Introduction
Peutz-Jeghers Syndrome [PJS; Mendelian Inheritance
in Man (MIM), 175200, www.ncbi.nlm.gov/OMIM] is a rare autosomal dominant
disorder, which is characterized by gastrointestinal hamartomatous
polyps, mucocutaneous pigmentation and an elevated risk of various
neoplasms (1,2). Mutations in the serine threonine kinase
11/liver kinase B1 (STK11/LKB1; MIM, 602216) gene on
chromosome 19p13.3 have been identified as the major cause of PJS
(3–5).
Previously, direct sequencing of the STK11
gene in combination with a multiplex ligation-dependent probe
amplification (MLPA) assay for deletion detection resulted in a
mutation detection rate of 67.3% (35/52) in a group of PJS patients
(6). This rate is consistent with the
50 to 90% frequency reported by various research groups (3,7). The
majority of mutations have been shown to be either frameshift or
nonsense, resulting in an abnormally truncated protein and a
subsequent loss of kinase activity (5). However, a number of STK11
missense mutations have been identified in PJS with unknown
pathogenicity (5,8).
STK11 has been implicated as an important regulator
of cell proliferation and apoptosis (8,9) via
multiple signaling pathways (10),
which may involve its tumor suppressor function and/or catalytic
activity (11). The mammalian target
of rapamycin (mTOR) pathway is one of the major downstream pathways
that can be regulated by STK11 (12–14). The
ribosomal protein S6 kinase 1 (S6K1) is the first mTOR substrate
and has been an extensively studied effector of mTOR complex 1
(mTORC1) (15,16). Furthermore, S6K1 activation requires
mTORC1-mediated phosphorylation, which acts to improve S6K1
activity towards its S6 substrate (16,17).
Signals from S6K1 activation are involved in a variety of cellular
functions, including the regulation of protein translation, cell
growth, angiogenesis and metabolism (18,19).
Therefore, mutations in STK11 that inactivate its endogenous
activity can negatively regulate mTORC1 signaling, resulting in
phosphorylation and activation of its downstream targets. In turn,
this is able to relieve inhibition on protein synthesis, and
promote cell growth and tumorigenesis (20–22).
In the present study, STK11 genetic mutations
were analyzed from PJS patients in 11 unrelated families, and 8
(72.7%, 8/11) different mutations were found, including 6 point
mutations and 2 large deletions. Notably, 3 of the mutations were
novel. Of these 3 mutations identified, 2 were missense mutations
[c.88G>A (p.Asp30Asn) and c.869T>C (p.Leu290Pro)].
Furthermore, the missense mutations were able to disrupt the
function of the tumor suppressor STK11, which led to
phosphorylation of S6K1 and S6 and promoted protein synthesis and
cell proliferation.
These findings demonstrated the pathogenicity of the
two novel missense mutations and extended the current understanding
of STK11 mutations. Understanding how the function of
STK11 is disrupted may lead to an improved understanding of
the molecular mechanisms implicated in the pathogenesis of PJS and
carcinogenesis. This may also provide potential therapeutic targets
for future treatment.
Materials and methods
Patients
Peripheral blood samples of PJS patients (n=21) from
6 PJS families and 5 sporadic cases were collected between January
2012 and September 2015 at Nanfang Hospital (Guangzhou, China). The
patients were diagnosed according to previously published criteria
(6). Endoscopic or surgical
polypectomy and histopathological examination (by ≥2 pathologists)
were performed in all patients. Endoscopic or surgical polypectomy
were performed in all patients. The tissues were fixed in 10%
formalin solution for 24 h at 4°C and embedded in paraffin for 0.5
h at 65°C. Serial sections (4 µm) were cut and prepared for
hematoxylin-eosin (HE) staining and immunohistochemistry, as
previously described (6). All
HE-stained sections were reviewed by two experienced pathologists
independently, who defined the hamartomatous polyps. Surgical
resection was performed on other organs, including the breasts and
cervix, in 3 patients due to the presence of benign or malignant
tumors. The family cancer history was collected from the patients'
medical records. All patients provided informed consent for this
study, and the principles outlined in the Declaration of Helsinki
were followed. Ethical approval was obtained from the Medical
Ethics Committee at Nanfang Hospital.
Detection of STK11 germline
mutations
Genomic DNA was extracted from peripheral blood
using the commercially available QiAamp DNA Blood Midi kit (Qiagen
GmbH, Hilden, Germany). PCR primers were designed using the
software program Primer (version 3.0; http://frodo.wi.mit.edu/primer3/) to amplify the
STK11 exons (RefSeq accession number NM_000455.4) and
intron-exon boundaries. Sanger sequencing was performed for each
index case in the 6 familial and 5 sporadic cases, as previously
described (6).
All STK11 exons (exon1-9) were amplified (for
primers see Table I) using regular
Taq polymerase (Takara Ex Taq; Takara Bio, Inc., Otsu, Japan). PCR
amplification comprised an initial denaturation cycle at 95°C for
15 min, followed by 38 amplification cycles consisting of 95°C for
30 sec, annealing at 60°C for 45 sec, and extension at 72°C for 7
min. Sanger sequencing was performed for each index case in the 6
familial and 5 sporadic cases, as previously described (6). In familial cases, identified mutations
were further tested in all available family members to confirm
segregation of the mutation with the disease. All variants
identified were screened against the single nucleotide polymorphism
database (www.ncbi.nlm.nih.gov/SNP) to exclude the possibility
of these variants being polymorphisms.
 | Table I.The polymerase chain reaction primers
of 9 exons of STK11 gene. |
Table I.
The polymerase chain reaction primers
of 9 exons of STK11 gene.
| Primer | Primer sequence
(5–3) |
|---|
| Exon 1A | F:
CTCAGGGCTGGCGGCGGGACT |
|
| R:
CTTGCGGCGCGGCTGGTAGATGA |
| Exon 1B | F:
ACGTTCATCCACCGCATCGAC |
|
| R:
GCACAGCGTCTCCGAGTCCAG |
| Exon1C | F:
TCTTACGGCAAGGTGAAGGAGGTG |
|
| R:
CCGACCCCAGCAAGCCATACTTA |
| Exon 2 | F:
ATACACCCCTGTCCTCTCTG |
|
| R:
AGGCCCCGCGGTCCCAAC |
| Exon 3 | F:
CTGAGCTGTGTGTCCTTAGCG |
|
| R:
GTGTGGCCTCACGGAAAGGAG |
| Exon 4 | F:
CGGCCCCAGGACGGGTG |
|
| R:
GTGCAGCCCTCAGGGAG |
| Exon 5 | F:
ACTCCCTGAGGGCTGCAC |
|
| R:
CCCCTCGGAGTGTGCGTGTGG |
| Exon 6 | F:
TCAACCACCTTGACTGACCA |
|
| R:
ACACCCCCAACCCTACATTT |
| Exon 7 | F:
TCACCCAGGGCCTGACAACAGAG |
|
| R:
GCAGCCTCGGCCCCACTG |
| Exon 8 | F:
GGCGCCACTGCTTCTGGGC |
|
| R:
CGAGTCAGCAGAGCCGGGC |
| Exon 9A | F:
TGTAAGTGCGTCCCCGTGGTG |
|
| R:
CGCCCTGGATTTGGTGCTA |
| Exon 9B | F:
GTGTATGAACGGCACAGAGGC |
|
| R:
CAGGCGTTGTCCCCACAT |
An MLPA assay was performed for large intragenic
deletions using the MLPA test kit (SALSA P101-B1 STK11;
MRC-Holland, Amsterdam, Netherlands) as previously described
(6). Deletion screening was performed
according to the manufacturer's instructions, and the results were
analyzed using the GeneMarker®HID STR Human Identity
software (version 3.0; SoftGenetics, LLC., State College, PA, USA).
Values of 0.85 to 1.15 indicate normal results (presence of two
copies), and values of 0.35 to 0.65 or 1.35 to 1.65 indicate a
deletion or duplication, respectively. All identified deletions
were confirmed in a second independent reaction and confirmed to
segregate within the relevant family members.
Predicting the effect of the
identified mutations
The novel missense mutations were all predicted
using the PolyPhen-2 web tool (PolyPhen2: Http://genetics.bwh.harvard.edu/pph2/). The PolyPhen-2
score represents the probability that a substitution is damaging.
The novel missense mutation (p.Leu290Pro) is predicted to be highly
likely to be damaging with a score of 1.0, and the other
(p.Asp30Asn) is predicted to be possibly damaging with a score of
0.775. Variants with scores of 0.0 are predicted to be benign.
Values closer to 1.0 are more confidently predicted to be
deleterious.
Tissue staining
Immunohistochemistry (IHC) was performed as
previously described (6). Tissue
sections were deparaffinized and incubated with a phosphorylated
(p)-S6 (Ser240/244) primary antibody (1:400; catalog no. 2903; Cell
Signaling Technology, Inc., Danvers, MA, USA) overnight at 4°C.
After three washes with PBST (2,000 ml PBS+4 ml Tween 20), the
sections were incubated with biotin-conjugated goat anti-rabbit
immunoglobulin G (1:300; catalog no. SPN-9001; Invitrogen; Thermo
Fisher Scientific, Inc.) for 10 min at room temperature, and then
incubated with streptavidin-peroxidase for 10 min at room
temperature. A known p-S6 positive colorectal cancer tissue, from
one patient who had undergone endoscopic at Nanfang Hospital. HE
stained specimens were reviewed by a senior pathologist, who
determined the polyp histology and tumor type, was used as a
positive control for p-S6 staining. p-S6 staining was scored as
positive when >10% of the cells exhibited nuclear
expression.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using Trizol (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), and cDNA was
synthesized by reverse transcription from 1 µg total RNA using
oligo(dT) primers (Promega Corporation, Madison, WI, USA). RT-qPCR
was performed using the All-in-One™ qPCR mix
(GeneCopoeia Inc., Rockville, MD, USA) on a LightCycler 480 system
(Roche Diagnostics, Basel, Switzerland). Primer sequences for
STK11 were as follows: Forward, 5′-AGGGCCGTCAAGATCCTCAA-3′
and reverse, 5′-GCATGCCACACACGCAGTA-3′. Primer sequences for
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were as follows:
Forward, 5′-GAAGGTGAAGGTCGGAGTC-3′ and reverse,
5′-GAAGATGGTGATGGGATTTC-3′. The housekeeping gene GAPDH was used as
an internal control for normalization. The relative expression of
STK11 was calculated using the 2−ΔΔCq method
(23). Experiments were repeated
three times and the mean of the data was determined.
Western blotting
Total protein was extracted using
radioimmunoprecipitation assay lysis buffer with protease and
phosphatase inhibitors (both, 1:100; Roche, Nutley, NJ, USA). Total
protein was quantified using bicinchoninic acid assay (Thermo
Fisher Scientific, Inc.). Total protein (25 µg) was resolved by 10%
SDS-PAGE (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and
transferred to polyvinylidene difluoride membranes (EMD Millipore,
Billerica, MA, USA). Membranes were blocked with 5% BSA (Bio-Rad
Laboratories, Inc.) in PBS with 0.1% Tween-20 for 2 h at room
temperature. Membranes were incubated with primary antibodies
against STK11 (1:1,000; catalog no. 3047) S6K1 (1:1,000; catalog
no. 2903) S6 (1:1,000; catalog no. 1155), p-S6K1 (Thr389; 1:1,000;
catalog no. 9234) and p-S6 (Ser240/244; 1:1,000; catalog no. 2903)
(all Cell Signaling Technology, Inc.) overnight at 4°C. Mouse
polyclonal anti-GAPDH antibody (1:3,000; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) was used as the loading control. The
membranes were subsequently incubated for 2 h at room temperature
with either horseradish peroxidase (HRP)-conjugated goat
anti-rabbit IgG (1:2,000; catalog no. 2903; Cell Signaling
Technology, Inc.) or HRP-conjugated goat anti-mouse IgG (1:5,000;
catalog no. sc-2005; Santa Cruz Biotechnology, Inc.). Protein
expression was detected using an enhanced chemiluminescence kit
(Thermo Fisher Scientific, Inc.).
Cell lines and culture
Human colon cancer (SW1116, HT29, SW620, SW480,
LoVo, Caco2, and HCT116), human cervical carcinoma (HeLa), normal
human gastric epithelial (GES-1), and human kidney (293T) cell
lines were cultured in Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.) and maintained in a
humidified atmosphere of 5% CO2 at 37°C. All cell lines
used in the present study were obtained from the American Type
Culture Collection, Rockville, MD, USA.
Cell lines transfection
SW117 and HeLa cells were used for transfection and
subsequent experiments. The sequences of all STK11 gene mutant
plasmids were confirmed by genetic sequencing. The constructs
pGCMV-green fluorescent protein (GFP) (empty vector),
pGCMV-GFP-STK11 WT (wild-type), pGCMV-GFP-STK11 p.Leu290Pro and
pGCMV-GFP-STK11 p.Asp30Asn were synthesized by Shanghai GenePharma
Co., Ltd., (Shanghai, China). Cells were seeded (4×105)
in tissue culture plates and grown to 70 to 80% confluence.
Subsequently, the cells were transfected with the vectors using
Lipofectamine 3000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Gene overexpression
was examined by RT-qPCR, and protein expression was analyzed by
western blotting at 24 to 48 h post-transfection. Stable
transfections were performed at 48 h post-transfection, and the
transfected cells were grown with G418 (400 mg/ml; Sigma-Aldrich;
Merck Millipore, Darmstadt, Germany) for 4 to 8 weeks to allow for
positive selection.
Cell Counting Kit-8 (CCK-8) and colony
formation assays
To determine the effect of novel STK11
mutants on cell proliferation, transfected cells were measured
using CCK-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan)
according to the manufacturer's protocols. Proliferation was
assessed by colony formation assay. Transfected cells were counted
and seeded into 6-well plates (1,000 cells/well). The number of
colonies containing >50 cells were stained with 0.5% crystal
violet in isopropanol and counted 2 weeks after seeding. The
relative cell colony numbers were evaluated as the ratio of the
last visible colonies to those at initialization.
Flow cytometric analysis
Cell apoptosis was quantitated using the Annexin
V-APC and 7-aminoactinomycin D (7-AAD) double dye cell
apoptosis kit (BD Biosciences, Franklin Lakes, NJ, USA) according
to the manufacturer's protocol. Cells (1×106 cells/well)
were collected at 48 h post-transfection with the aforementioned
STK11 plasmids and washed twice with ice-cold PBS. The cells
were re-suspended in 500 µl 1X binding buffer, stained with 5 µl
Annexin V-APC and 5 µl 7-AAD and incubated on ice for 15 min in the
dark. Apoptotic cells (10,000 cells/test) were quantified by flow
cytometer (BD Biosciences).
Statistical analysis
All statistical analyses were performed using SPSS
(version 17.0; SPSS Inc., Chicago, IL, USA). The data are expressed
as the mean ± standard deviation unless otherwise noted.
Comparisons among multiple groups were determined using one-way
analysis of variance, followed by least significance difference or
Dunnett's T3 test. P<0. 05 was considered to indicate a
statistically significant difference.
Results
Identification of STK11 mutations
The present study identified 6 different germline
point mutations in 6 out of 11 index cases (52.4%; Table II). A number of mutations (3/6, 50%)
were associated with colon or esophagus cancer in either index
patients or in relatives with PJS. A total of 4 mutations were
identified in familial cases (4/6, 66.7%) and 2 in sporadic cases
(2/5, 40%) of PJS. Of the 6 mutations identified, 4 were truncation
mutations and 2 were missense mutations. To the best of our
knowledge, 3 of the mutations [c.143_144insA (p.Try49Valfs*114),
c.869T>C (p.Leu290Pro) and c.88G>A (p.Asp30Asn)] have
previously been unreported and are therefore novel (Fig. 1; Table
II).
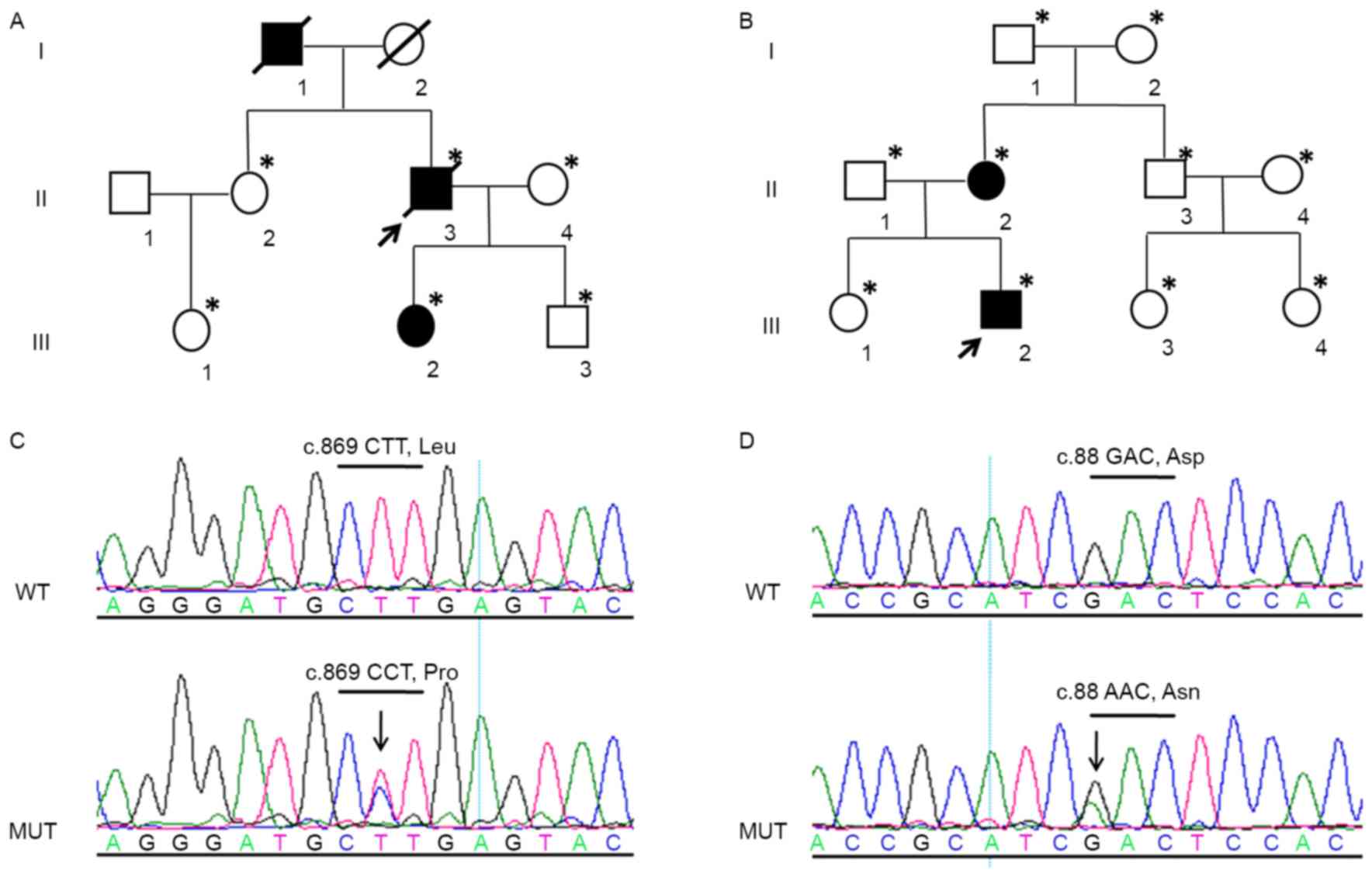 | Figure 1.Segregation of 2 novel STK11
missense mutations in 2 PJS families. Pedigree charts in 2
unrelated families with PJS (PJS4 and PJS5). (A) PJS4 II-2, PJS4
II-4, PJS4 III-1 and PJS4 III-3 carried the wild-type c.869T
(p.Leu290); while PJS4 III-2 carried the mutation c.869T>C
(p.Leu290Pro). Proband (PJS4 II-3) carried the mutation c.869T>C
(p.Leu290Pro) and succumbed to colon adenocarcinoma at 56 years of
age. (B) PJS5 I-1, PJS5 I-2, PJS5 II-1, PJS5 II-3, PJS5 II-4, and
PJS5 III-1, PJS5 III-3, PJS5 III-4 carried the wild-type c.88G
(p.Asp30); while PJS5 II-2 and PJS5 III-2 carried the mutation
c.88G>A (p.Asp30Asn). Arrow, proband; square, males; circle,
females; open symbols, unaffected individuals; solid symbols,
affected individuals; diagonal line, deceased; *genotypes of
individuals examined by DNA analysis. Sequencing chromatograms of
genomic DNA from the proband (C) PJS4 II-3 and (D) PJS5 III-2.
Arrows indicate the position of the mutation and the codon
containing the mutation is underlined. Asp, aspartic acid; Asn,
Asparagine; Leu, leucine; MUT, mutated; PJS, Peutz-Jeghers
syndrome; Pro, proline; STK11, serine threonine kinase 11; WT,
wild-type. |
| Table II.Spectrum of STK11 mutations
and clinical characteristics of 8 unrelated Peutz-Jeghers syndrome
families. |
Table II.
Spectrum of STK11 mutations
and clinical characteristics of 8 unrelated Peutz-Jeghers syndrome
families.
|
|
|
|
|
|
| Malignancy |
|
|---|
|
|
|
|
|
|
|
|
|
|---|
| Family no. | Family history | Exon | Nucleotide
change | Amino acid
alteration | Type of
mutation | Family member | Age, years | Type | Previously reported
mutation (reference) |
|---|
| 1 | F | 3 | c.454C>T | p.Gln152* | Point | Father | 42 | Colon | Y (7) |
| 2 | S | 1 | c.143_144insA |
p.Try49Valfs*114 | Point | − | − | − | N |
| 3 | F | 6 | c.842_843insC |
p.Leu282Alafs*3 | Point | − | − | − | Y (24) |
| 4 | F | 7 | c.869T>C | p.Leu290Pro | Point | Proband | 56 | Colon | N |
| 5 | F | 1 | c.88G>A | p.Asp30Asn | Point | − | − | − | N |
| 6 | S | 2 | c.334C>T | p.Gin112* | Point | Father | 30 | Esophageal | Y (25) |
| 7 | F | 1 |
c.1114-?_290+?del | Not identified | Exonic
deletion | Mother | 45 | Breast | Y (6) |
| 8 | S | 3 |
c.375-?_464+?del | Not identified | Exonic
deletion | − | − | − | Y (6) |
MLPA was used to determine the presence or absence
of exonic rearrangements in the 5 PJS probands, where no mutations
were identified by Sanger sequencing. This resulted in the
identification of two large deletions, with one being associated
with the presence of breast cancer in the relative of the proband
(PJS7; Table II). The two deletions
were predicted to affect the kinase domain of the protein. The
total mutation detection rate in the index cases was 72.7% (8/11),
which indicated that genetic alterations in STK11 in
individuals with PJS may increase the risk of developing
PJS-associated cancer.
In total, 3 of the truncation mutations identified
in the present study have previously been described (7,24,25), and 1 is novel [c.143_144insA
(p.Try49Valfs*114)] (PJS2; Table
II). The novel mutation involves an insertion of nucleotide A
between nucleotides 143 and 144 in exon 1. This results in a
433-amino acid sequence with a frameshift after nucleotide 49 and
premature termination at nucleotide 114. Therefore, the alteration
leads to a complete loss of the kinase domain of STK11 (26).
The novel missense mutation [c.869T>C
(p.Leu290Pro)] leads to an amino acid change at codon 290 in exon
7, which is located in the catalytic kinase region of STK11 (PJS4
II-3; Fig. 1C; Table II). The other novel missense mutation
[c.88G>A (p.Asp30Asn)] leads to an alteration at nucleotide 88
(G<A) and an amino acid change at codon 30 in exon 1, which is
located at the N-terminal of STK11 (PJS5 III-2; Fig. 1D; Table
II). This results in the substitution of aspartate by
asparagine. The two mutations were predicted to be pathogenic by
PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/).
Novel STK11 missense mutations induce
p-S6 expression in tissues from PJS patients
To investigate whether the novel missense mutations
affect the phosphorylation of mTOR pathway key downstream target
gene S6, IHC was performed to detect p-S6 (Ser240/244)
expression in colon hamartomatous polyp and colorectal mucosal
epithelium tissues obtained from individuals with or without
STK11 mutations. p-S6 staining was observed in the nuclei of
epithelial cells in colonic hamartoma obtained from patient PJS4
II-3 with the mutation c.869T>C (p.Leu290Pro) (Fig. 2B) and in the breast intraductal
papillary tumor from the patient PJS5 III-2 with the mutation
c.88G>A (p.Asp30Asn) (Fig. 2E). By
contrast, p-S6 staining was not observed in colorectal mucosa
derived from PJS4 III-3, who is a relative of PJS4 II-3 (Fig. 2C) and in breast hyperplasia glandular
epithelial obtained from PJS5 II-2, who is a relative of PJS5 III-2
(Fig. 2F). PJS4 III-3 and PJS5 II-2
did not carry STK11 mutations. These findings suggest that
the two novel missense mutations may disrupt the protein function
of STK11 serine/threonine kinase and induce S6 phosphorylation.
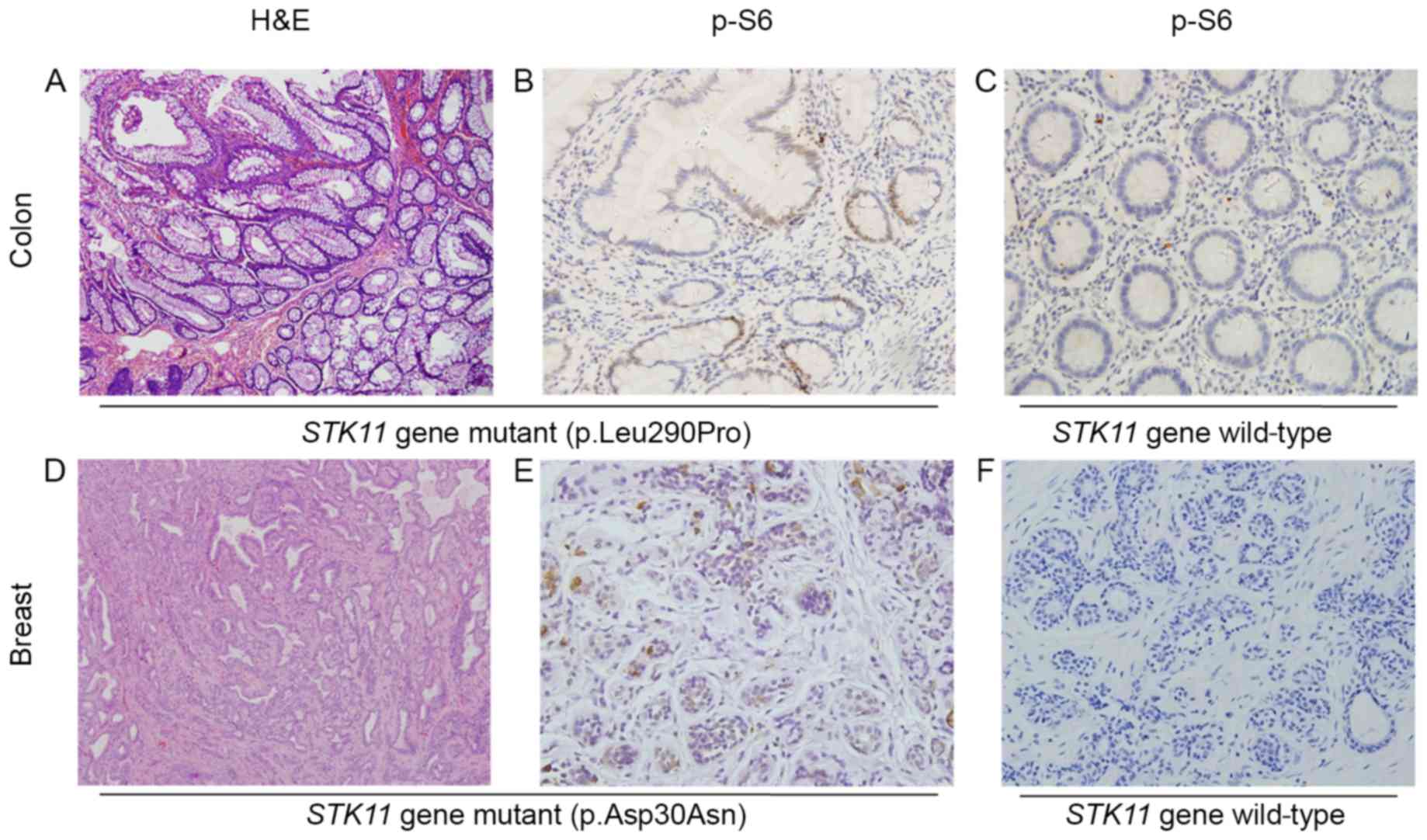 | Figure 2.Staining of tissue sections from
patients with PJS. (A) Hematoxylin and eosin staining of a colon
hamartomatous polyp obtained from the patient PJS4 II-3 carrying
the STK11 missense mutation c.869T>C (p.Leu290Pro).
Micrograph shows branched bundles of smooth muscle fibers.
Magnification, ×100. (B) Immunohistochemical staining of a colon
hamartomatous polyp obtained from patient PJS5 III-2 with the
STK11 missense mutation c.88G>A (p.Asp30Asn). Image shows
the localization of p-S6 (Ser240/244) to the epithelial cell
nuclei. Magnification, ×400. (C) p-S6 staining is absent in the
colorectal mucosa epithelium samples without STK11
mutations. Magnification, ×400. (D) Hematoxylin and eosin staining
of breast intraductal papillary tumor tissues obtained from patient
PJS5 III-2. Image shows marked cellular heterogeneity in pathology.
Magnification, ×100. (E) Immunohistochemical staining of breast
intraductal papillary tumor tissues obtained from patient PJS5
III-2. Image shows localization of p-S6 to the nuclei of glandular
epithelial cells. Magnification, ×400. (F) p-S6 staining is absent
in the ductal epithelium of breast hyperplasia samples without
STK11 mutations. Magnification, ×400. Asp, aspartic acid;
Asn, Asparagine; Leu, leucine; PJS, Peutz-Jeghers syndrome; p,
phosphorylated; Pro, proline; STK11, serine threonine kinase
11. |
Expression of novel STK11 mutations in
HeLa and SW1116 cells
RT-qPCR and western blotting were used to detect
endogenous expression levels of STK11 in human cell lines.
As shown in Fig. 3A, STK11
expression levels were nearly undetectable in HeLa and SW1116 cells
when compared with all other tested cell lines. Subsequently, HeLa
and SW1116 cells were used for further studies, and the effects of
the novel STK11 mutations were investigated.
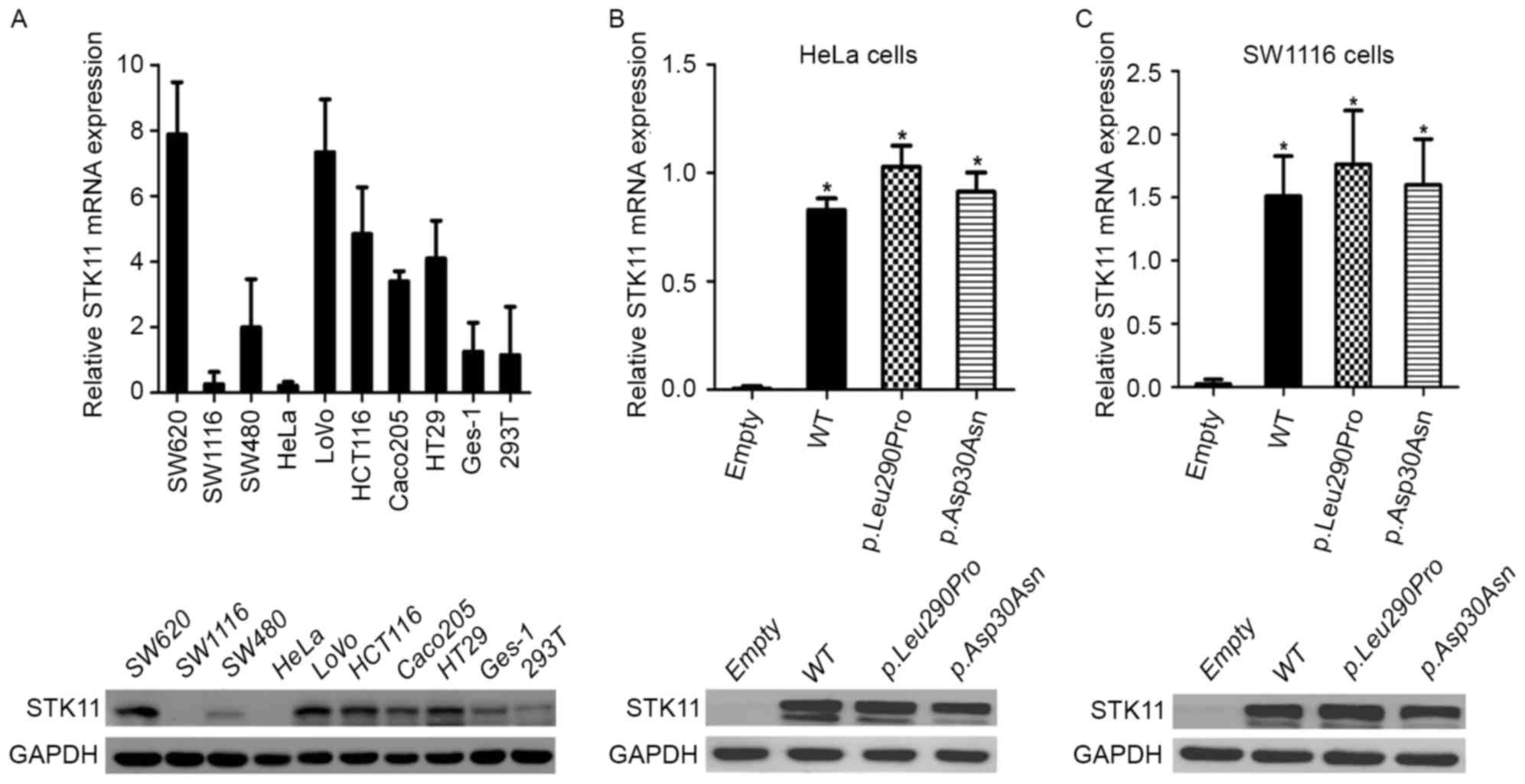 | Figure 3.Expression of STK11 in human
cell lines and STK11 mutants in HeLa and SW1116 cells. (A) Relative
STK11 mRNA expression levels were markedly reduced in SW1116
and HeLa cells. (B) STK11 protein expression in HeLa and
SW1116 cells was not detected. mRNA and protein expression levels
were significantly increased in (C) HeLa and (D) SW1116 cell lines
transfected with WT, STK11 mutant p.Leu290Pro and
STK11 mutant p.Asp30Asn vectors compared with cells
transfected with the empty vector. No significant differences
between the WT vector and the STK11 mutant vectors were
observed. *P<0.05. Asp, aspartic acid; Asn, Asparagine; GAPDH,
glyceraldehyde 3-phosphate dehydrogenase; Leu, leucine; PJS,
Peutz-Jeghers syndrome; p, phosphorylated; Pro, proline; STK11,
serine threonine kinase 11; WT, wild-type. |
STK11 plasmids (empty vector, wild-type
vector, STK11 mutant p.Leu290Pro vector and STK11
mutant p.Asp30Asn vector) were transfected into HeLa and SW1116
cells, and STK11 expression was examined by RT-qPCR and
western blot analysis. As shown in Fig.
3B and C, the expression of STK11 in HeLa and SW1116
cells transfected with the STK11 mutants and the wild-type
vector was significantly increased when compared with cells
transfected with the empty vector. However, there were no
significant differences in STK11 expression in cells
transfected with the wild-type vector and the STK11 mutants.
Taken together, these results suggest that the novel STK11
mutants have negligible effects on STK11 transcription and
translation.
Novel STK11 missense mutations induce
phosphorylation of S6K1 and S6 in cell lines
The effects of the two novel STK11 missense
mutations on gene function and whether the mutations result in
dysregulation of the downstream mTOR-mediated pathway was
investigated. S6K1 and S6 are substrates of mTORC1. Phosphorylation
of S6K1 at Thr389 and S6 at Ser240/244 was analyzed using western
blotting. As shown in Fig. 4, there
was increased expression of p-S6K1 (Thr389) and p-S6 (Ser240/244)
in HeLa and SW1116 cells transfected with the two novel missense
STK11 mutants compared with cells transfected with the empty
vector. By contrast, cells transfected with the wild-type vector
exhibited a marked decrease in the levels of p-S6K1 and p-S6. These
results demonstrated that the novel STK11 missense mutations
disrupted the endogenous protein kinase activity of STK11, which
led to the activation of the mTORC1 signaling pathway.
Consequently, this results in the dysregulation of the STK11-S6K1
axis. We hypothesized that this disruption leads to abnormal
de-repression of protein synthesis and promotion of cell
growth.
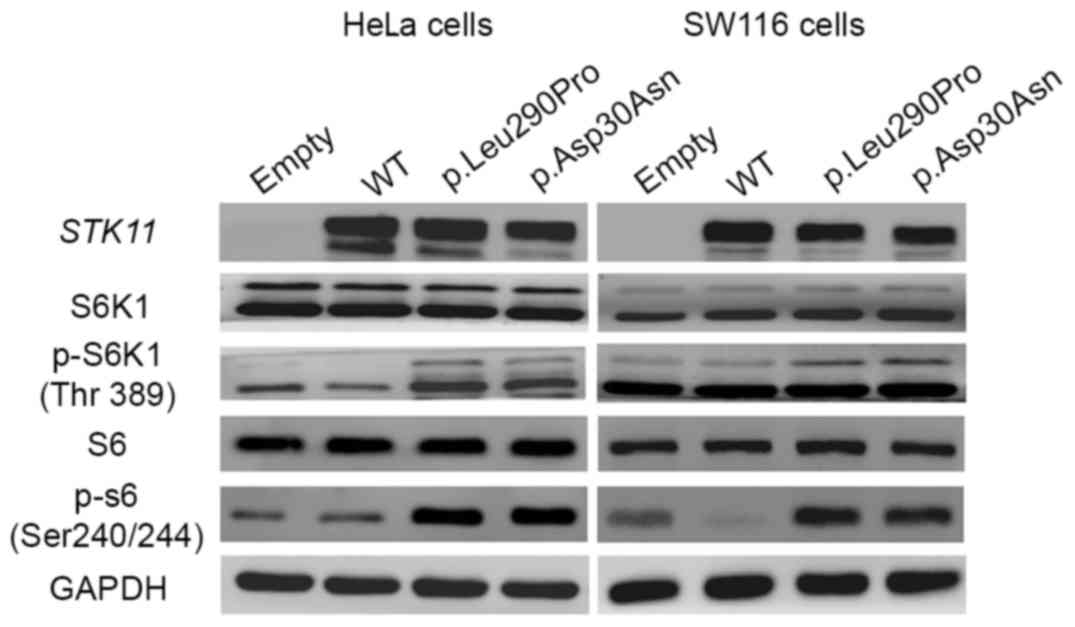 | Figure 4.Missense STK11 mutations
[c.869T>C (p.Leu290Pro) and c.88G>A (p.Asp30Asn)] induce
phosphorylation of S6K1 and S6. Western blotting of key
mTOR-mediated target genes in transfected cells. Expression of
p-S6K1 and p-S6 in cells transfected with STK11 mutant
p.Leu290Pro and STK11 mutant p.Asp30Asn vectors increased
compared with cells transfected with empty vectors and WT vectors.
Total levels of S6K1 and S6 remained unchanged. Asp, aspartic acid;
Asn, Asparagine; GAPDH, glyceraldehyde 3-phosphate dehydrogenase;
GFP, green fluorescent protein; Leu, leucine; mTOR, mammalian
target of rapamycin; p, phosphorylated; Pro, proline; Ser, serine;
STK11, serine threonine kinase 11; S6K1, ribosomal protein S6
kinase 1; Thr, threonine; WT, wild-type. |
Cell proliferation is promoted by two
novel STK11 missense mutations
To further determine whether the novel missense
STK11 mutations are able to effectively suppress cell growth and
proliferation, CCK-8 and colony formation assays were performed. As
shown in Fig. 5, HeLa and SW1116
cells transfected with wild-type vectors exhibited a marked
reduction in cell growth compared with cells transfected with the
STK11 mutant vectors. The growth of HeLa and SW1116 cells
transfected with the STK11 mutants (p.Leu290Pro and p.Asp30Asn) was
significantly increased (P<0. 01) when compared with the empty
vector control (Fig. 5A and C). These
results suggest that STK11 gene is a tumor suppressor gene
in the two cell lines. The two novel STK11 mutations
abrogated cellular growth suppression function of the STK11 gene,
which contributes to tumorigenesis. Furthermore, the effect of the
novel STK11 missense mutations on cell apoptosis was
investigated. Compared with the empty vector control, the wild-type
vector promoted apoptosis. However, there was no statistically
significant difference between the two novel missense STK11 mutants
and the control (data not shown). These findings indicate that
although the STK11 gene exerts a role in promoting apoptosis
in HeLa and SW1116 cells, the two novel STK11 missense
mutations have no significant effect.
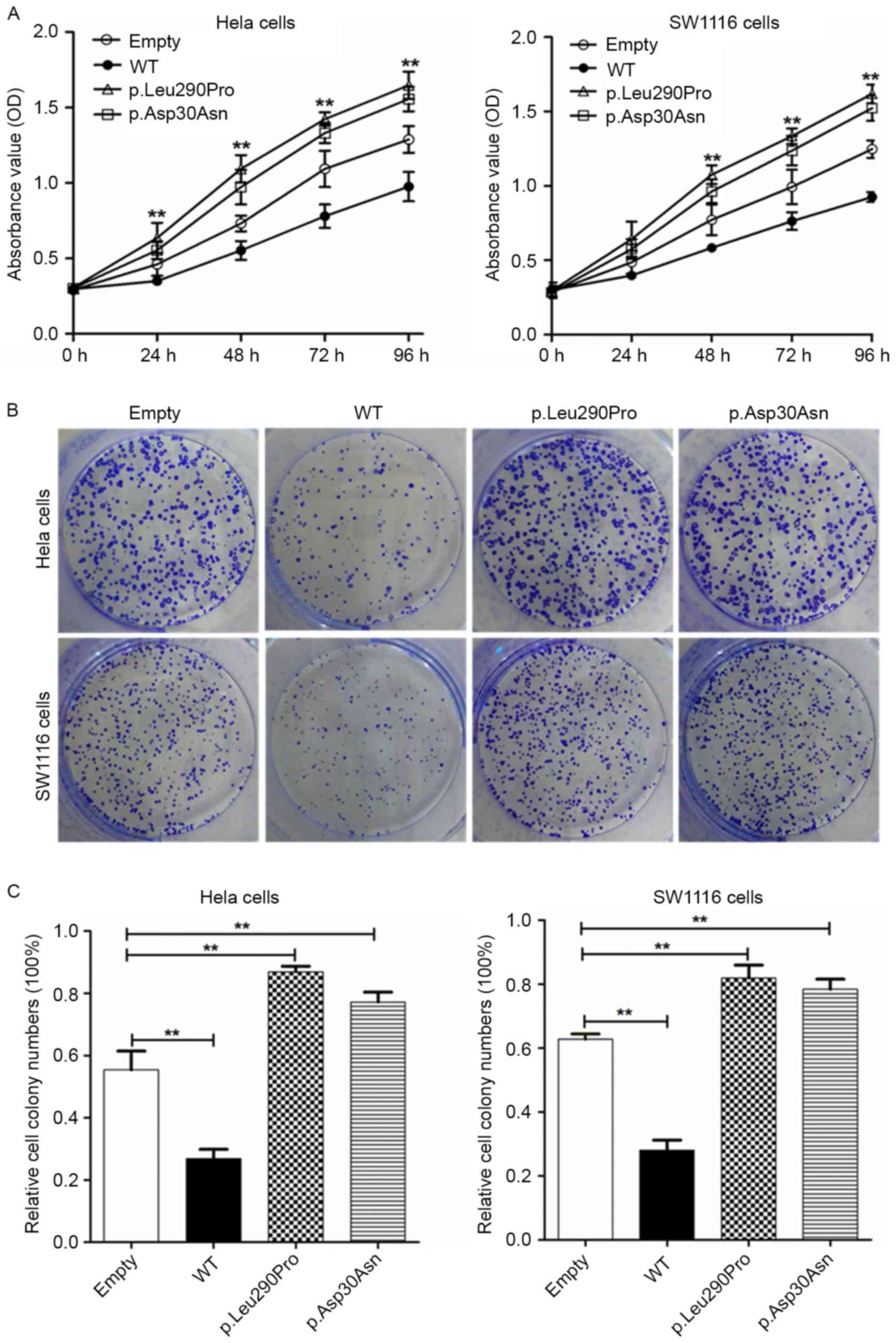 | Figure 5.Missense STK11 mutations
[c.869T>C (p.Leu290Pro) and c.88G>A (p.Asp30Asn)] promote
proliferation of HeLa and SW1116 cells. (A) Cell counting kit-8
assay revealed that the proliferative capacity of cells transfected
with STK11 mutant p. Leu290Pro vector was significantly
increased compared with cells transfected with WT vectors. (B)
Images of colony formation assays in HeLa and SW1116 cells
transfected with empty vector, WT, STK11 mutant p.Leu290Pro
and STK11 mutant p.Asp30Asn vectors. (C) Colony counts were
evaluated by determining the ratio between HeLa and SW1116 cells
transfected with empty, WT, STK11 mutant p.Leu290Pro or
STK11 mutant p.Asp30Asn vectors. Statistical analysis was
performed using a one-way analysis of variance between all groups.
The data are represented as the mean values of three independent
experiments. **P<0. 01. Asp, aspartic acid; Asn, Asparagine;
Leu, leucine; Pro, proline; STK11, serine threonine kinase 11; WT,
wild-type. |
Discussion
In the present study, a cohort of 21 affected PJS
patients were analyzed, which included index cases and family
members. Direct sequencing of the STK11 gene combined with
the MLPA assay for the detection of deletions resulted in a
mutation detection rate of 72.7% (8/11). This rate is consistent
with the 67.3% frequency reported previously (6). Notably, to the best of our knowledge, 3
of the mutations identified were novel, and therefore the present
study has extended the spectrum of known disease-causing
STK11 mutations.
The STK11 gene contains nine exons (27) encoding a 433-amino acid protein that
has a single catalytic kinase domain for adenosine 5′-triphosphate
binding, two N-terminal nuclear leading sequences that contain
nuclear localization signals and a C-terminal regulatory domain
that contains a prenylation motif (CAAX-box) (26,28). The
catalytic kinase domain is located at nucleotides 44 to 309
(26,28).
The novel PJS missense mutation, c.869T>C
(p.Leu290Pro), identified in the proband PJS4 II-3 in the present
study, leads to an amino acid alteration at codon 290 in exon 7,
which is located in the catalytic kinase region of the STK11
protein. The proband was diagnosed with colon adenocarcinoma at 56
years of age. It has been reported that the majority of reported
PJS-associated missense mutations are located in the STK11
kinase domain and are crucial to the function of the STK11
protein (3,7,29). These
mutations may lead to the loss of inhibition on cell growth
(30,31), which suggests that c.869T>C may be
pathogenic.
The other novel PJS missense mutation, c.88G>A,
identified in the proband PJS5 III-2, leads to an amino acid
alteration at codon 30 in exon 1 located at the N-terminal of the
STK11 protein. The proband PJS5 III-2 was diagnosed with a
breast intraductal papillary tumor and has a family with a history
of PJS. This change results in the substitution of aspartate by
asparagine. The N-terminal of the STK11 protein contains a
nuclear localization signal domain, which is required for the
interaction with other proteins in order to be actively exported
out of the nucleus (30,31). The translocation of STK11 from
the nucleus to the cytoplasm is associated with the activity of
STK11 kinase, which is closely dependent on the formation of
the STK11/Ste20-related adaptor/calcium-binding protein 39 complex
(29,31). Notably, a number of previously
reported PJS mutants are retained in the nucleus (3,32). This
suggests that that the mutation, c.88G>A, may affect the
translocation of STK11 from the nucleus to the cytoplasm,
and therefore, it is predicted to be pathogenic. Consistent with
the existing studies, a number of PJS patients carry germline
mutations that inactivate the kinase activity of STK11
(33).
The inactivation of STK11 kinase activity has also
been associated with an increased risk for the development of
cancer (34). This relative cancer
risk was estimated to be 9 to 18 times higher in PJS patients
compared with the general population (35,36). By
contrast, Hearle et al (1)
indicated that there is no significant correlation between the
incidence of a tumor and its type from an analysis of clinical data
of 419 patients with PJS.
The development of the PJS phenotype is speculated
to be due to the loss of the kinase activity of STK11, which is
associated with the loss of its growth suppression function
(32). Therefore, mutations that
affect the kinase activity of the STK11 protein are most likely to
contribute to the development of the PJS phenotype and potentially
to malignancy.
The tumor suppressor gene STK11 has an
important role in cell proliferation through a multitude of
targets, all of which may require the tumor suppressor function of
this kinase and/or its catalytic activity (8,11). Loss of
STK11 is able to activate certain downstream signaling
pathways to promote the growth and metastasis of tumors (14). Furthermore, it has been reported that
mTOR is a central node that acts to integrate different cell
signals to regulate cell growth (18). The kinase activity of STK11 protein
primarily downregulates the mTORC1 signaling pathway and prevents
the phosphorylation of the major downstream targets S6K1 and S6.
S6K1 and S6 have been identified as important cell proliferation
effectors, and phosphorylation of these targets leads to inhibition
of cell proliferation and tumorigenesis (10,12,13).
Furthermore, it has been reported that phosphorylation of S6K1
leads to an increased number of hamartomatous gastrointestinal
polyps in STK11 mutant mice (21,37),
suggesting that mTOR overactivation contributes to hamartomatous
tumor growth when STK11 is inactivated.
In the present study, immunohistochemical analysis
of two novel missense STK11 mutant tissue samples exhibited
an increased immunostaining signal for p-S6. This finding suggests
that inactivation of STK11 protein may result from novel missense
mutations, leading to dysregulation of the mTOR signaling pathway
and induction of S6 phosphorylation to promote protein synthesis
and cell proliferation. In order to further investigate the effects
of two novel missense mutations, cell viability was determined
in vitro. The results indicated that the two novel
STK11 missense mutants abrogated the tumor suppression
function of the STK11 gene, and promoted cell growth and
proliferation. In addition, western blotting demonstrated increased
phosphorylation of S6K1 and S6 proteins. These results indicated
that loss-of-function mutations in STK11 serine/threonine
kinase are able to hyperactivate the STK11-mTORC1 axis, which leads
to aberrant mTORC1-mediated phosphorylation of S6K1 and S6. This,
in turn, promotes cell proliferation and malignant transformation
(21,38).
Furthermore, STK11 has also been found to suppress
anti-apoptotic factors, including signal transducer and activator
of transcription 3, c-Jun N-terminal kinase 1, c-Myc, k-Ras,
mitogen-activated protein kinase and cyclooxygenase-2, to inhibit
cell survival (39,40). STK11 is able to induce cell
apoptosis under metabolic stresses, including hypoxia and energy
deprivation (8,12). Previous studies reported the absence
of STK11 staining and reduced numbers of apoptotic cells in
polyps derived from PJS patients (41,42). In
the present study, it was demonstrated that transfection of the
wild-type STK11 vector was able to promote cell apoptosis.
The two novel STK11 missense mutations, located at the
catalytic kinase region and the N-terminal of the STK11
protein, are associated with the impaired function of STK11
protein and are therefore pathogenic.
Although the detailed mechanisms of STK11
gene function as a tumor suppressor remains to be fully understood,
STK11 has been shown to control cell growth via regulating
mTOR-associated signaling pathways and cellular responses (18). For example, activation of mTORC1
stimulates angiogenesis by stabilizing hypoxia-inducible factor 1α
under hypoxic conditions (43).
Furthermore, activation of mTORC1 inhibits autophagy via
phosphorylation of autophagy-related protein 13 and ULK1/2
(44). Rapamycin, a specific mTOR
inhibitor, has been developed as an effective drug for the
treatment of PJS and its associated tumors (22,45).
Taken together, the two novel missense mutations of
STK11 identified in the present study are able to disrupt
the tumor suppression function of STK11 and impair
STK11 serine/threonine protein kinase activity. Therefore,
the major downstream molecules of the mTOR signaling pathway, S6K1
and S6, may be potential therapeutic targets for
STK11-associated cancer.
Acknowledgements
The present study was supported by the Guangzhou
Pilot Project of Clinical and the Translational Research Center
(Early Gastrointestinal Cancers; grant no. 7415696196402), the
Guangdong Provincial Bioengineering Research Center for
Gastroenterology Diseases, the National Natural Science Foundation
of China (grant no. 81401925), the Natural Science Foundation of
Guangdong Province (grant no. 2015A030310102) and the President
Foundation of Nanfang Hospital, Southern Medical University (grant
no. 2013B005).
Glossary
Abbreviations
Abbreviations:
|
PJS
|
Peutz-Jeghers syndrome
|
|
STK11
|
serine threonine kinase 11
|
|
mTOR
|
mammalian target of rapamycin
|
|
S6K1
|
ribosomal protein S6 kinase 1
|
References
|
1
|
Hearle N, Schumacher V, Menko FH,
Olschwang S, Boardman LA, Gille JJ, Keller JJ, Westerman AM, Scott
RJ, Lim W, et al: Frequency and spectrum of cancers in the
Peutz-Jeghers syndrome. Clin Cancer Res. 12:3209–3215. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McKay V, Cairns D, Gokhale D, Mountford R
and Greenhalgh L: First report of somatic mosaicism for mutations
in STK11 in four patients with Peutz-Jeghers syndrome. Fam Cancer.
15:57–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jenne DE, Reomann H, Nezu J, Friedel W,
Loff S, Jeschke R, Müller O, Back W and Zimmer M: Peutz-Jeghers
syndrome is caused by mutations in a novel serine threonine kinase.
Nat Genet. 18:38–43. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hemminki A, Markie D, Tomlinson I,
Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M,
Höglund P, et al: A serine/threonine kinase gene defective in
Peutz-Jeghers syndrome. Nature. 391:184–187. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang Z, Ellis I, Zauber P, Iwama T,
Marchese C, Talbot I, Xue WH, Yan ZY and Tomlinson I: Allelic
imbalance at the LKB1 (STK11) locus in tumours from patients with
Peutz-Jeghers' syndrome provides evidence for a
hamartoma-(adenoma)-carcinoma sequence. J Pathol. 188:9–13. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Z, Wu B, Mosig RA, Chen Y, Ye F,
Zhang Y, Gong W, Gong L, Huang F, Wang X, et al: STK11 domain XI
mutations: Candidate genetic drivers leading to the development of
dysplastic polyps in Peutz-Jeghers syndrome. Hum Mutat. 35:851–858.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ylikorkala A, Avizienyte E, Tomlinson IP,
Tiainen M, Roth S, Loukola A, Hemminki A, Johansson M, Sistonen P,
Markie D, et al: Mutations and impaired function of LKB1 in
familial and non-familial Peutz-Jeghers syndrome and a sporadic
testicular cancer. Hum Mol Genet. 8:45–51. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao RX and Xu ZX: Targeting the LKB1
tumor suppressor. Curr Drug Targets. 15:32–52. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boudeau J, Sapkota G and Alessi DR: LKB1,
a protein kinase regulating cell proliferation and polarity. FEBS
Lett. 546:159–165. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vaahtomeri K and Mäkelä TP: Molecular
mechanisms of tumor suppression by LKB1. FEBS Lett. 585:944–951.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Korsse SE, Peppelenbosch MP and van Veelen
W: Targeting LKB1 signaling in cancer. Biochim Biophys Acta.
1835:194–210. 2013.PubMed/NCBI
|
|
12
|
Corradetti MN, Inoki K, Bardeesy N,
DePinho RA and Guan KL: Regulation of the TSC pathway by LKB1:
Evidence of a molecular link between tuberous sclerosis complex and
Peutz-Jeghers syndrome. Gene Dev. 18:1533–1538. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shaw RJ, Bardeesy N, Manning BD, Lopez L,
Kosmatka M, DePinho RA and Cantley LC: The LKB1 tumor suppressor
negatively regulates mTOR signaling. Cancer Cell. 6:91–99. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou W, Marcus AI and Vertino PM:
Dysregulation of mTOR activity through LKB1 inactivation. Chin J
Cancer. 32:427–433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Magnuson B, Ekim B and Fingar DC:
Regulation and function of ribosomal protein S6 kinase (S6K) within
mTOR signalling networks. Biochem J. 441:1–21. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fenton TR and Gout IT: Functions and
regulation of the 70kDa ribosomal S6 kinases. Int J Biochem Cell
Biol. 43:47–59. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meyuhas O and Dreazen A: Ribosomal protein
S6 kinase: From TOP mRNAs to cell size. Progress in molecular
biology and translational science. Hershey J: 109–153. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zoncu R, Efeyan A and Sabatini DM: mTOR:
From growth signal integration to cancer, diabetes and ageing. Nat
Rev Mol Cell Biol. 12:21–35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Inoki K, Kim J and Guan K: AMPK and mTOR
in cellular energy homeostasis and drug targets. Annu Rev Pharmacol
Toxicol. 52:381–400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shaw RJ: LKB1 and AMP-activated protein
kinase control of mTOR signalling and growth. Acta Physiol (Oxf).
196:65–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wei C, Amos CI, Zhang N, Wang X, Rashid A,
Walker CL, Behringer RR and Frazier ML: Suppression of
Peutz-Jeghers polyposis by targeting mammalian target of rapamycin
signaling. Clin Cancer Res. 14:1167–1171. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van Veelen W, Korsse SE, van de Laar L and
Peppelenbosch MP: The long and winding road to rational treatment
of cancer associated with LKB1/AMPK/TSC/mTORC1 signaling. Oncogene.
30:2289–2303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boardman LA, Couch FJ, Burgart LJ,
Schwartz D, Berry R, McDonnell SK, Schaid DJ, Hartmann LC,
Schroeder JJ, Stratakis CA and Thibodeau SN: Genetic heterogeneity
in Peutz-Jeghers syndrome. Hum Mutat. 16:23–30. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mehenni H, Resta N, Guanti G, Mota-Vieira
L, Lerner A, Peyman M, Chong KA, Aissa L, Ince A, Cosme A, et al:
Molecular and clinical characteristics in 46 families affected with
peutz-jeghers syndrome. Dig Dis Sci. 52:1924–1933. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boudeau J, Kieloch A, Alessi DR, Stella A,
Guanti G and Resta N: Functional analysis of LKB1/STK11 mutants and
two aberrant isoforms found in Peutz-Jeghers Syndrome patients. Hum
Mutat. 21:1722003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hemminki A, Tomlinson I, Markie D,
Järvinen H, Sistonen P, Björkqvist AM, Knuutila S, Salovaara R,
Bodmer W, Shibata D, et al: Localization of a susceptibility locus
for Peutz-Jeghers syndrome to 19p using comparative genomic
hybridization and targeted linkage analysis. Nat Genet. 15:87–90.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sanchez-Cespedes M: A role for LKB1 gene
in human cancer beyond the Peutz-Jeghers syndrome. Oncogene.
26:7825–7832. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boudeau J, Baas AF, Deak M, Morrice NA,
Kieloch A, Schutkowski M, Prescott AR, Clevers HC and Alessi DR:
MO25alpha/beta interact with STRADalpha/beta enhancing their
ability to bind, activate and localize LKB1 in the cytoplasm. EMBO
J. 22:5102–5114. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smith DP, Spicer J, Smith A, Swift S and
Ashworth A: The Mouse Peutz-Jeghers Syndrome gene Lkbl encodes a
nuclear protein kinase. Hum Mol Genet. 8:1479–1485. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Baas AF, Boudeau J, Sapkota GP, Smit L,
Medema R, Morrice NA, Alessi DR and Clevers HC: Activation of the
tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD.
EMBO J. 22:3062–3072. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mehenni H, Gehrig C, Nezu J, Oku A,
Shimane M, Rossier C, Guex N, Blouin JL, Scott HS and Antonarakis
SE: Loss of LKB1 kinase activity in Peutz-Jeghers syndrome, and
evidence for allelic and locus heterogeneity. Am J Hum Genet.
63:1641–1650. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alessi DR, Sakamoto K and Bayascas JR:
LKB1-dependent signaling pathways. Annu Rev Biochem. 75:137–163.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miyoshi H, Nakau M, Ishikawa T, Seldin MF,
Oshima M and Taketo MM: Gastrointestinal hamartomatous polyposis in
Lkb1 heterozygous knockout mice. Cancer Res. 62:2261–2266.
2002.PubMed/NCBI
|
|
35
|
Mehenni H, Resta N, Park JG, Miyaki M,
Guanti G and Costanza MC: Cancer risks in LKB1 germline mutation
carriers. Gut. 55:984–990. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van Lier MGF, Westerman AM, Wagner A,
Looman CW, Wilson JH, de Rooij FW, Lemmens VE, Kuipers EJ,
Mathus-Vliegen EM and van Leerdam ME: High cancer risk and
increased mortality in patients with Peutz-Jeghers syndrome. Gut.
60:141–147. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shackelford DB, Vasquez DS, Corbeil J, Wu
S, Leblanc M, Wu CL, Vera DR and Shaw RJ: mTOR and
HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of
Peutz-Jeghers syndrome. Proc Natl Acad Sci USA. 106:pp.
11137–11142. 2009; View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sanchez-Cespedes M, Parrella P, Esteller
M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG and Sidransky
D: Inactivation of LKB1/STK11 is a common event in adenocarcinomas
of the lung. Cancer Res. 62:3659–3662. 2002.PubMed/NCBI
|
|
39
|
Partanen JI, Nieminen AI, Mäkelä TP and
Klefstrom J: Suppression of oncogenic properties of c-Myc by
LKB1-controlled epithelial organization. Proc Natl Acad Sci USA.
104:pp. 14694–14699. 2007; View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee JH, Koh H, Kim M, Park J, Lee SY, Lee
S and Chung J: JNK pathway mediates apoptotic cell death induced by
tumor suppressor LKB1 in Drosophila. Cell Death Differ.
13:1110–1122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zeng PY and Berger SL: LKB1 is recruited
to the p21/WAF1 promoter by p53 to mediate transcriptional
activation. Cancer Res. 66:10701–10708. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Karuman P, Gozani O, Odze RD, Zhou XC, Zhu
H, Shaw R, Brien TP, Bozzuto CD, Ooi D, Cantley LC and Yuan J: The
Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell
death. Mol Cell. 7:1307–1319. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hudson CC, Liu M, Chiang GG, Otterness DM,
Loomis DC, Kaper F, Giaccia AJ and Abraham RT: Regulation of
hypoxia-inducible factor 1alpha expression and function by the
mammalian target of rapamycin. Mol Cell Biol. 22:7004–7014. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Moschetta M, Reale A, Marasco C, Vacca A
and Carratu MR: Therapeutic targeting of the mTOR-signalling
pathway in cancer: Benefits and limitations. Br J Pharmacol.
171:3801–3813. 2014. View Article : Google Scholar : PubMed/NCBI
|














