Introduction
It is estimated that >1.6 million new cases of
cancer were diagnosed in the United States of America in 2015
(1). Therefore, cancer is one of the
most common diseases and a major cause of mortality in modern
society. Distant metastases, rather than cell proliferation itself,
are lethal and contribute to >90% of mortalities among patients
with cancer (2). Although numerous
breakthroughs in this field allow for the clinical management
cancer, the underlying molecular mechanisms of metastasis remain
poorly understood. Notably, numerous publications have reported
that different types of cancer induce metastases preferentially at
specific distal sites, which supports the concept of the ‘seed and
soil’ theory proposed by Stephen Paget (3). According to this theory, the organ
microenvironment serves a vital role in the formation of
metastases. The organ in which metastasis from a certain type of
cancer may or may not occur depends on the interaction between the
disseminating cancer cells and the microenvironment of the organ
itself.
Following the lungs and liver, bone is the third
most common site of metastasis (4).
According to data from autopsy studies, as many as 30–70% of
patients with cancer have spinal metastases (5), which demonstrates that the spine is a
common site for skeletal metastases. Spinal metastases destroy the
stability of the spine, leading to refractory pain, fractures and
devastating neurologic consequences (6). This process results in a significant
negative impact on morbidity and survival (7). Although patients have an increased
number of therapeutic options because of improvements in
multidisciplinary treatments, patients with spinal metastases still
have a poor quality of life for the remaining course of their
disease (8). This may be ascribed to
the failure to appreciate that prevention, rather than the cure of
spinal metastasis, may be a more successful approach. To achieve
this, a comprehensive knowledge of the underlying molecular
mechanisms of spinal metastasis is required.
To investigate the underlying molecular mechanisms
of organ-specific metastasis, particularly in the bone and spine,
researchers have examined the genes potentially involved in this
process in different primary cancers, including breast (9), prostate (10) and lung (11,12)
cancer. However, the majority of existing studies investigating
spinal metastasis focus primarily on the analysis of the genetic
alterations in the metastatic cells, rather than the
microenvironment of the sites of metastasis in the spinal tissue.
Given the interaction between disseminated cancer cells and the
microenvironment of the spine, certain features of the gene
expression of cancellous bone tissue in the spine may have changed
to mediate and favor the colonization of cancer cells. Hence,
investigating the gene expression profile of cancellous bone in
spinal metastases may aid in future studies into the underlying
molecular mechanisms of spinal metastasis.
In the present study, a microarray analysis was
performed to identify differentially expressed genes (DEGs) in
cancellous bone tissue from patients with spinal metastases
compared with that from normal control patients. To explore
different gene expression signatures and the underlying molecular
mechanisms associated with spinal metastases, 5 patients with
different primary cancers (lung, breast, liver, prostate and kidney
cancer) were included. Additionally, gene ontology (GO) term and
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analyses were performed, and a protein-protein interaction (PPI)
network was constructed to identify key (hub) genes. The current
study aimed to provide a comprehensive perspective into the
underlying molecular mechanisms, and the prevention and treatment,
of spinal metastases.
Materials and methods
Patients and specimens
The present study was reviewed and approved by the
Ethics Committee of Zhongshan Hospital (Fudan University, Shanghai,
China). Written informed consent was obtained from all patients or
their families. In total, 8 participants were enrolled, including 5
patients with spinal metastases (Group 1) and 3 normal controls
(Group 2; Table I). Each of the 5
patients had a different primary cancer, including lung, breast,
liver, prostate and kidney cancer, confirmed by pathological
diagnosis. Between January 2012 and December 2015, each patient
received a total en bloc spondylectomy, and cancellous bone tissue
specimens 0.5 mm away from the metastatic spinal tumors were
obtained and snap frozen in liquid nitrogen. The specimens were
stored at −80°C until gene expression analysis was performed. The 3
normal controls were patients who had undergone spinal surgery at
Zhongshan Hospital due to non-cancerous diseases. The cancellous
bone from their spines was collected via procedures that were
already required during the surgery, such as decompression. These
specimens were also stored at −80°C.
 | Table I.Clinical profiles of the patients with
spinal metastasis and normal patients. |
Table I.
Clinical profiles of the patients with
spinal metastasis and normal patients.
| Patient group | Sex | Age (years) | Primary disease | Location of
specimen |
|---|
| Group 1a |
|
|
|
|
| 1 | Female | 75 | Lung cancer | T4 |
| 2 | Female | 39 | Breast cancer | T9 |
| 3 | Male | 50 | Liver cancer | L2 |
| 4 | Male | 57 | Prostate cancer | T10 |
| 5 | Male | 80 | Kidney cancer | C4 |
| Group 2b |
|
|
|
|
| 1 | Female | 65 | Cervical
spondylopathy | C5 |
| 2 | Male | 33 | Spine fracture | L4 |
| 3 | Male | 63 | Disc
herniation | T6 |
RNA isolation
Total RNA was extracted from each specimen using the
RNeasy Protect Mini kit (Qiagen, Inc., Valencia, CA, USA) according
to the manufacturer's protocol. The concentration of the RNA
obtained was detected using a NanoDrop 1000 spectrophotometer
(NanoDrop; Thermo Fisher Scientific, Inc., Wilmington, DE, USA).
The RNA integrity was assessed via a Bioanalyzer 2100 (Agilent
Technologies, Inc., Santa Clara, CA, USA). Specimens with an
absorbance (A) 260/A280 ratio >1.9 and RNA integrity values
>8.0 were used for further analysis.
Microarray analysis
Gene expression profiles were assessed using
Illumina HumanHT-12_V4 BeadChip arrays (cat. no. 9479628056;
Illumina Inc., San Diego, CA, USA). Each array contained >47,000
probes, including specific gene probes or probe sets derived from
the National Center for Biotechnology Information RefSeq and
UniGene databases (13). According to
the manufacturer's protocol, reverse transcription to synthesize
first strand complementary (c) DNA was primed with T7 Oligo (dT)
Primer in order to synthesize cDNA containing a T7 promoter
sequence. Single-stranded cDNA was subsequently converted into a
double-stranded DNA, providing the template for transcription.
During the amplification and labeling step, multiple copies of
biotinylated cRNA from the double-stranded cDNA templates were
generated. Following purification, the cRNA was ready for use with
the Illumina direct hybridization array kits (14). The cRNA was hybridized to the bead
arrays at 55°C for 18 h and then scanned using an Illumina iScan
reader (cat. no. 9479628056; Illumina Inc., San Diego, CA,
USA).
Data pre-processing, differential
expression analysis and clustering
The initial array scan intensity data were analyzed
using Illumina Genome Studio Gene Expression Module software
(v1.1.1; Illumina Inc.). Data pre-processing, such as background
adjustment, normalization and log transformation of the values, was
performed. Furthermore, the probe-level data were converted to gene
expression values. Where several probes corresponded to one gene,
the mean value of the probe-level data was taken as the gene
expression value. Cluster analysis was used to group the patients
into clusters. Patients assigned to the same cluster are more
closely related to one another compared with patients assigned to
different clusters. An unpaired t-test analysis was used to
identify the DEGs between the spinal metastasis and normal groups.
Then, the log2 fold change value was calculated. The raw
P-values were adjusted into false discovery rates (FDRs) using the
Benjamin and Hochberg method as described previously (15). An FDR <0.05 and
|log2FC|>1 were used as the cut-off criteria to
identify significantly DEGs. Finally, the cluster analysis was used
to group the cases into clusters according to the DEGs.
GO term and KEGG pathway enrichment
analyses
GO (16) is a tool for
the unification of biology in terms of biological processes,
molecular functions and cellular components. KEGG (17) is a knowledge database used for
classifying correlating gene sets into their respective signaling
pathways. The Database for Annotation, Visualization and Integrated
Discovery (DAVID) (18), a
comprehensive set of functional annotation tools, is used for the
systematic and integrative analysis of large gene lists. To analyze
the DEGs at the functional level, GO term and KEGG pathway
enrichment analyses were performed using the DAVID online tool to
obtain the enriched GO terms and pathways via a clustering
algorithm. P<0.05 was set as the threshold value.
PPI network construction
The Search Tool for the Retrieval of Interacting
Genes (STRING) database (19) is a
pre-computed global resource, which was designed to explore and
evaluate PPI information. In the present study, the PPI of the DEGs
identified was screened with a required confidence (combined) score
>0.4 using the STRING online tool (version 10.0). Then, the PPI
network was constructed and visualized using Cytoscape (20), which is a general bioinformatics
package to aid in visualizing biological networks and integrating
PPI data. Given that the majority of the networks were scale-free,
hub genes with a connectivity degree >5 were selected, as
described previously (21).
Results
Identification of DEGs in spinal
metastasis
The genes that were significantly upregulated or
downregulated in the cancellous bone samples from patients with
spinal metastases compared with the samples from normal patients
were identified with a FDR <0.05 and a |log2FC|>1.
As a result, a total of 540 DEGs were obtained following data
processing (data not shown). Among the DEGs, 152 were significantly
upregulated and 388 were significantly downregulated.
Cluster analysis
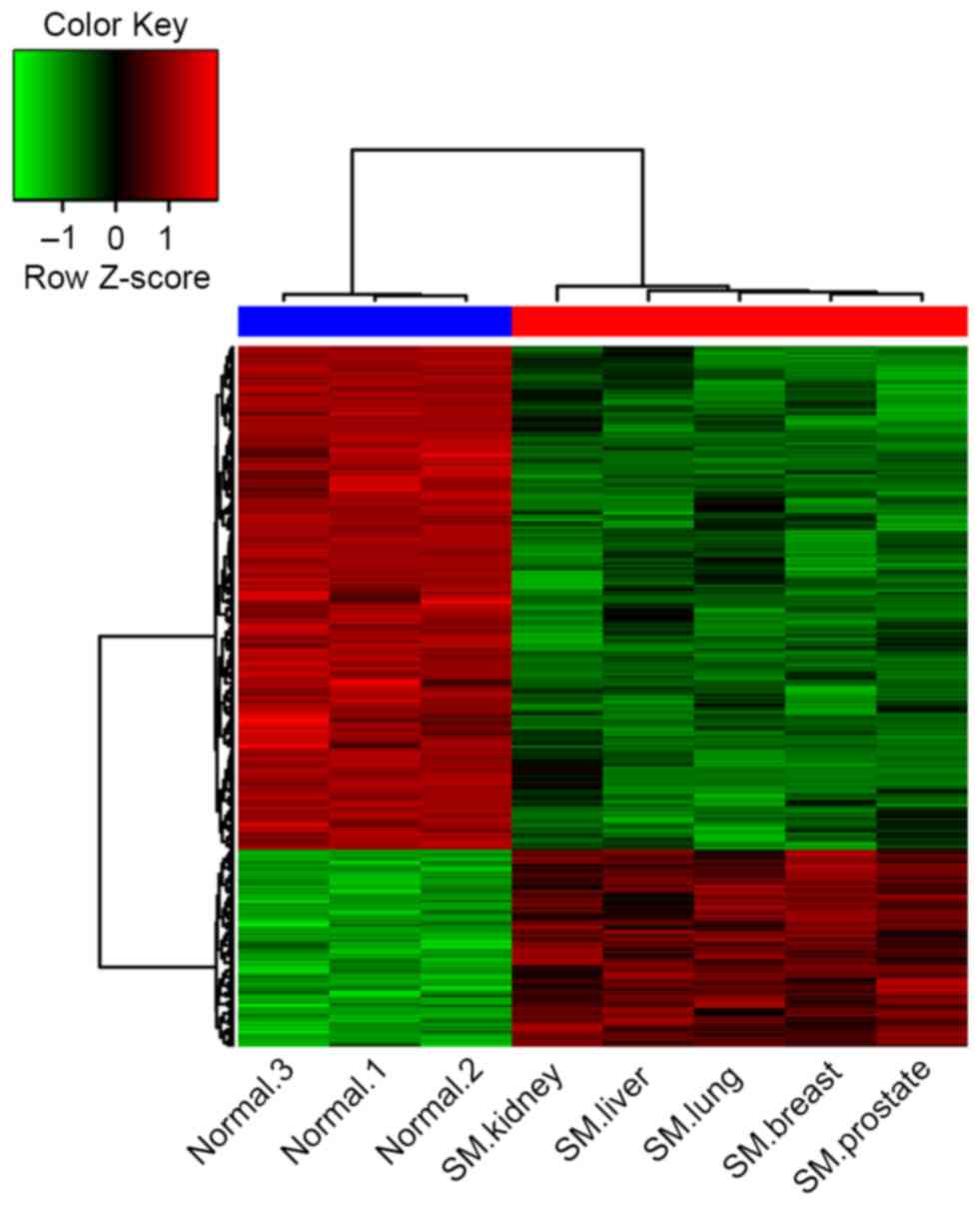
The clustering of the DEGs demonstrated that the
gene expression signature in samples from patients with spinal
metastases more closely resembled each other compared with the
normal controls (Fig. 1). There were
notable differences between the cancellous bone from patients with
spinal metastases and the normal controls according to their gene
expression signatures.
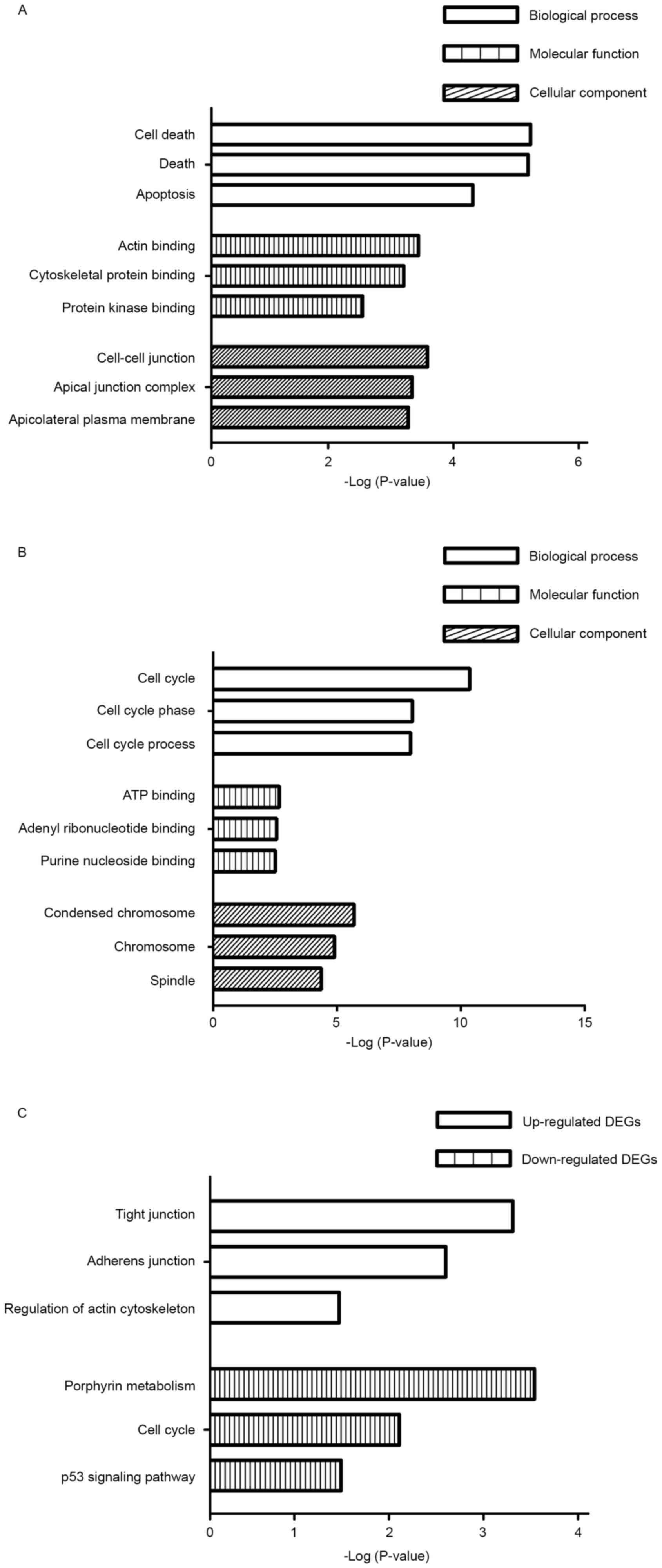
GO term enrichment analysis
Based on GO term enrichment analysis using the DAVID
tool, the DEGs were categorized into the following three major
terms: Biological processes, molecular functions and cellular
components. The top three GO terms of each of the categories are
illustrated in Fig. 2. The enriched
terms of the upregulated genes in samples from patients with spinal
metastases were significantly associated with cell death, actin
binding and cell-cell junctions in the three categories (Fig. 2A). The enriched terms identified among
the downregulated genes in samples from patients with spinal
metastases were significantly associated with the cell cycle, ATP
binding and condensed chromosomes in the three categories (Fig. 2B).
KEGG pathway enrichment analysis
The DAVID tool was used to identify the KEGG
biological pathways associated with the DEGs in the samples from
patients with spinal metastases. The upregulated genes were
significantly associated with tight junctions, adherence junctions
and regulation of the actin cytoskeleton (Fig. 2C). By contrast, the downregulated
genes from the spinal metastases were significantly associated with
porphyrin metabolism, the cell cycle and tumor protein p53
signaling pathway (Fig. 2C).
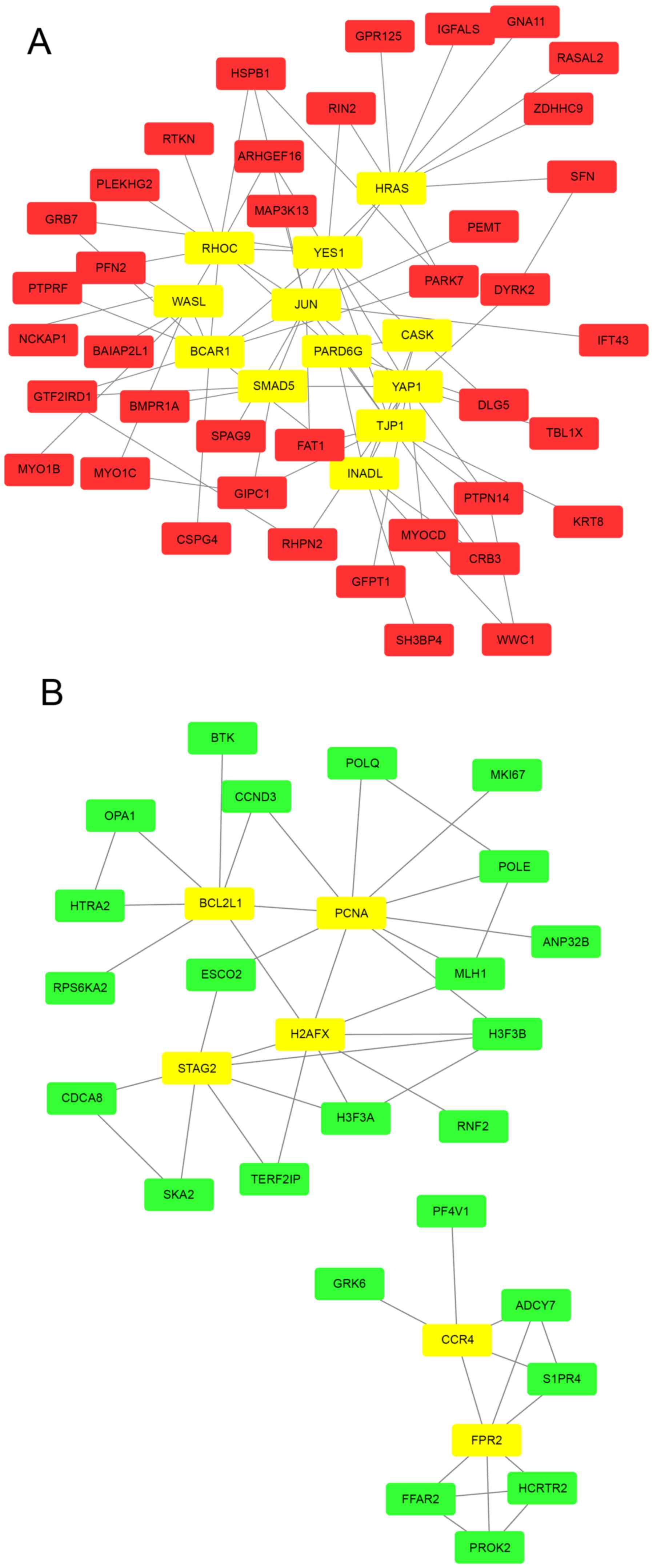
PPI network construction
Based on the STRING database, the PPI networks with
hub genes possessing a connectivity degree >5 were constructed
using Cytoscape. Networks with 51 and 30 nodes were obtained using
the proteins encoded by the upregulated (Fig. 3A) and downregulated (Fig. 3B) genes, respectively. Within a PPI
network, each node indicates a protein, and the lines between nodes
indicate PPIs. The connectivity degree represents the number of
lines linked to a given node, and nodes with a high connectivity
degree (≥5) are defined as hub genes that possess important
biological functions. A total of 12 hub genes were selected from
the upregulated PPI network, which included transcription factor
AP-1 (JUN), GTPase HRas (HRAS) and Rho-related GTP-binding protein
RhoC (RHOC) with connectivity degrees of 13, 10 and 10,
respectively (Fig. 3A) Meanwhile, 6
hub genes, including proliferating cell nuclear antigen (PCNA),
histone H2AX (H2AFX) and cohesion subunit SA-2 (STAG), with
connectivity degrees of 10, 8 and 7, respectively, were identified
from the downregulated PPI network (Fig.
3B).
Discussion
Although the advancement of surgical techniques has
improved the quality of life of patients with spinal metastases
(22), the underlying molecular
mechanisms of this condition are not well understood. Therefore,
studies investigating spinal metastases are required to develop
effective prevention and therapy strategies. Recently, the bone
marrow microenvironment has become an area of intense preclinical
and clinical investigation. The bone microenvironment is composed
of a mineralized extracellular matrix and specific cell types,
which provide a unique and fertile ‘soil’ for cancer metastases.
Cancer cells modify the bone microenvironment during their invasion
and expansion by recruiting and modulating osteoclasts,
osteoblasts, immune cells, vascular elements and bone matrix
(23). Therefore, a better
characterization of the interactions between cancer cells and the
spinal microenvironment is essential for developments in this
field.
In the present study, the microarray data generated
from the cancellous bone tissue of 5 patients with spinal
metastases and 3 normal patients was analyzed and 540 DEGs were
identified. The different gene expression signatures demonstrate
that the microenvironment of the bone marrow in spinal metastases
is altered compared with the normal condition. The DEGs were
subjected to an integrative systematic bioinformatics approach,
including functional and pathway enrichment analyses, in addition
to a PPI network construction. Based on these results, the
underlying molecular mechanisms of spinal metastases could be
explored at genetic and molecular levels, in order to provide
further insights into spinal metastasis prevention and
treatment.
The results of the cluster analysis, based on the
DEGs, demonstrated marked differences between the cancellous bone
from spinal metastases and that from normal patients. This result
indicates that a change in the underlying gene activity is
associated with spinal metastases. Notably, the gene expression
signatures for breast cancer and prostate cancer were similar. In
breast cancer, bone metastases are predominantly osteolytic
(24), while bone metastases are
predominantly osteoblastic in prostate cancer (25). They are thus theorized to represent
two extremes of a continuum. However, data from the present study
and previous research (26,27) indicates that bone metastases typically
have osteolytic and osteoblastic elements as a mixed phenotype.
This may be ascribed to the interaction between osteoblastic and
osteoclastic cells (28).
Functional enrichment analysis, based on GO, was
performed in order to identify the underlying biological processes
that the DEGs were associated with. In the present study, the
enriched GO terms from the upregulated genes were primarily
associated with cell death and actin binding in the cell-cell
junctions. This may reflect the interaction between cancer cells
and immune cells in spinal metastases, which indicates that tumor
cells may acquire the ability to escape immune control or even
eliminate immune cells, such as cluster of differentiation
(CD)4+ and CD8+ T cells (29). The enriched GO terms from the
downregulated genes were primarily associated with the cell cycle
and ATP binding in condensed chromosomes. This suggests that immune
cells persist around cancer cells in the bone marrow in a quiescent
state (30). Therefore, cancer cells
may escape immune surveillance via altering intrinsic tumor
suppressor mechanisms in spinal metastases.
The pathway enrichment analysis based on KEGG
evaluates differential expression patterns of gene groups rather
than those of individual genes, and in cases in which the
individual genes exhibit subtle biological function or property
changes they are omitted by typical individual gene analysis
(31). In the present study, the
enriched pathways in the upregulated genes were predominantly
associated with tight junctions, while the downregulated genes were
associated with porphyrin metabolism. These results overlapped with
the GO term enrichment analyses. Therefore, the data from the
current study indicates an immunocompromised status in patients
with spinal metastasis, which supports the findings from a previous
study that a decline in the ability of the immune cells to
recognize and destroy the tumor drives the dissemination of cancer
cells to the bone (32).
A PPI network is necessary to understand the
underlying molecular mechanisms of spinal metastasis, as the signal
transduction network that responds to external and internal
environmental stimuli is based upon interactions between proteins.
The hub genes identified serve a vital role in this signal
transduction network. The top hub genes identified in the present
study were JUN and PCNA from the upregulated and downregulated
genes, respectively. JUN interacts directly with specific target
DNA sequences to regulate gene expression. For example, JUN
participates in the receptor activator of nuclear factor kappa-β
(RANK)-RANK ligand (RANKL) system in osteolytic bone metastases.
Briefly, signaling through RANK in osteoclast progenitors activates
JUN, resulting in the differentiation of osteoclast progenitors
into mature osteoclasts, which are responsible for bone resorption
(33). Other hub genes, such as HRAS
and RHOC, also act as signaling proteins with GTPase activity in
activating osteoclasts (34). Thus,
the activation of the RANK-RANKL system may serve a role in spinal
metastases. PCNA is a cofactor of DNA polymerase δ and exists in
the nucleus. PCNA acts as a homotrimer and increases the
processivity of leading strand synthesis during DNA replication.
Furthermore, in response to DNA damage, PCNA is ubiquitinated and
is involved in the ubiquitin-conjugating enzyme E2 2-dependent DNA
repair signaling pathway (35).
Therefore, the relative lack of PCNA in the metastatic
microenvironment could inhibit the maturation of immune cells while
promoting the heterogeneity of cancer cells. H2AFX and STAG are
also responsible for DNA replication, and their downregulation
further exacerbates perturbations in the metastatic
microenvironment.
The traditional view ascribes the frequency of
spinal metastases to the specialized structure of the bone marrow
in the spine. The first part of the structure is the vascular
sinusoids that are lined with endothelial cells in the vertebra,
have fenestrae of 60 Å in diameter and lack a basement membrane
(36). The second part is the marrow
blood flow, which is relatively abundant in the vertebral bodies
(37). However, the data from the
present study revealed genetic changes in the metastatic
microenvironment, which may be favorable to the metastasis,
survival and growth of cancer cells in the spine, independent of
the cancer type. This concept is in accordance with a previous
study characterizing the importance of the interaction between
cancer cells and the bone marrow in the vicinity of future
metastatic sites (38). The extent to
which DEGs in spinal metastasis are produced by the cancer cells
themselves and by the microenvironment in response to the cancer
cells requires further research.
There are a number of limitations to the present
study. Firstly, the sample size for the microarray was small.
Secondly, the results from the array and bioinformatics analysis
lack corresponding in vitro experiments. Thus, genetic and
experimental studies with a larger sample size are required to
confirm the results from the current study.
In conclusion, based on the comprehensive set of
bioinformatics analyses of microarray data, the results of the
present study identified DEGs that are potentially associated with
the molecular mechanisms of spinal metastasis in a number of cancer
types. This will provide new insights into the underlying molecular
mechanisms, prevention and treatment of spinal metastases. However,
further experiments are required to confirm these results.
Acknowledgements
The present study was funded by the National Natural
Science Foundation of China (grant no. 81572629).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sporn MB: The war on cancer. Lancet.
347:1377–1381. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Paget S: The distribution of secondary
growths in cancer of the breast. Cancer Metastasis Rev. 8:98–101.
1989.PubMed/NCBI
|
|
4
|
Aaron AD: The management of cancer
metastatic to bone. JAMA. 272:1206–1209. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jacobs WB and Perrin RG: Evaluation and
treatment of spinal metastases: An overview. Neurosurg Focus.
11:e102001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sciubba DM, Petteys RJ, Dekutoski MB,
Fisher CG, Fehlings MG, Ondra SL, Rhines LD and Gokaslan ZL:
Diagnosis and management of metastatic spine disease. A review. J
Neurosurg Spine. 13:94–108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tokuhashi Y, Matsuzaki H, Oda H, Oshima M
and Ryu J: A revised scoring system for preoperative evaluation of
metastatic spine tumor prognosis. Spine (Phila Pa 1976).
30:2186–2191. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ibrahim A, Crockard A, Antonietti P,
Boriani S, Bünger C, Gasbarrini A, Grejs A, Harms J, Kawahara N,
Mazel C, et al: Does spinal surgery improve the quality of life for
those with extradural (spinal) osseous metastases? An international
multicenter prospective observational study of 223 patients.
Invited submission from the Joint Section Meeting on Disorders of
the Spine and Peripheral Nerves, March 2007. J Neurosurg Spine.
8:271–278. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kang Y, Siegel PM, Shu W, Drobnjak M,
Kakonen SM, Cordón-Cardo C, Guise TA and Massagué J: A multigenic
program mediating breast cancer metastasis to bone. Cancer Cell.
3:537–549. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jamieson WL, Shimizu S, D'Ambrosio JA,
Meucci O and Fatatis A: CX3CR1 is expressed by prostate epithelial
cells and androgens regulate the levels of CX3CL1/fractalkine in
the bone marrow: Potential role in prostate cancer bone tropism.
Cancer Res. 68:1715–1722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cai X, Luo J, Yang X, Deng H, Zhang J, Li
S, Wei H, Yang C, Xu L, Jin R, et al: In vivo selection for
spine-derived highly metastatic lung cancer cells is associated
with increased migration, inflammation and decreased adhesion.
Oncotarget. 6:22905–22917. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dat le T, Matsuo T, Yoshimaru T, Kakiuchi
S, Goto H, Hanibuchi M, Kuramoto T, Nishioka Y, Sone S and Katagiri
T: Identification of genes potentially involved in bone metastasis
by genome-wide gene expression profile analysis of non-small cell
lung cancer in mice. Int J Oncol. 40:1455–1469. 2012.PubMed/NCBI
|
|
13
|
Wheeler DL, Church DM, Federhen S, Lash
AE, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E,
Tatusova TA and Wagner L: Database resources of the National Center
for Biotechnology. Nucleic Acids Res. 31:28–33. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wozniak MB, Le Calvez-Kelm F,
Abedi-Ardekani B, Byrnes G, Durand G, Carreira C, Michelon J,
Janout V, Holcatova I, Foretova L, et al: Integrative genome-wide
gene expression profiling of clear cell renal cell carcinoma in
Czech Republic and in the United States. PLoS One. 8:e578862013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Reiner A, Yekutieli D and Benjamini Y:
Identifying differentially expressed genes using false discovery
rate controlling procedures. Bioinformatics. 19:368–375. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI
|
|
19
|
von Mering C, Huynen M, Jaeggi D, Schmidt
S, Bork P and Snel B: STRING: A database of predicted functional
associations between proteins. Nucleic Acids Res. 31:258–261. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kou Y, Zhang S, Chen X and Hu S: Gene
expression profile analysis of colorectal cancer to investigate
potential mechanisms using bioinformatics. Onco Targets Ther.
8:745–752. 2015.PubMed/NCBI
|
|
22
|
Wu J, Zheng W, Xiao JR, Sun X, Liu WZ and
Guo Q: Health-related quality of life in patients with spinal
metastases treated with or without spinal surgery: A prospective,
longitudinal study. Cancer. 116:3875–3882. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weilbaecher KN, Guise TA and McCauley LK:
Cancer to bone: A fatal attraction. Nat Rev Cancer. 11:411–425.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Coleman RE and Seaman JJ: The role of
zoledronic acid in cancer: Clinical studies in the treatment and
prevention of bone metastases. Semin Oncol. 28(2 Suppl 6): S11–S16.
2001. View Article : Google Scholar
|
|
25
|
Charhon SA, Chapuy MC, Delvin EE,
Valentin-Opran A, Edouard CM and Meunier PJ: Histomorphometric
analysis of sclerotic bone metastases from prostatic carcinoma
special reference to osteomalacia. Cancer. 51:918–924. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guise TA: The vicious cycle of bone
metastases. J Musculoskelet Neuronal Interact. 2:570–572.
2002.PubMed/NCBI
|
|
27
|
Pavelic S Kraljevic, Sedic M, Bosnjak H,
Spaventi S and Pavelic K: Metastasis: New perspectives on an old
problem. Mol Cancer. 10:222011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ren G, Esposito M and Kang Y: Bone
metastasis and the metastatic niche. J Mol Med (Berl).
93:1203–1212. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang K, Kim S, Cremasco V, Hirbe AC,
Collins L, Piwnica-Worms D, Novack DV, Weilbaecher K and Faccio R:
CD8+ T cells regulate bone tumor burden independent of osteoclast
resorption. Cancer Res. 71:4799–4808. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schreiber RD, Old LJ and Smyth MJ: Cancer
immunoediting: Integrating immunity's roles in cancer suppression
and promotion. Science. 331:1565–1570. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nam D and Kim SY: Gene-set approach for
expression pattern analysis. Brief Bioinform. 9:189–197. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Capietto AH and Faccio R: Immune
regulation of bone metastasis. Bonekey Rep. 3:6002014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mundy GR: Metastasis to bone: Causes,
consequences and therapeutic opportunities. Nat Rev Cancer.
2:584–593. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Russell RG: Bisphosphonates: The first 40
years. Bone. 49:2–19. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hoege C, Pfander B, Moldovan GL,
Pyrowolakis G and Jentsch S: RAD6-dependent DNA repair is linked to
modification of PCNA by ubiquitin and SUMO. Nature. 419:135–141.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
De Bruyn PP: Structural substrates of bone
marrow function. Semin Hematol. 18:179–193. 1981.PubMed/NCBI
|
|
37
|
Kahn D, Weiner GJ, Ben-Haim S, Ponto LL,
Madsen MT, Bushnell DL, Watkins GL, Argenyi EA and Hichwa RD:
Positron emission tomographic measurement of bone marrow blood flow
to the pelvis and lumbar vertebrae in young normal adults. Blood.
83:958–963. 1994.PubMed/NCBI
|
|
38
|
Kaplan RN, Psaila B and Lyden D: Bone
marrow cells in the ‘pre-metastatic niche’: within bone and beyond.
Cancer Metastasis Rev. 25:521–529. 2006. View Article : Google Scholar : PubMed/NCBI
|