LA, Ocampo E
Arechaga, Gariglio P, García Mena J, Sepúlveda N Villegas, Martínez
Castillo M and López-Camarillo C: Resveratrol inhibits cell cycle
progression by targeting Aurora kinase A and Polo-like kinase 1 in
breast cancer cells. Oncol Rep. 35:3696–3704. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park HS, Park WS, Bondaruk J, Tanaka N,
Katayama H, Lee S, Spiess PE, Steinberg JR, Wang Z, Katz RL, et al:
Quantitation of Aurora kinase A gene copy number in urine sediments
and bladder cancer detection. J Natl Cancer Inst. 100:1401–1411.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu X, Gu J, Grossman HB, Amos CI, Etzel C,
Huang M, Zhang Q, Millikan RE, Lerner S, Dinney CP and Spitz MR:
Bladder cancer predisposition: A multigenic approach to DNA-repair
and cell-cycle-control genes. Am J Hum Genet. 78:464–479. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
de Martino M, Shariat SF, Hofbauer SL,
Lucca I, Taus C, Wiener HG, Haitel A, Susani M and Klatte T: Aurora
a kinase as a diagnostic urinary marker for urothelial bladder
cancer. World J Urol. 33:105–110. 2015. View Article : Google Scholar : PubMed/NCBI
|