Introduction
Worldwide, there were 429,800 new cases of and
165,100 deaths due to bladder cancer in 2012 (1). Gemcitabine
(2′,2′-difluoro-2′-deoxycytidine) is an important drug for treating
cancers including bladder cancer. The combination of gemcitabine
and cisplatin (GC) has been standard chemotherapy for metastatic
bladder cancer and for muscle invasive bladder cancer as a
neoadjuvant chemotherapy. GC is effective for about half of
patients with advanced or metastatic bladder cancer. In a phase III
study (2), the response rate was 49%.
In our hospital, the response rate for GC was reported as 44%
(3). However, the many of these
patients later developed progressive disease.
Biomarkers associated with and molecular mechanism
of gemcitabine resistance acquisition in bladder cancer are not
fully understood. It is possible that a protein that is more highly
expressed in gemcitabine-resistant bladder cancer may be a
gemcitabine-resistant biomarker or therapeutic target. Proteomic
analysis is an ideal method for identification of such a protein
and indeed, in recent years, proteins associated with
chemoresistance have been successfully identified using proteomic
analysis (4–6). Here, we used the method of isobaric tags
for relative and absolute quantification (iTRAQ method), which can
compare the protein levels of more than three samples in proteomic
analysis (7).
In the present study, to identify a protein
associated with gemcitabine resistance, we established
gemcitabine-resistant human bladder cancer cell lines (UMUC3GR,
HT1376GR) that we derived from human bladder cancer cell lines
(UMUC3 and HT1376). We analyzed these cell lines at the protein
level using the iTRAQ method, liquid chromatography, and tandem
mass spectrometry (MS/MS). In addition, we further analyzed a
protein that was found to be more highly expressed in
gemcitabine-resistant cell lines, using biochemical and molecular
biological techniques.
Materials and methods
Cell culture
The human bladder cancer cell lines, UMUC3 and
HT1376, which were used in this study, were purchased from DS
Pharma Biomedical (Osaka, Japan). UMUC3 cells were maintained in
minimum essential medium (MEM) supplemented with MEM non-essential
amino acids (NEAA) and sodium pyruvate (Gibco, St. Louis, MO, USA).
HT1376 cells were maintained in RPMI-1640 medium (Wako, Osaka,
Japan). Both media were supplemented with 10% FBS (Sigma-Aldrich,
St. Louis, MO, USA). The cells were incubated in a humidified
incubator at 37°C in an atmosphere of 5% CO2 and 95%
air. Each gemcitabine-resistant cell line (GR) was obtained from
the parental UMUC3 or HT1376 cells.
The UMUC3 and HT1376 cells were grown in cell
culture media containing gemcitabine (Wako), starting with a
concentration of 10-2 µM. The cells were then passaged through
stepwise increasing concentrations of gemcitabine up to a
concentration of 50 µM. The cells were repeatedly passaged at each
gemcitabine concentration in the stepwise gradient.
iTRAQ proteomic analysis with Liquid chromatography
tandem mass spectrometry (LC-MS/MS) analysis. The four human
bladder cancer cell lines HT1376, HT1376GR, UMUC3, and UMUC3GR were
each grown to 80% confluency, following which the cellular proteins
were extracted using Mammalian Protein Extraction Reagents (M-PER;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). Protein
concentration was determined using the BCA protein assay kit
(Thermo Fisher Scientific, Inc.). The protein concentration of the
cellular lysate was adjusted to a concentration of 1 µg/µl using
dissolution buffer. Each sample was digested with 1 µg/µl trypsin
solution (AB SCIEX, Framingham, MA, USA) at 37°C for 24 h and was
then desalted. Peptide samples from each of the cell lines were
labelled using the iTRAQ® Reagent-multiple Assay kit (AB
SCIEX) as follows: HT1376 with the 116 tag, HT1376GR with the 117
tag, UMUC3 with the 118 tag, and UMUC3GR with the 119 tag. All
samples were mixed and fractionated using strong cation exchange
chromatography (SCX) with a Cation Exchange Buffer Pack (AB SCIEX).
The peptide sample from each SCX fraction was enriched using a trap
column (HiQ sil C18; KYA Technologies, Tokyo, Japan) and was then
separated on an electrospray ionization (ESI) column (HiQ sil
C18P-3; KYA Technologies, Tokyo, Japan) at a flow rate of 150
nl/min. MS/MS analysis of peptide samples was carried out using the
Triple TOF™ 5600 system (AB SCIEX) interfaced with the DiNa system
(LC) (KYA Technologies, Tokyo, Japan).
MS and MS/MS data searches were carried out using
ProteinPilot™ software 4.5 (AB SCIEX). Searches used the UniProtKB
(http://www.uniprot.org) database. The false
discovery rate (FDR) was calculated and high-confidence protein
identifications were obtained by using a Global FDR from Fit 1.0%
at the peptide level. Quantitative estimates provided for each
protein by ProteinPilot were utilized: the fold change ratios of
differential expression between labeled protein extracts, and the
P-value representing the probability that the observed ratio is
different than 1 by chance. We selected 1.5-fold-change as a cutoff
to classify upregulated proteins.
Western blot analysis
Cells were lysed with cell lysis buffer containing
phenylmethanesulfonyl fluoride (Cell Signaling Technology, Inc.,
Danvers, MA, USA) and protease inhibitor cocktails (Sigma-Aldrich).
Samples were centrifuged at 14,000 × g for 10 min at 4°C,
and supernatants were electrophoresed on sodium dodecyl
sulfate-polyacrylamide gels and transferred to polyvinylidene
difluoride membranes (Millipore, Bedford, MA, USA). After blocking
with 5% skimmed milk, the membranes were probed with primary
antibodies against β-actin, p27 (Cell Signaling Technology, Inc.)
and ethylmalony-CoA decarboxylase (ECHDC1, Abcam) overnight at 4°C,
followed by horseradish peroxidase-conjugated secondary antibody
(Cell Signaling Technology, Inc.) for 1 h at room temperature. The
immune complexes were visualized with the Enhanced
Chemiluminescence Plus detection system (GE Healthcare, Piscataway,
NJ, USA) according to the manufacturer's instructions. The signal
was quantified using ImageJ and normalized to that of β-actin.
Immunofluorescence
Cells were seeded in an 8-well chamber slide (Thermo
Fisher Scientific, Inc.) and incubated for 24 h. The cells were
fixed with 4% paraformaldehyde followed by blocking with 1% bovine
serum albumin. The cells were incubated with an anti-ECHDC1
antibody (Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h.
Thereafter, they were incubated with fluorescein
isothiocyanate-conjugated secondary antibody (Jackson
ImmunoResearch Laboratories, West Grove, PA, USA) for 30 min.
Hoechst 33342 (Invitrogen Life Technologies, Carlsbad, CA, USA) was
used for nuclear staining. Fluorescence was photographed using a
BZ9000 Fluorescence microscope (Keyence Corporation, Osaka,
Japan).
Transfection
Silencer Select Negative control #1 siRNA (Life
Technologies Corp., Carlsbad, CA, USA) or Silencer Select
ECHDC1: Ethylmalonyl-CoA decarboxylase1 siRNA (s229273; Life
Technologies Corp.) was added to the adherent cells at a final
concentration of 5 nM using Lipofectamine® RNAiMAX
(Invitrogen Life Technologies) as the transfection reagent for 24
h.
Drug cytotoxicity analysis and real
time analysis of cell proliferation
Cells were seeded in 96-well plates at a density of
3×103 cells/well and were cultured with graded
concentrations of gemcitabine in at least three replicate wells at
37°C. At 72 h after gemcitabine exposure, the relative effect of
gemcitabine on the proliferation of each cell line was assessed by
using the Cell Counting kit-8 (CCK-8: Dojindo, Kumamoto, japan).
Absorbance at 450 nm was determined using a spectrophotometer
(Thermo Scientific Multiskan FC; Thermo Fischer Scientific, Inc.).
The absorbance of cells not treated with gemcitabine was considered
to be 100%.
Real-time analysis of cell proliferation was
performed using impedance measurement with the xCELLigence system.
Cell proliferation (Cell Index) was checked using the xCELLigence
Real-Time Cell Analyzer (RTCA) instrument according to the
instructions of the supplier (Roche Applied Science and ACEA
Biosciences, San Diego, CA, USA). This system has been extensively
used in other studies (8,9). The xCELLigence system can quantify the
electrical impedance across electrodes at the bottom of each well
of the tissue culture plates. Impedance changes reflect cell
numbers, and cell viability is expressed as Cell Index values.
Cells were seeded at a density of 8×103 cells/well in a
specialized 8-well plate (E-plate) used with the RTCA instrument.
After leaving the E-plates at room temperature for 30 min to allow
for cell attachment, they were locked into the RTCA xCELLigence
instrument and the experiment was allowed to run for 96 h at 37°C.
Cell Index values were recorded at 15 min interval sweeps until the
end of the experiment. We normalized the Cell Index at 24 h after
seeding the cells.
Cell cycle analysis
Cells were collected using Accutase (Innovative Cell
Technologies, San Diego, CA, USA). Cell cycle analysis was
performed using the CycleTest Plus DNA reagent kit (BD Biosciences,
San Jose, CA, USA). The data were analyzed using ModFitLT (Verity
software House, Topsham, ME, USA) to generate percentages of cells
in G0/G1, S and G2/M phases. At least 18,000 cells were analyzed in
each experiment.
Statistical analysis
Quantitative data are expressed as means ± standard
deviation (SD). Statistical significance was assessed using
Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Establishment of gemcitabine-resistant
bladder cancer cell lines
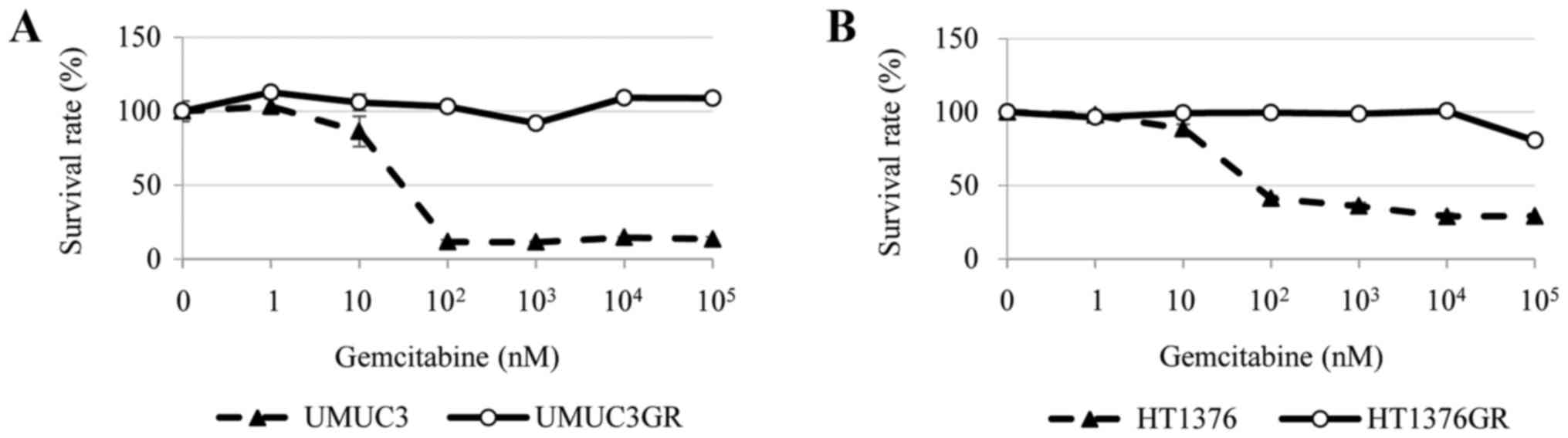
The cytotoxicity of gemcitabine towards the four
human bladder cancer cell lines HT1376, HT1376GR, UMUC3, and
UMUC3GR was examined. Analysis of cell viability using CCK-8
indicated that, while the parental cell lines showed a
dose-dependent sensitivity to gemcitabine, the HT1376GR and UMUC3GR
cell lines derived from them had acquired gemcitabine resistance
(Fig. 1A and B).
Identification of proteins involved in
gemcitabine resistance using iTRAQ proteomic analysis
A total of 3,930 proteins were identified in the
cell lines using iTRAQ proteomic analysis. Of these proteins, the
expression of several proteins was increased more than 1.5-fold in
the gemcitabine-resistant cells (UMUC3GR and HT1376GR), compared to
the corresponding gemcitabine-sensitive parental cells (UMUC3 and
HT1376). Only expression of the ECHDC1 protein was significantly
increased (P<0.05) in both of the gemcitabine-resistant cell
lines (Table I).
 | Table I.Proteins with increased expression in
gemcitabine-resistant cells identified using iTRAQ proteomic
analysis. |
Table I.
Proteins with increased expression in
gemcitabine-resistant cells identified using iTRAQ proteomic
analysis.
| Protein name | Gene | Fold-change
HT1376GR/HT1376 | P-value | Fold-change
UMUC3GR/UMUC3 | P-value | %Cov(95) | Accession number |
|---|
| Ethylmalonyl-CoA
decarboxylase | ECHDC1 | 1.674 | 0.034 | 3.593 | 0.000 | 42.00 | sp|Q9NTX5 |
| Integrin α-2 | ITGA2 | 3.356 | 0.359 | 1.778 | 0.205 | 23.60 | sp|P17301 |
|
Vasodilator-stimulated phosphoprotein | VASP | 3.645 | 0.049 | 1.609 | 0.131 | 15.50 | sp|P50552 |
| Isoform 2 of Cleavage
stimulation factor subunit 2 | CSTF2 | 1.898 | 0.264 | 1.913 | 0.180 | 12.90 | sp|P33240-2 |
| Scaffold attachment
factor B2 | SAFB2 | 1.551 | 0.039 | 1.596 | 0.398 | 13.60 | sp|Q14151 |
| Serine incorporator
1 | SERINC1 | 2.475 | 0.139 | 1.593 | 0.286 | 6.60 | sp|Q9NRX5 |
| Histone deacetylase
3 | HDAC3 | 1.970 | 0.363 | 2.225 | 0.084 | 5.40 | sp|O15379 |
| ATP-dependent RNA
helicase SUPV3L1 | SUPV3L1 | 2.134 | 0.281 | 1.705 | 0.121 | 3.20 | sp|Q8IYB8 |
Western blotting and
immunofluorescence analysis of ECHDC1 protein expression in
gemcitabine-resistant cells
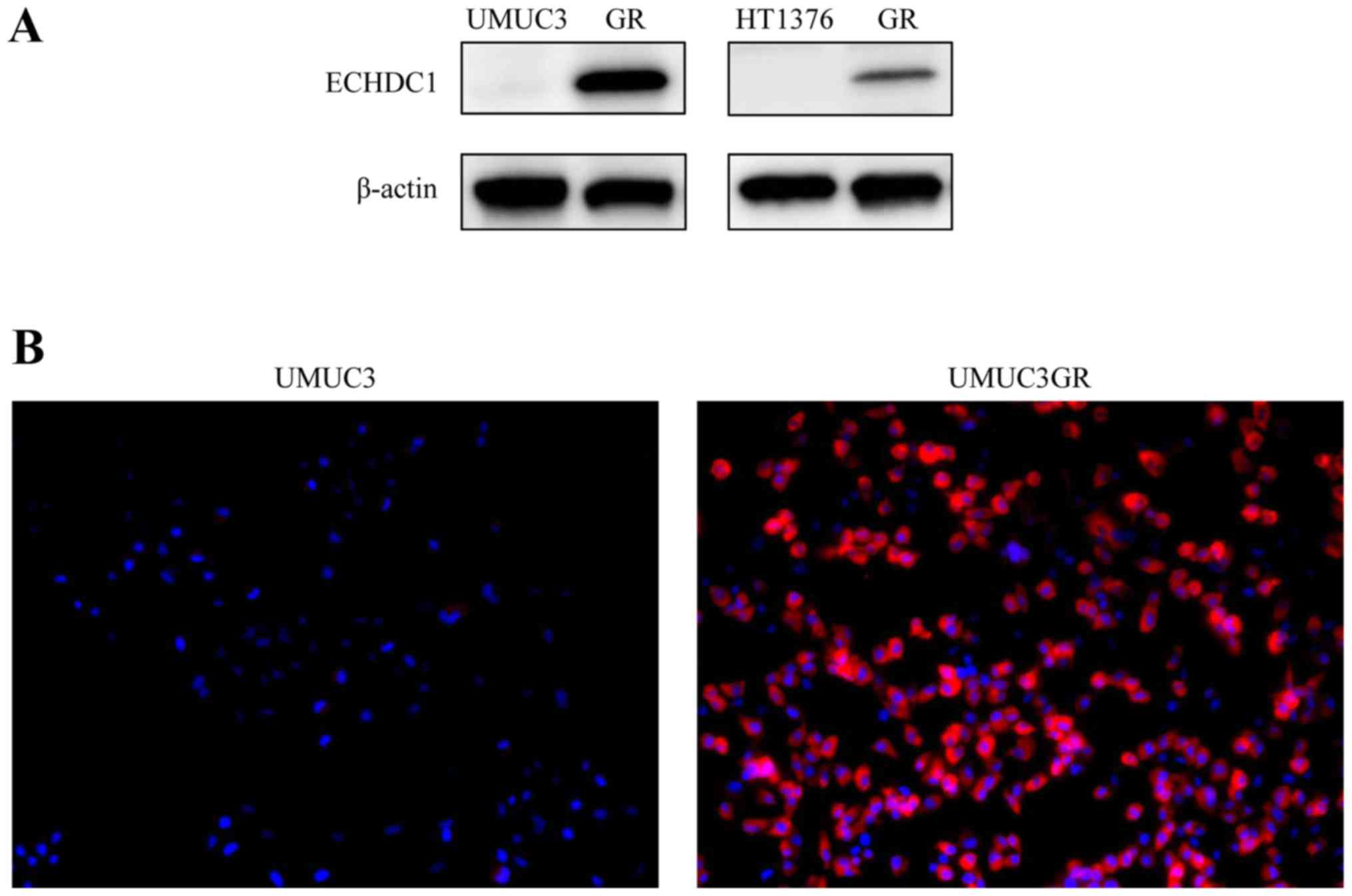
Western blotting of the four cell lines confirmed a
strong increase in ECHDC1 protein levels (34 kDa) in the
gemcitabine-resistant cells, UMUC3GR and HT1376GR, compared with
its expression in the respective parental UMUC3 and HT1376 cell
lines (Fig. 2A). Immunofluorescence
analysis also resulted in a much stronger cytoplasmic ECHDC1
protein signal in UMUC3GR cells than in the UMUC parental cells
(Fig. 2B).
Silencing of ECHDC1 significantly
inhibited cell proliferation in UMUC3GR cells
To examine the functional role of the ECHDC1 protein
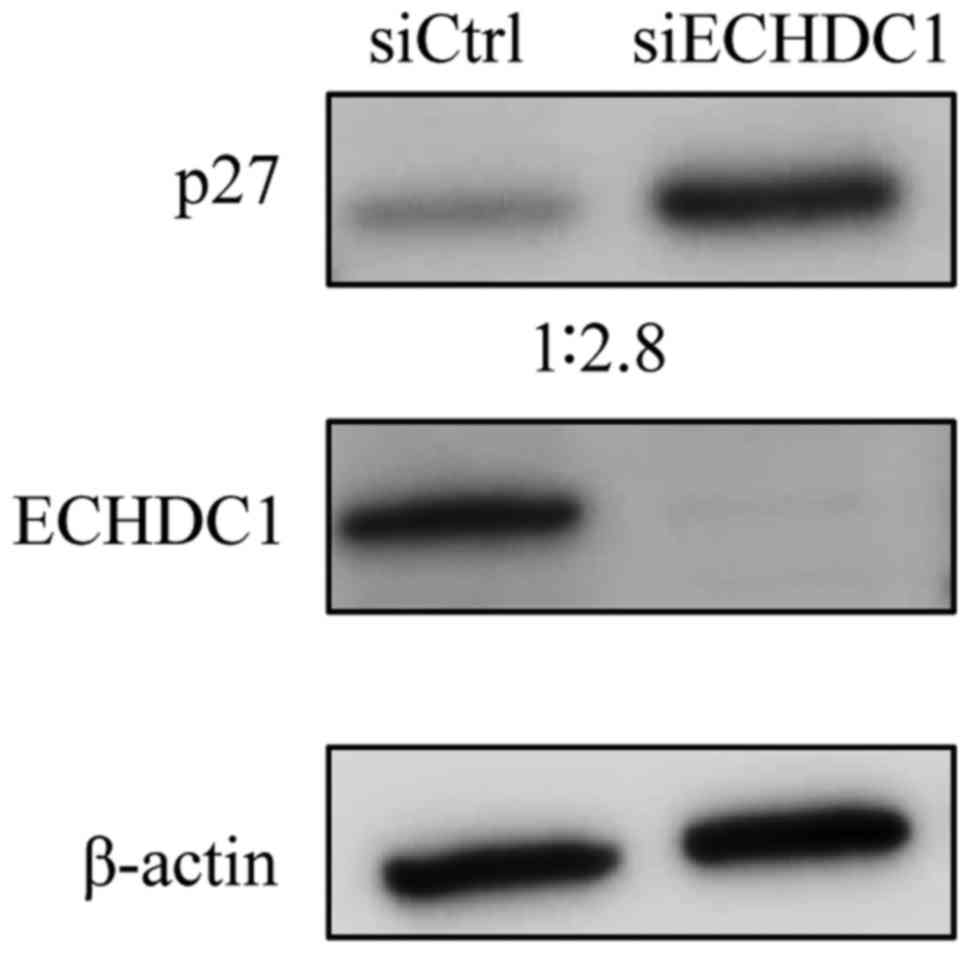
in bladder cancer cells, we knocked down ECHDC1 in UMUC3GR
cells using siRNA. We confirmed using western blotting that ECHDC1
protein expression was significantly reduced in cells transfected
with ECHDC1-siRNA compared with cells transfected with
control siRNA (Fig. 3). We then
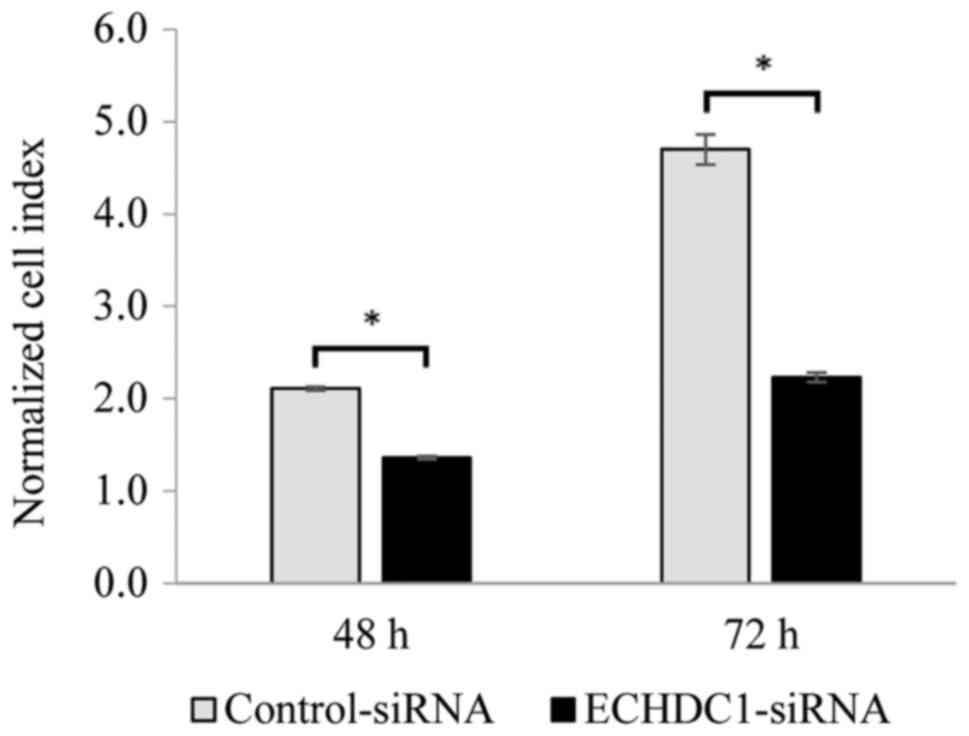
determined the effect of ECHDC1 silencing on cell
proliferation in vitro. RTCA analysis showed that the
proliferation of bladder cancer cells was significantly inhibited
(P<0.05) by silencing of ECHDC1 compared to cells
transfected with control siRNA (Fig.
4).
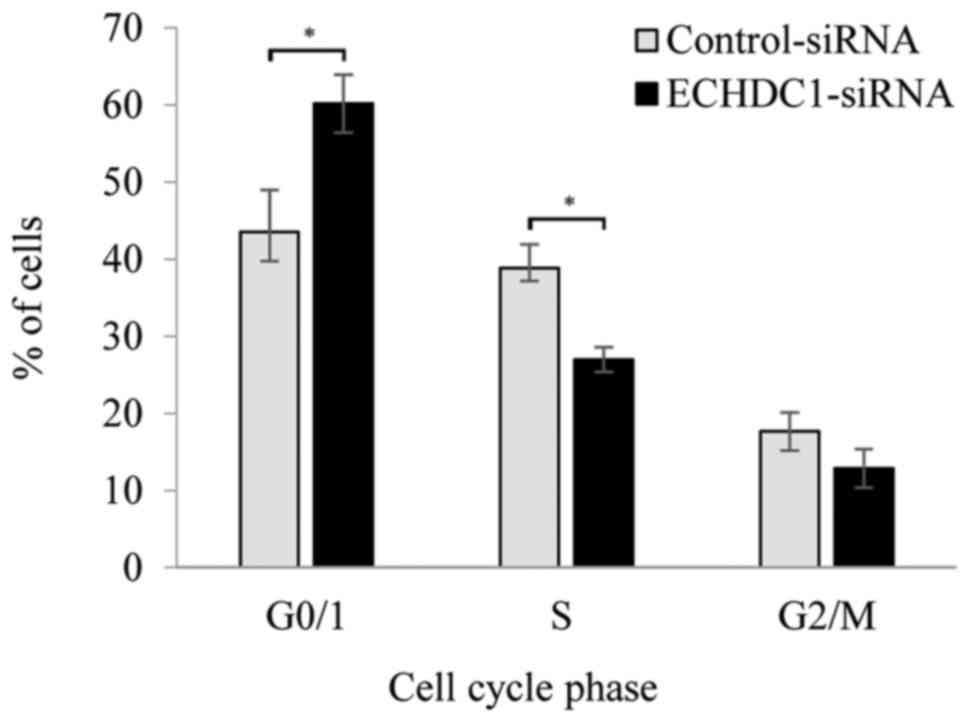
Knockdown of ECHDC1 induced G0/G1
phase cell cycle arrest and upregulation of p27
To investigate the mechanism of the
anti-proliferative effect of knockdown of ECHDC1 on UMUC3GR
cells, we measured the percentage of the cells in different cell
cycle phases using flow cytometry with propidium iodide (PI)
staining at 48 h after transfection with siRNA against
ECHDC1 or with control siRNA. Knockdown of ECHDC1
induced significantly higher accumulation of cells in the
G0/G1-phase cell compared with the control (P<0.05). Thus, the
percentage of G0/G1 phase cells was increased from 43.5% in the
control to 60.2% in the knockdown cells, and the percentage of S
phase cells was decreased from 38.8 to 27.0% (Fig. 5). To investigate the molecular basis
of these changes, we analyzed the expression of proteins involved
in cell cycle regulation. Western blotting indicated that
expression of the p27 protein (27 kDa), which is critical for cell
cycle arrest in the G1 phase, was increased in ECHDC1
knockdown cells compared to the control cells (Fig. 3).
Discussion
iTRAQ proteomic analysis has been shown to be a
useful technique for investigation of chemoresistant factors in
cancer (6,10). We therefore used iTRAQ proteomic
analysis to identify proteins associated with gemcitabine
resistance by comparing the expression of proteins in two
gemcitabine-resistant cell lines (UMUC3GR, HT1376GR) with that of
the two parental gemcitabine-sensitive cell lines (UMUC3, HT1376).
This analysis showed that expression of the ECHDC1 protein was
significantly increased in both of the gemcitabine-resistant cell
lines compared to the parental cells.
ECHDC1 has been identified as a new metabolite
proofreading enzyme, ethylmalonyl-CoA decarboxylase (11,12). It is
localized mainly in the cytosol and corrects a side activity of
acetyl-CoA carboxylase. Acetyl-CoA carboxylase synthesizes
malonyl-CoA from acetyl-CoA, and malonyl-CoA then feeds into the de
novo fatty acid synthesis pathway (13). However, Acetyl-CoA carboxylase
displays a lack of substrate specificity, and it is also able to
synthesize methylmalonyl-CoA and ethylmalonyl-CoA (14–16).
Ethylmalonyl-CoA could perturb lipid synthesis by trapping CoA and
inhibiting fatty acid synthesis, leading to the formation of
abnormal ethyl-branched fatty acids due to its structural
similarity with malonyl-CoA. Ethylmalonyl-CoA decarboxylase
(ECHDC1) can eliminate ethylmalonyl-CoA by converting it to
butyryl-CoA (11).
We observed that silencing of ECHDC1
significantly inhibited bladder cancer cell proliferation. This is
the first report to identify a function for ECHDC1 in cancer. The
ECHDC1 gene is included in a novel breast cancer risk locus
on 6q22.33 that was identified in a genome-wide association study
(17). However, the mechanism of
induced cancer risk is unknown. In human cells, silencing of
ECHDC1 decreased ethylmalonyl-CoA decarboxylase activity and
increased the formation of ethylmalonic acid (EMA) (11). EMA induces oxidative stress in
skeletal muscle and in the cerebral cortex (18,19). Human
cancer cells are more sensitive to oxidative stress, which inhibits
cell proliferation (20). Oxidative
stress regulates the intracellular level of p27 (21). The p27 protein is a member of the
Cip/Kip family of cyclin-dependent kinase inhibitors that bind to
cyclin/CDK complexes and inhibit their activities. p27 arrests the
cell cycle in the G1 phase (22–24). In
agreement with these reports, we observed increased p27 expression
and G1 arrest in bladder cancer cells in which ECHDC1 was
silenced.
The limitation of this study is that we could not
identify the detailed mechanism by which gemcitabine increased
ECHDC1 and by which reduction of ECHDC1 inhibited the growth of
bladder cancer cells. Accumulation of EMA or perturbation of lipid
synthesis by decreasing ethylmalonyl-CoA decarboxylase activity may
be the cause. Further studies including animal or clinical
specimens are required to understand the role of ECHDC1 in
cancer.
In conclusion, ECHDC1 was increased in
gemcitabine-resistant bladder cancer cells and silencing of
ECHDC1 inhibited cell proliferation. The present study
suggested that gemcitabine may have induced ECHDC1 expression and
that ECHDC1 may be a novel potential target for development of
gemcitabine-resistant bladder cancer treatment.
This article does not contain any studies with human
participants or animals performed by any of the authors.
Acknowledgements
We thank Kenji Kameda (Integrated center for
sciences) and Kazumi Kanno, Izumi Tanimoto, and Maria Mori for
their excellent technical assistance.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global Cancer Statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
von der Maase H, Hansen SW, Roberts JT,
Dogliotti L, Oliver T, Moore MJ, Bodrogi I, Albers P, Knuth A,
Lippert CM, et al: Gemcitabine and cisplatin versus methotrexate,
vinblastine, doxorubicin, and cisplatin in advanced or metastatic
bladder cancer: Results of a large, randomized, multinational,
multicenter, phase III study. J Clin Oncol. 18:3068–3077. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tanji N, Ozawa A, Miura N, Yanagihara Y,
Sasaki T, Nishida T, Kikugawa T, Ikeda T, Ochi T, Shimamoto K, et
al: Long-term results of combined chemotherapy with gemcitabine and
cisplatin for metastatic urothelial carcinomas. Int J Clin Oncol.
15:369–375. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee DH, Chung K, Song JA, Kim TH, Kang H,
Huh JH, Jung SG, Ko JJ and An HJ: Proteomic identification of
paclitaxel-resistance associated hnRNP A2 and GDI 2 proteins in
human ovarian cancer cells. J Proteome Res. 9:5668–5676. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun QL, Sha HF, Yang XH, Bao GL, Lu J and
Xie YY: Comparative proteomic analysis of paclitaxel sensitive A549
lung adenocarcinoma cell line and its resistant counterpart
A549-Taxol. J Cancer Res Clin Oncol. 137:521–532. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nishimura K, Tsuchiya Y, Okamoto H, Ijichi
K, Gosho M, Fukayama M, Yoshikawa K, Ueda H, Bradford CR, Carey TE
and Ogawa T: Identification of chemoresistant factors by protein
expression analysis with iTRAQ for head and neck carcinoma. Br J
Cancer. 111:799–806. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ross PL, Huang YN, Marchese JN, Williamson
B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, et
al: Multiplexed protein quantitation in Saccharomyces cerevisiae
using amine-reactive isobaric tagging reagents. Mol Cell
Proteomics. 3:1154–1169. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bang D, Wilson W, Ryan M, Yeh JJ and
Baldwin AS: GSK-3α promotes oncogenic KRAS function in pancreatic
cancer via TAK1-TAB stabilization and regulation of noncanonical
NF-κB. Cancer Discov. 3:690–703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Weiler M, Blaes J, Pusch S, Sahm F,
Czabanka M, Luger S, Bunse L, Solecki G, Eichwald V, Jugold M, et
al: mTOR target NDRG1 confers MGMT-dependent resistance to
alkylating chemotherapy. Proc Natl Acad Sci USA. 111:pp. 409–414.
2014; View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang Y, Chen Y, Saha MN, Chen J, Evans K,
Qiu L, Reece D, Chen GA and Chang H: Targeting phospho-MARCKS
overcomes drug-resistance and induces antitumor activity in
preclinical models of multiple myeloma. Leukemia. 29:715–726. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Linster CL, Noël G, Stroobant V, Vertommen
D, Vincent MF, Bommer GT, Veiga-da-Cunha M and Van Schaftingen E:
Ethylmalonyl-CoA decarboxylase, a new enzyme involved in metabolite
proofreading. J Biol Chem. 286:42992–43003. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Van Schaftingen E, Rzem R, Marbaix A,
Collard F, Veiga-da-Cunha M and Linster CL: Metabolite
proofreading, a neglected aspect of intermediary metabolism. J
Inherit Metab Dis. 3:427–434. 2013. View Article : Google Scholar
|
|
13
|
Mounier C, Bouraoui L and Rassart E:
Lipogenesis in cancer progression (Review). Int J Oncol.
45:485–492. 2014.PubMed/NCBI
|
|
14
|
Bettey M, Ireland RJ and Smith AM:
Purification and characterization of acetyl CoA carboxylase from
developing pea embryos. J Plant Physiol. 140:513–520. 1992.
View Article : Google Scholar
|
|
15
|
Miller AL and Levy HR: Acetyl-CoA
carboxylase from rat mammary gland. EC 6.4.1.2 acetyl-CoA: Carbon
dioxide ligase (ADP). Methods Enzymol. 35:11–17. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Waite M and Wakil SJ: Studies on the
mechanism of fatty acid synthesis: XII. Acetyl coenzyme A
carboxylase. J Biol Chem. 237:2750–2757. 1962.PubMed/NCBI
|
|
17
|
Gold B, Kirchhoff T, Stefanov S,
Lautenberger J, Viale A, Garber J, Friedman E, Narod S, Olshen AB,
Gregersen P, et al: Genome-wide association study provides evidence
for a breast cancer risk locus at 6q22.33. Proc Natl Acad Sci USA.
105:pp. 4340–4345. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schuck PF, Busanello EN, Moura AP, Tonin
AM, Grings M, Ritter L, Vargas CR, da Costa Ferreira G and Wajner
M: Promotion of lipid and protein oxidative damage in rat brain by
ethylmalonic acid. Neurochem Res. 35:298–305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schuck PF, Milanez AP, Felisberto F,
Galant LS, Machado JL, Furlanetto CB, Petronilho F, Dal-Pizzol F,
Streck EL and Ferreira GC: Brain and muscle redox imbalance
elicited by acute ethylmalonic acid administration. PLoS One.
10:e01266062015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Albright CD, Klem E, Shah AA and Gallagher
P: Breast cancer cell-targeted oxidative stress: Enhancement of
cancer cell uptake of conjugated linoleic acid, activation of p53,
and inhibition of proliferation. Exp Mol Pathol. 79:118–125. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Quintos L, Lee IA, Kim HJ, Lim JS, Park J,
Sung MK, Seo YR and Kim JS: Significance of p27 as potential
biomarker for intracellular oxidative status. Nutr Res Pract.
4:351–355. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee J and Kim SS: The function of p27 KIP1
during tumor development. Exp Mol Med. 41:765–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Polyak K, Kato JY, Solomon MJ, Sherr CJ,
Massague J, Roberts JM and Koff A: p27Kip1, a cyclin-Cdk inhibitor,
links transforming growth factor-beta and contact inhibition to
cell cycle arrest. Genes Dev. 8:9–22. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Toyoshima H and Hunter T: p27, a novel
inhibitor of G1 cyclin-Cdk protein kinase activity, is related to
p21. Cell. 78:67–74. 1994. View Article : Google Scholar : PubMed/NCBI
|